

Abstract
Achromatopsia is a genetic disorder of cones, and one of the most common forms is a channelopathy caused by mutations in the β-subunit, CNGB3, of the cone cyclic nucleotide-gated (CNG) channel. Recombinant adeno-associated virus of serotype 5 (rAAV5)-mediated gene transfer of human CNGB3 cDNA to mutant dog cones results in functional and structural rescue in dogs <0.5 years of age, but treatment is minimally effective in dogs >1 year. We now test a new therapeutic concept by combining gene therapy with the administration of ciliary neurotrophic factor (CNTF). Intravitreal CNTF causes transient dedifferentiation of photoreceptors, a process called deconstruction, whereby visual cells become immature with short outer segments, and decreased retinal function and gene expression that subsequently return to normal. Cone function was successfully rescued in all mutant dogs treated between 14 and 42 months of age with this strategy. CNTF-mediated deconstruction and regeneration of the photoreceptor outer segments prepares the mutant cones optimally for gene augmentation therapy.
Introduction
Recent breakthroughs in retinal gene therapy provide hope of effective treatments for inherited blinding disorders once considered incurable. For example, Leber congenital amaurosis, caused by loss of function of the RPE65 gene, is being successfully treated by delivery of RPE65 to the retinal pigment epithelium via recombinant adeno-associated virus (rAAV)-based vectors.1,2,3,4,5 This promising therapy evolved from proof of concept studies using rAAV-based viral vectors that established efficacy and preliminary safety in directing gene expression to the retinal pigment epithelium in dogs, mice, and primates.6,7,8,9 The same technology is now used successfully to treat primary photoreceptor disorders in animal models, among which include achromatopsia and different forms of retinitis pigmentosa.10,11,12,13,14,15,16,17,18
Gene augmentation therapy, however, has been marginally effective or ineffective when the photoreceptor degeneration starts too early or progresses too rapidly.19,20,21 To circumvent this problem, rAAV vectors of different serotypes have been developed, e.g., rAAV8 versus rAAV5, with tyrosine-capsid mutations and/or self-complementary variants that minimize the interval between transduction and transgene expressions.18,22,23,24
An intermediate situation occurs when the degeneration rate is slow, but functional and/or structural rescue of the photoreceptor disease is not possible. Such is the case in CNGB3-achromatopsia, a channelopathy affecting cone photoreceptors in humans,25,26 dogs,27 and mice,28 where treatment outcomes were age and disease stage dependent.10,14 In dogs, we found that successful restoration of cone function decreased from 11 of 14 eyes treated at <0.5 year of age, to 1 of 3 in those >1 year.10 The decreased rescue of cone function could not be explained by the absence of cones or the lack of CNGB3 transgene expression. As successfully treated cones showed localization of cyclic nucleotide-gated (CNG) channel and G-protein expression in the outer segments, in contrast to their absence in treated but non-functional retinas, we suspected that the failure possibly resulted from the improper reassembly of the components of the phototransduction machinery when retinas were treated at later stages of disease.10
In this study, we tested the hypothesis that regenerating the older CNGB3-mutant cone outer segments with ciliary neurotrophic factor (CNTF) could enable restoration of cone phototransduction following gene augmentation therapy with the rAAV5-PR2.1-hCNGB3 therapeutic vector. CNTF is one of the most studied neurotrophic factors in retinal degenerative disorders and has profound effects on the retina and photoreceptors.29 When administered intravitreally, the outer segments shorten significantly before regrowing, and photoreceptor gene expression is transiently downregulated.30 Because the changes are reversible, and the treated photoreceptors transiently become more immature, we have termed the process transient photoreceptor deconstruction.31,32 Recent studies indicate that CNTF also promotes cone outer segment regeneration in the degenerating rat retina.33
Our present results clearly show that intravitreal injection of human recombinant CNTF one week before treatment with rAAV5-PR2.1-hCNGB3 in CNGB3-mutant dogs >1 year of age permitted restoration of cone function in all treated eyes, whereas no functional recovery was found in control eyes pretreated with phosphate-buffered saline (PBS). Our results provide proof of concept for a novel combination therapy to facilitate restoration of cone function in future clinical application. They also provide an insight into the mechanism that mediates the assembly of CNG channels, and their targeting to the cone outer segments in the disease.
Results
Subretinal rAAV5-PR2.1-hCNGB3 transduces older mutant cones but fails to restore function
Subretinal administration of the therapeutic rAAV serotype 5 vector with hCNGB3 regulated by the full-length human red cone opsin promoter, rAAV5-PR2.1-hCNGB3, resulted in cone functional rescue in only 1 of 10 treated eyes of mutant dogs >1 year of age (Table 1). This finding was the same in dogs with either the genomic deletion or D262N missense mutation,27 and was observed with treatment by subretinal injection alone (n = 3; prior study),10 or one week following intravitreal PBS injection (n = 7; Figure 1a). The failure of functional rescue was not due to lack of cones. Retinal flat mounts at 1 year of age showed an ~25% average loss of both L/M- and S-cones (Figure 2a). Loss is greater in the inferior and nasal quadrants, and cones are best preserved in the area centralis region that was targeted for treatment (Figure 2a).34 Over a 7-year time period, we find a continuous, but slow loss of cones. Even in older animals, however, some intact cones with shortened outer segments remain, confirming previous observations (Figure 2b).35
Table 1. Treatment of CNGB3-mutant dogs.
Figure 1.
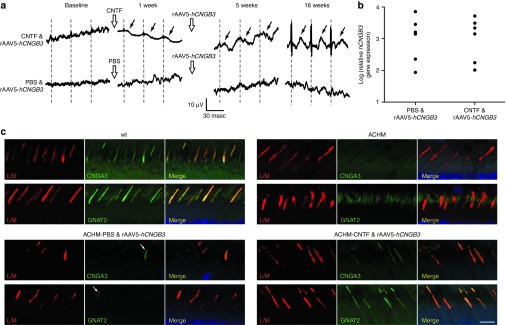
Effect of intravitreal CNTF administration and subretinal rAAV5-PR2.1-hCNGB3 on the CNGB3-mutant retina. (a) Gene augmentation in a CNGB3−/−-mutant dog (28.3 months) rescues 29-Hz flicker cone responses when preceded by intravitreal CNTF injection (top row). No rescue of cone function was achieved when intravitreal PBS was used before gene therapy (bottom row). The dashed lines indicate stimulus onset and oblique arrows the individual cone-mediated flicker responses. (b) The relative amounts of retinal hCNGB3 mRNA expression were comparable and not significantly different when subretinal rAAV injections were preceded by either intravitreal PBS (no cone function) or CNTF (cone function). (c) Immunolabeling with CNGA3 and GNAT2 shows absence of cone outer segment labeling in untreated mutants (illustrated for a 10-month-old CNGB3m/m retina) even though L/M-cone opsin label is present. With few exceptions (arrows) cone outer segments following the treatment with PBS & rAAV5-PR2.1-hCNGB3 are also negative. Normal retinas and those treated with CNTF & rAAV5-PR2.1-hCNGB3 have normal outer segment immunolabeling. Calibration bar = 10 µm. ACHM, achromatopsia-affected; CNTF, ciliary neurotrophic factor; msec, milli second; wt, wild-type.
Figure 2.

Changes in cones with age and disease progression. (a) Calculated loss of L/M- and S-cone densities at 1 year of age are shown separately for four quadrants based on cone counts from selected regions of retinal flat mounts from wild-type (wt) and CNGB3−/−-mutant dogs. The decrease in cone density for both classes is most significant in the peripheral regions of the inferior, nasal, and superior quadrants. The loss is ≤10% and not significant in the area centralis located temporal to the optic disc. (b) Compared with wt controls, mutant (ACHM) retinas show gradual loss of cones (labeled red for human cone arrestin, hCAR), preceded by shortening and disruption of their inner and outer segments. There is migration of hCAR-labeled cone nuclei into the subretinal space adjacent to the retinal pigment epithelium (arrows, ectopic cone nuclei). Cell nuclei are shown in blue with DAPI. *P < 0.05, **P < 0.0001. Calibration bar = 20 µm. Dog fundus drawing reprinted with permission from Dr Kerry Ketring. IS, inner segments; T, temporal; N, nasal; OS, outer segments; ONL, outer nuclear layer; OPL, outer plexiform layer.
Likewise, the lack of functional rescue in older mutants was not the result of low or absent transduction efficiency. When incorporated into the rAAV5 vector, the PR2.1 promoter results in specific expression of reporter gene in normal or CNGB3-mutant L/M cones.10,36 Furthermore, quantitative real-time PCR (qRT-PCR) analysis of hCNGB3 expression 18 to 46 weeks after treatment that resulted in no cone functional recovery showed high expression levels (Figure 1b), and comparable with those previously reported for successfully treated younger eyes.10
Intravitreal CNTF enhances rAAV5-mediated cone functional rescue in older mutant retinas
To determine if pretreatment with CNTF improved functional rescue in older retinas, we injected CNTF intravitreally 1 week before rAAV5-PR2.1-hCNGB3 administration in 14.3- to 42-month-old mutants, and assessed functional outcomes by recording the isolated cone electroretinogram (ERG) responses. All seven dogs treated in this manner had a robust and stable recovery of cone function detected by 4–5 (n = 4) and 7.5–9 (n = 3) weeks after gene therapy (Figure 1a and Table 1). Expression of the hCNGB3 transgene in successfully treated (CNTF and therapeutic vector) older mutant retinas was comparable with successfully treated younger eyes,10 and to retinas pretreated with PBS that showed no functional cone rescue (Figure 1b). Hence, the restoration of cone function in older CNGB3-mutant dogs is not solely depend on the expression of the hCNGB3 transgene mRNA.
We have not been able to determine the expression of hCNGB3 transgene product in treated retinas as multiple antibodies produced have not been specific. However, surrogate markers of effective cone transduction, e.g., GNAT2, CNGA3, can be used.10 In the normal retina, both show localization restricted to the outer segments, whereas in mutants, the proteins are expressed but delocalized from the cone outer segments.10,35,37 We find that restored cone function in CNTF/rAAV5-treated CNGB3-mutant adult retinas is associated with normal localization of both GNAT2 and CNGA3 in cone outer segments, but this is not found in PBS/rAAV5-treated cones (Figure 1c).
Intravitreal CNTF and reversible photoreceptor deconstruction
To better understand the effect of CNTF in enhancing functional rescue in CNGB3-mutant cones, we first examined the photoreceptor effects and potential signaling pathways involved in normal canine retinas (see Supplementary Table S1 for details of procedures). Consistent with previous observations in rodents,30 intravitreal CNTF had a dramatic effect on retinal function, with decreases in both rod- and cone-mediated ERG response amplitudes (Figure 3a). This effect was first noted at 3 days, peaked at 1 week, and recovered to normal by 5 weeks after injection (Figure 3b). The ERG effects corresponded temporally with changes in photoreceptor structure and gene product expression. At 1 week, both rod and cone outer segments shortened dramatically throughout the retina. S-cones appeared more severely affected at the 1-week time point as hardly any S-cone outer segments were identifiable (Figure 4). Electron microscopy demonstrated that, in addition to outer segment shortening, there was membrane disorganization and vesiculation of both rod and cone outer segments (Supplementary Figure S1). By 2 weeks, outer segments were longer but appeared slightly disorganized, and photoreceptors were normal 5 weeks after injection (Figure 4).
Figure 3.

Intravitreal CNTF results in a transient decrease in rod- and cone-mediated ERG responses in normal retina. (a) In comparison with intravitreal PBS that had no effect on the ERG (gray traces), intravitreal CNTF (black traces) markedly reduced rod and cone amplitudes. Retinal function was most severely affected at 1 week, partially restored at 2 weeks, and returned to normal at 5 weeks after injection. (b) Scatterplots showing relative ERG amplitudes over time after injection (relative amplitude = amplitude CNTF-treated eye/amplitude contralateral PBS-treated eye). The depression of retinal responses was first noted at 3 days after CNTF injection, peaked at 1 week, and recovered to normal by 5 weeks. The linear traces connect the median values between each time point. CNTF, ciliary neurotrophic factor; msec, milli second.
Figure 4.

Intravitreal CNTF leads to a transient shortening of photoreceptor outer segments in the normal canine retina. Compared with PBS-injected control eyes, CNTF causes a uniform shortening of cone (hCAR) and rod (Rho) outer segments at 1 week. This effect was more dramatic for the S-cones (S) than for the L/M-cones (L/M) since hardly any labeled S-cone outer segments could be identified at 1 week. At 2 weeks, the photoreceptors appear partially recovered, and are normal at 5 weeks. Cell nuclei are shown in blue with DAPI. Calibration bar = 20 µm. CNTF, ciliary neurotrophic factor; hCAR, human cone arrestin; INL, inner nuclear layer; IS, inner segments; L/M, L/M-opsin; ONL, outer nuclear layer; OPL, outer plexiform layer; OS, outer segments; S, S-opsin.
We then examined the gene expression levels of outer segment visual pigment proteins and CNG channel subunits of rods and cones, and found a marked decrease in photoreceptor gene expression 1 week following CNTF that appeared to fully recover by 2 weeks (Figure 5a,b). With the exception of S-opsin expression which was ~0.1% of the biological control group, cone-specific gene expression decreased to ~24–39% and rod-specific gene expression to ~14–19% at 1 week (Figure 5a,b).
Figure 5.

Intravitreal CNTF leads to a transient reduction of photoreceptor-specific gene expression. Relative (a) cone and (b) rod gene expression measured by qRT-PCR is shown following either intravitreal CNTF or PBS injection compared with a normal biologic control group (n = 3). CNTF treatment results in gene expression levels that are ~14–39% of normal for most genes at 1 week. S-cone opsin levels were affected to the greatest extent (~0.1% of normal). Expression was comparable with PBS injected eyes by 2 and 5 weeks. CNTF, ciliary neurotrophic factor; qRT-PCR, quantitative real-time PCR.
We found no increased labeling of cells in normal and CNGB3-mutant retinas for phosphohistone-H3 (PHH3),38 ruling out CNTF-induced cell proliferation. Furthermore, TUNEL labeling38,39 was negative following CNTF treatment, emphasizing that there was no cell death associated with treatment despite the dramatic structural changes.
As the Jak/Stat pathway is presumably activated following treatment with CNTF29 in the Müller glia and photoreceptor cells that express the α-subunit of the CNTF receptor (CNTFRα40), we next assessed this pathway. Both STAT1 and STAT3 mRNA expression levels were elevated at 1 week, remained elevated at 2 weeks, and returned to normal at 5 weeks (Figure 6a). Levels of phosphorylated STAT1 and STAT3 were elevated at 1 week, but by 2 weeks had returned to baseline (pSTAT1) or remained slightly elevated (pSTAT3) (Figure 6b,c). CNTF also led to upregulation of STAT1 expression at 1, 2, and 5 weeks (Figure 6b,c). In the retina, phosphorylated STAT was present in the vitreal margin of the inner nuclear layer, and in photoreceptor cells. As with western blots, peak expression was at 1 week, and decreased to control levels by 5 weeks (Figure 6d). These results show that the intravitreal injection of CNTF in the normal canine retina results in activation of the Jak/STAT pathway in photoreceptors and other inner nuclear layer cells.
Figure 6.

Intravitreal CNTF injection activates the Jak/Stat pathway in the normal canine retina. (a) Compared with intravitreal PBS, both STAT1 and STAT3 mRNA expression levels were elevated 1 week after CNTF, remained elevated at 2 weeks, and returned to normal levels at 5 weeks. At the protein level, total (b) STAT1 expression was upregulated 1, 2, and 5 weeks after CNTF injection, and both (b) STAT1 and (c) STAT3 became phosphorylated 1 week after CNTF. By 2 weeks, the levels had returned to baseline (pSTAT1) or remained slightly elevated (pSTAT3). (d) Both proteins were present in the inner margin of the inner nuclear layer, presumably in Müller cells, and in the outer nuclear layer. As with western blots, peak expression was at 1 week, and decreased to control levels by 5 weeks. Calibration bar = 20 µm. CNTF, ciliary neurotrophic factor; GCL, ganglion cell layer; INL, inner nuclear layer; IPL, inner plexiform layer; NFL, nerve fiber layer; ONL, outer nuclear layer; OPL, outer plexiform layer; PBS, phosphate-buffered saline; PR, photoreceptors; RPE, retinal pigment epithelium.
In the mutant retina, surprisingly, we found that CNTF by itself also induced a partial restoration of cone function in older animals. One to 2.5 weeks after CNTF treatment, four of four eyes had low amplitude cone flicker ERG responses with prolonged implicit times (Table 1 and Figures 1a and 7). This effect was short-lived, and cone signals were lost after 5 weeks in the absence of staged rAAV5-PR2.1-hCNGB3 treatment (Table 1 and Figure 7).
Figure 7.

Full-field ERGs recorded from developing CNGB3 mutants, and adult CNGB3 mutants treated with CNTF. (a) Responses recorded from a mature wild-type dog for comparison. (b) Responses were comparable during retinal development between the dogs with the genomic deletion (CNGB3−/−) and the D262N missense mutation (CNGB3m/m) of CNGB3. Cone-mediated function (oblique arrows) is present at 4 and 6 weeks, but nonrecordable at 8 weeks. Rod function development is normal. (c) CNTF treatment of mutant dogs results in transient recovery of the 1 and 29-Hz cone flicker responses (oblique arrows) by 1 week, but these are lost by 5 weeks. Note that intravitreal CNTF results also in dramatic decreases in rod and mixed rod-cone response amplitudes. The arrows point to cone-mediated function elicited under light adaptation with single flashes (Cone—1 Hz) or fast repeating flashes (Cone—29 Hz). Note that the µV scales are different in c. CNTF, ciliary neurotrophic factor; msec, milli second.
In the dog, retinal development occurs postnatally with gradual maturation of ERG responses.41 A similar process occurs in CNGB3 mutants, and immature, albeit abnormal, cone responses are present only between 4 and 6 weeks of age before being permanently lost (Figure 7). In adult mutants, CNTF treatment resulted in cone responses that were similar to, but of lower amplitude, than those recorded from mutant retinas in the early phases of postnatal differentiation. We posit that this transient appearance of cone function occurs from partially functional CNGA3 homotetramer channels that form following the regrowth of cone outer segments after CNTF administration.42,43 Indeed, the immunohistochemical evaluation of a CNGB3-mutant retina with partially restored cone-mediated function 1 week following CNTF treatment (M635 in Table 1) revealed restoration of weak expression of both CNGA3 and GNAT2 in some of the deconstructed cone outer segments (Supplementary Figure S2).
Discussion
Our present work clearly shows that a combined staged therapy of CNTF and rAAV5-PR2.1-hCNGB3 successfully restores cone function in two canine models of CNGB3-achromatopsia at older ages (14–42 months) when, in the majority of cases, gene augmentation therapy alone cannot. Our data thus provide proof of concept for potential CNTF and gene augmentation combination therapy for restoring cone function in older patients with CNGB3-achromatopsia if standard gene augmentation therapy is not effective. In addition, understanding the mechanistic basis for the CNTF effect will provide essential insights for future understanding of the disease progression in CNGB3-achromatopsia, and the mechanism that mediates the CNG channel assembly and targeting to the cone outer segments.
Although only ~5% of the photoreceptors in the human retina are cones,44 they are essential for color vision, central visual acuity and day vision, and loss of their function leads to severe visual dysfunction and impaired quality of life. To this end, we have selected achromatopsia, an autosomal-recessive cone visual function disorder, as a test platform to develop gene therapy that broadly may be applicable to cone-specific diseases. The majority of human achromatopsia patients have a channelopathy caused by mutations of either the α- or β-subunits of the cone CNG channels, responsible for the last step in phototransduction.25,26,42,45 The present studies were carried out in canine models with either a missense mutation in exon 6 or a genomic deletion of CNGB3,27 both of which are functional nulls based on disease phenotype and response to therapy.10 As in human patients with achromatopsia,46 the loss of the non-functional cones in canine CNGB3-achromatopsia is slow, and adequate numbers of cones are present, even at advanced ages, to permit functional recovery provided treatment is effective. Thus, preclinical studies, including our present work on the CNGB3-achromatopsia models, may have broad implication not only on treating CNGB3-achromatopsia in humans, but also other cone-specific diseases.
Proof of principle studies have demonstrated successful cone-directed therapy in mouse and canine achromatopsia models (mouse: GNAT2,12 CNGA3,13,15 and CNGB3;14 dog: CNGB3).10 However, despite the very slow cone loss in CNGB3-mutant dogs and the successful treatment by gene augmentation in young animals, treatment with the same therapeutic vector is ineffective in animals >1 year.10 Carvalho and colleagues have demonstrated a small age effect when assessing outcome of gene therapy in CNGB3-mutant mice; there was both a decrease in visual function and cone ERG amplitude recovery with increasing age at the time of treatment.14 Similar to the CNGB3-mutant dogs,10 cone function was restored in mice up to 6 months of age with gene therapy alone.14 Older mice were not treated, and visual acuity could not be improved in the 6-month old animals.14 Our present work demonstrates that the lack of functional restoration in older animals is not due to the loss of cones. Neither is it because of lack of therapeutic transgene expression, indicating that viral vector tropism and transduction efficiency are not limiting factors (Komáromy and colleagues10 and present study). Instead, our results point to an age-related defect in the ability of the hCNGB3 transgene protein to either assemble with CNGA3 or promote localization of the heterodimeric complexes to the outer segment. Alternatively, the deterioration of cone outer segments found in some older mutant retinas indicates that those cells are in the early stages of degeneration, and not capable of assembling structural components of the outer segment.33 The two explanations are not mutually exclusive.
As a means of developing a treatment for older animals that had adequate numbers of viable cones whose function potentially could be rescued, we used intravitreal bolus administration of CNTF to transiently deconstruct the mutant cones before gene transfer. We reasoned that such treatment could potentially “rejuvenate” the cone outer segment, allowing for proper assembly of the membrane subunits of the CNG channel, thereby permitting functional recovery following therapy. In support, intravitreal bolus administration of CNTF successfully modified cone cells in the retinas of older mutants which then functioned, albeit abnormally, like those of younger mutants. After rAAV-PR2.1-hCNGB3 treatment, assembled channels were correctly targeted to the outer segment, leading to successful functional recovery. We posit that deconstruction induced by CNTF, either concurrent with or after vector administration, is likely to also lead to recovery of cone function. This hypothesis needs to be tested experimentally, and investigations are currently underway to establish the most effective treatment method. Although our current data suggest that the transient photoreceptor deconstruction may be mediated by the Jak/Stat pathway, the exact mechanism leading to the restoration of cone function with CNTF treatment in the older animals requires more investigation. As an example, the use of CNTF could be combined with Jak or Stat inhibitors to support or exclude the Jak/Stat pathway as a mediator of this finding.
The partially and transiently restored cone ERG in some of the CNGB3-mutant canine retinas with weak expression of CNGA3 and GNAT2 following CNTF treatment indicates that the phototransduction components were at least temporarily reassembled in some of the regenerated cone outer segments, providing the optimal conditions for successful outcome for gene augmentation. Because of the design of our ERG testing protocol, and the equal deconstruction of rod and cone outer segments, it is unlikely that the observed photopic responses in eyes treated only with CNTF were rod mediated rather than cone mediated. The light adaptation levels were more than sufficient to suppress rod signals, and the flash frequencies used for 29-Hz flicker stimuli and for averaging of single responses were too fast for rods.
The concept of combining retinal gene augmentation therapy with the application of neurotrophic factors to enhance treatment outcome has been used previously as a means of delaying photoreceptor cell death prior therapeutic transgene expression.47 However, deconstructing and regenerating the photoreceptor outer segments in preparation for gene augmentation is a novel concept that may complement more traditional approaches when remaining photoreceptors, either rods or cones, are refractory to therapy. Future studies are needed to examine the broader applicability and molecular mechanisms of CNTF-induced photoreceptor deconstruction, and evaluate if it allows for improvement of gene therapy outcomes in other inherited photoreceptor-specific diseases. As CNTF delivery via Encapsulated Cell Technology implants have been tested experimentally in animals48 and are undergoing clinical trials for age-related macular degeneration and retinitis pigmentosa, it is possible that such Encapsulated Cell Technology implants combined with therapeutic vectors may further facilitate translational applications.29
In summary, we have shown that a combination therapy of CNTF and gene augmentation can successfully restore cone function in two CNGB3-achromatopsia dog models when treated at older ages when gene therapy alone is ineffective. This novel approach could also help in the treatment of other inherited photoreceptor-specific diseases. Further studies are needed to inform on the molecular mechanism that underlying the effects of CNTF in facilitating gene augmentation therapy for CNGB3-achromatopsia.
Materials and Methods
Animals. Achromatopsia-affected dogs were homozygous for either a CNGB3 genomic deletion (CNGB3−/−; n = 7) or a D262N missense mutation (CNGB3m/m; n = 4), both of which result in an identical phenotype.10,27 To evaluate the effect of intravitreal CNTF injection on the normal retina, 12 unaffected dogs were used. At the conclusion of the study, the dogs were euthanatized with an overdose of euthanasia solution (Euthasol; Virbac, Fort Worth, TX), and the eyes enucleated for molecular and histologic studies. In addition, eyes were collected from seven mutant dogs and six normal controls to determine cone densities and topographic distribution with disease progression on retinal flat mounts, and for comparison of immunohistochemical labeling. Retinal RNA was extracted from the eyes of three additional normal, untreated dogs as biologic controls for analysis of the qRT-PCR data. All the dogs were part of a research colony maintained at the Retinal Disease Studies Facility (Kennett Square, PA) and supported by National Eye Institute, NIH (EY-06855) and Foundation Fighting Blindness Center grants. The studies were done in accordance with the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research, and approved by the University of Pennsylvania IACUC.
Recombinant CNTF protein. Recombinant protein was generated by cloning the human CNTF cDNA ORF into expression vector pQE30 (Qiagen, Valencia, CA), and expressed in E. coli (XL-blue; Stratagene, La Jolla, CA) as previously described.30 The CNTF solution was filtered (Millex-GV 0.22 µm filter unit; Millipore, Carrigtwohill, Ireland) and stored in 30 µl PBS aliquots at −80 °C, each containing 12 µg of CNTF. For control injections, sterile 30-µl PBS aliquots were stored at −80 °C.
Vector. The therapeutic vector, rAAV5-PR2.1-hCNGB3, was constructed, titered, and purified as previously described.10 This vector provides the most robust rescue of cone function in achromatopsia, but only targets the canine L/M-cones.10,36 Initially, the vector was injected at concentrations of 1.02–4.02 × 1013 vector genomes/ml (vg/ml). Because of mild multifocal chorioretinitis and slight retinal thinning in the treatment area, an indication of vector-associated retinal toxicity,49 the concentration was lowered to 7.96 × 1011 vg/ml and all adverse effects prevented.
Intraocular injections. Intravitreal injection of either CNTF (12 µg of CNTF in 30 µl of PBS) or PBS (30 µl) was performed under general anesthesia (Table 1 and Supplementary Table S1). On the basis of the vitreous volume of adult dogs (2.5–3.0 ml50), the injected dose of CNTF was ~ 4–5 µg/ml. In 14 eyes of 8 dogs, the intravitreal injection of CNTF or PBS was followed 1 week later by the subretinal administration of rAAV5-PR2.1-hCNGB3 vector (Table 1). Between 120 and 200 µl of vector was injected with a RetinaJect subretinal injector (SurModics, Eden Prairie, MN) through a transvitreal approach as previously described.10,36 The area treated was in the tapetal zone superior to the optic disc, a region with higher cone density,34 and was ~25% of the photoreceptor surface area. Immediately following surgery, the retinal location and extent of the subretinal blebs were documented by fundus drawings for reference in the morphologic studies. Flattening of the subretinal bleb and retinal reattachment occurred within 24–36 hours.
Electroretinography. Standard Ganzfeld scotopic and photopic ERGs were recorded from anesthetized dogs using a modified Ganzfeld dome fitted with the LED stimuli of a ColorDome stimulator (Diagnosys LLC, Lowell, MA).10 Rod and mixed rod-cone–mediated responses were recorded after 20 minutes of dark adaptation with single white flash stimuli of increasing intensities (from 0.000577 to 10.26 cd.seconds/m2). Following 10 minutes of light adaptation (34.26 cd/m2), cone-mediated signals were recorded to 1-Hz single flash (from 0.00577 to 10.26 cd.seconds/m2) and 29.41-Hz flicker (from 0.00577 to 5.77 cd.seconds/m2) stimuli. The combination of light adaptation with short flash intervals led to saturation of rods and isolation of pure cone-mediated responses. Except for the brighter scotopic light stimuli (≥ 0.577 cd.seconds/m2) multiple responses were averaged.
Retinal morphology and immunohistochemistry. Enucleated eyes were fixed for 3 hours in 4% paraformaldehyde followed by 21 hours in 2% paraformaldehyde, both in 0.1 mol/l PBS at 4 °C. Subsequently, the tissue was processed for embedding in optimal cutting temperature medium.11,39,40,49 Retinas that were only treated by intravitreal CNTF or PBS injections were sectioned in the four cardinal meridians through the optic nerve head. Retinas that underwent subretinal injections were oriented and sectioned based on the location of the treated area to compare treated and untreated regions. Cryosections of 10µm were examined by immunohistochemistry for protein expression and H&E staining. The primary antibodies and cell markers used are detailed in Supplementary Table S2. Six different custom made canine CNGB3 antibodies were made, but lacked specificity for either the canine or human proteins even though they shared sequence homology for the targeted epitopes. Alexa Fluor–labeled goat antirabbit IgG, donkey antigoat IgG, chicken antimouse IgG, or donkey antimouse IgG (1:200; Molecular Probes, Eugene, OR) were used as secondary antibody. For pSTAT1 and pSTAT3 immunohistochemistry, heat-induced epitope retrieval was done before overnight incubation with primary antibodies. HRP-labeled goat antirabbit secondary antibodies were used in combination with the ABC immunoenzymatic detection method. With fluorescence immunohistochemistry, DAPI stain was used as a nuclear stain. The In Situ Cell Death Detection kit (Roche Applied Science, Indianapolis, IN) was used to visualize apoptotic nuclei following CNTF injection by TUNEL (terminal deoxynucleotidyl transferase-mediated biotinylated UTP nick end labeling). Sections were examined with a Zeiss Axioplan microscope (Carl Zeiss Meditec, Dublin, CA), images were digitally captured (Spot 4.0 camera; Diagnostic Instruments, Sterling Heights, MI) and imported into a graphics program (Photoshop; Adobe, Mountain View, CA) for display. Selected tissues (Figure 1c and Supplementary Figure S2) were imaged by laser scanning confocal microscopy (Olympus FluoView FV1000; Olympus America, Center Valley, PA).
The effect of intravitreal delivery of CNTF or PBS on photoreceptor ultrastructure was evaluated by electron microscopy 1 week after injection in a normal dog. Immediately after enucleation, the posterior segments were isolated and fixed, using a triple fixative protocol as previously reported.41 The posterior segments were then trimmed, dehydrated, and embedded in epoxy resin (PolyBed 812; Polysciences, Warrington, PA). Thin sections (70–80 nm) were cut and picked up on Formvar coated copper grids, stained with uranyl acetate and lead citrate, and examined with a JEOL 1010 electron microscope (JEOL USA, Peabody, MA) fitted with a Hamamatsu digital camera (Hamamatsu Corporation, Bridgewater, NJ) and AMT Advantage image capture software (Advanced Microscopy Techniques, Corp., Woburn, MA).
Mapping of cone photoreceptors in CNGB3-achromatopsia. To determine the cone density in mutant retinas, selected retinal regions were examined in 2 affected and 3 control dogs at 1 year of age. Eyes were collected following euthanasia, fixed between 20 min and 1 h in 4% paraformaldehyde in 0.1 mol/l phosphate-buffered saline (PBS) at 4 °C, and each retina divided into quadrants along the horizontal and vertical meridians. Beginning 2 mm from the optic disc, two consecutive 4-mm circular retinal punches without retinal pigment epithelium were collected with a disposable biopsy punch from each quadrant; these were fixed for 2 hours in 2% paraformaldehyde in 0.1 mol/l PBS and then stored in PBS at 4 °C for immunolabeling with antibodies for S- and L/M-cone opsin and hCAR (Supplementary Table S2). Alexa Fluor–labeled goat antirabbit IgG, donkey antigoat IgG, or chicken antirabbit IgG (1:200; Molecular Probes) secondary antibodies were used. The cone subclasses were identified by double labeling with either S- or L/M- and hCAR-antibodies. Double-labeled retinal patches were flat mounted on a glass slide in 0.1 mol/l PBS with the photoreceptor outer segments pointing up, cover slipped, and sealed with nail polish.
The immunolabeled cone subclasses were imaged within a 2-mm diameter circle centered inside each 4-mm retinal trephine using the same Zeiss Axioplan microscope (Carl Zeiss Meditec) and Spot 4.0 camera (Diagnostic Instruments) as described above. The labeled cone outer segments were manually marked on the digital images using Photoshop software (Adobe) and counted with image analysis software (MCID Analysis version 7.0 grain counting software; Interfocus, Linton, UK).
qRT-PCR analysis of retinal gene expression. Whole retinas were isolated from the eyecups within 1–2 minutes of euthanasia, flash frozen in liquid nitrogen, and stored at −80 °C until used. The retinal tissue was homogenized, and total RNA extracted with TRIzol Reagent (Invitrogen, Life Technologies, Carlsbad, CA). RNA concentrations were determined using an ND1000 spectrophotometer (Nanodrop Products, Wilmington, DE). Retinal cDNA was synthesized by reverse transcription using the High Capacity cDNA Reverse Transcription kit (Applied Biosystems, Life Technologies, Carlsbad, CA) and the manufacturer's instructions. qRT-PCR was done as previously described using primers and TaqMan MGB probes designed for the hCNGB3 transgene, and canine-specific probes for cCNGA3, cCNGB3, cL/M-opsin, cS-opsin, cCNGA1, cCNGB1, and cRHO (Supplementary Table S3).10 TaqMan expression assays were commercially available for STAT1 and STAT3 mRNA (Cf02662970_m1 and Cf02666647_m1; Applied Biosystems). Relative gene expression for each gene compared with the 18S rRNA product (Hs99999901_s1; Applied Biosystems) was calculated as 1/[2^(CtGene − Ct18S)]. Only for the hCNGB3 transgene in retinas injected with rAAV5-PR2.1-hCNGB3 was relative gene expression used to directly compare CNTF and PBS-treated eyes by unpaired t-test. For all the other genes, the ratio of relative gene expressions was calculated between the treated eye and an untreated biological control group using the ΔΔCT method.38 Any ratio considerably different from 1 was relevant and indicated either an increase (>1) or decrease (<1) of gene expression. Because of the small group size for each time point in normal animals undergoing intravitreal injections alone, comparisons were made by descriptive statistics. In contrast, the sample sizes were larger for rAAV5-PR2.1-hCNGB3-treated eyes, allowing the comparison of these gene expression ratios between PBS- and CNTF-injected eyes by one-way analysis of variance.
Western blotting. Retinal samples were harvested as described for qRT-PCR analysis and frozen at −80 °C. The retinas were homogenized in a modified RIPA buffer (1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS, 158 mmol/l NaCl, and 10 mmol/l Tris) containing a protease inhibitor cocktail (Complete-EDTA; Roche Diagnostics, Indianapolis, IN), followed by sonication on ice. The homogenate was centrifuged at 13,000g for 15 minutes at 4 °C, and the supernatant containing the proteins was collected. Total protein levels were determined by the Bradford method (ABC protein assay; Bio-Rad, Hercules, CA). For western blot analysis, 10 µg of protein were loaded onto each lane. Membranes were blocked for 1 hour at 4 °C (Blocking buffer; Li-Cor Biosciences, Lincoln, NE), and incubated for 1.5 hours at room temperature in primary antibodies pSTAT1 or pSTAT3 and actin to control for equal loading of each well (Supplementary Table S2). pSTAT1 and pSTAT3 antibodies were detected with a near infrared fluorescent goat antirabbit secondary antibody (IRDye 800 CW goat antirabbit IgG; Li-Cor Biosciences), and STAT1 and actin antibodies with a goat antimouse antibody (IRDye 680 LT goat antimouse IgG; Li-Cor Biosciences) diluted to 1:15,000 for 1 hour at room temperature. The bands were quantified by densitometry using Odyssey infrared imaging system software (Odyssey version 2.1; Li-Cor Biosciences). Detection of STAT3 levels was performed on a separate blot using the enhanced chemiluminescence technique as we were unsuccessful in detecting both pSTAT3 and STAT3 antibodies with the Odyssey technique on the same blot. Levels of STAT3 and pSTAT3 were normalized to actin before calculating the pSTAT3/STAT3 ratio.
SUPPLEMENTARY MATERIAL Figure S1. Electron micrographs of normal canine retinas 1 week following intravitreal injection of PBS or CNTF. Figure S2. Partial restoration of CNGA3 and GNAT2 protein localization in the CNGB3-mutant retina 1 week following intravitreal CNTF injection. Table S1. Intravitreal CNTF injections in normal dogs. Table S2. Antibodies and reagents used for immunohistochemistry and western blotting. Table S3. Primers and probes for qRT-PCR and exon junction crossed by PCR product.
Acknowledgments
This work was supported by grants from NIH (EY-01583, 06855, 07132, 08571, 11123, 15398, 17549, 19304, 18586, P30EY14801, R24EY022012, 2PNEY018241; the content of this publication is solely the responsibility of the authors and does not necessarily represent the official views of the National Eye Institute or the National Institutes of Health), Department of Defense (W81XWH-09-1-0674), the Foundation Fighting Blindness (Center and Individual Investigator grants), the Macula Vision Research Foundation, the ONCE International Prize, Hope for Vision, Van Sloun Fund, and Research to Prevent Blindness (an unrestricted grant to Bascom Palmer Eye Institute). The authors thank Bernd Wissinger (University Clinics Tübingen) for the hCNGB3 cDNA; Jeremy Nathans (Johns Hopkins University) for the pR2.1-LacZ plasmid; Cheryl Craft (University of Southern California) for the hCAR antibody; Vince Chiodo, Tom Doyle, and Min Ding (University of Florida) for assistance in vector production; J Huang and G-S Ying (University of Pennsylvania) for statistical support; Melinda Frame (Michigan State University) for assistance with confocal microscopy; Mary Leonard (University of Pennsylvania) for illustrations; Leslie King (University of Pennsylvania) for editorial support; Lydia Melnyk (University of Pennsylvania) for research coordination; and Simone Iwabe, Kendra McDaid and Karla Carlisle and the staff at the Retinal Disease Studies Facility (University of Pennsylvania) for veterinary or technical support with the animals. W.W.H. and the University of Florida have a financial interest in the use of rAAV therapies, and own equity in a company (AGTC Inc.) that may commercialize some aspects of this work. All remaining authors have declared that no conflict of interest exists. This work was done in Philadelphia, Pennsylvania, USA; Gainesville, Florida, USA; and Miami, Florida, USA.
Supplementary Material
References
- Maguire AM, Simonelli F, Pierce EA, Pugh EN, Jr, Mingozzi F, Bennicelli J, et al. Safety and efficacy of gene transfer for Leber's congenital amaurosis. N Engl J Med. 2008;358:2240–2248. doi: 10.1056/NEJMoa0802315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bainbridge JW, Smith AJ, Barker SS, Robbie S, Henderson R, Balaggan K, et al. Effect of gene therapy on visual function in Leber's congenital amaurosis. N Engl J Med. 2008;358:2231–2239. doi: 10.1056/NEJMoa0802268. [DOI] [PubMed] [Google Scholar]
- Cideciyan AV, Aleman TS, Boye SL, Schwartz SB, Kaushal S, Roman AJ, et al. Human gene therapy for RPE65 isomerase deficiency activates the retinoid cycle of vision but with slow rod kinetics. Proc Natl Acad Sci USA. 2008;105:15112–15117. doi: 10.1073/pnas.0807027105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson SG, Cideciyan AV, Ratnakaram R, Heon E, Schwartz SB, Roman AJ, et al. Gene therapy for leber congenital amaurosis caused by RPE65 mutations: safety and efficacy in 15 children and adults followed up to 3 years. Arch Ophthalmol. 2012;130:9–24. doi: 10.1001/archophthalmol.2011.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett J, Ashtari M, Wellman J, Marshall KA, Cyckowski LL, Chung DC, et al. AAV2 gene therapy readministration in three adults with congenital blindness. Sci Transl Med. 2012;4:120ra15. doi: 10.1126/scitranslmed.3002865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson SG, Acland GM, Aguirre GD, Aleman TS, Schwartz SB, Cideciyan AV, et al. Safety of recombinant adeno-associated virus type 2-RPE65 vector delivered by ocular subretinal injection. Mol Ther. 2006;13:1074–1084. doi: 10.1016/j.ymthe.2006.03.005. [DOI] [PubMed] [Google Scholar]
- Jacobson SG, Boye SL, Aleman TS, Conlon TJ, Zeiss CJ, Roman AJ, et al. Safety in nonhuman primates of ocular AAV2-RPE65, a candidate treatment for blindness in Leber congenital amaurosis. Hum Gene Ther. 2006;17:845–858. doi: 10.1089/hum.2006.17.845. [DOI] [PubMed] [Google Scholar]
- Acland GM, Aguirre GD, Ray J, Zhang Q, Aleman TS, Cideciyan AV, et al. Gene therapy restores vision in a canine model of childhood blindness. Nat Genet. 2001;28:92–95. doi: 10.1038/ng0501-92. [DOI] [PubMed] [Google Scholar]
- Pang JJ, Chang B, Kumar A, Nusinowitz S, Noorwez SM, Li J, et al. Gene therapy restores vision-dependent behavior as well as retinal structure and function in a mouse model of RPE65 Leber congenital amaurosis. Mol Ther. 2006;13:565–572. doi: 10.1016/j.ymthe.2005.09.001. [DOI] [PubMed] [Google Scholar]
- Komáromy AM, Alexander JJ, Rowlan JS, Garcia MM, Chiodo VA, Kaya A, et al. Gene therapy rescues cone function in congenital achromatopsia. Hum Mol Genet. 2010;19:2581–2593. doi: 10.1093/hmg/ddq136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltran WA, Cideciyan AV, Lewin AS, Iwabe S, Khanna H, Sumaroka A, et al. Gene therapy rescues photoreceptor blindness in dogs and paves the way for treating human X-linked retinitis pigmentosa. Proc Natl Acad Sci USA. 2012;109:2132–2137. doi: 10.1073/pnas.1118847109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander JJ, Umino Y, Everhart D, Chang B, Min SH, Li Q, et al. Restoration of cone vision in a mouse model of achromatopsia. Nat Med. 2007;13:685–687. doi: 10.1038/nm1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalakis S, Mühlfriedel R, Tanimoto N, Krishnamoorthy V, Koch S, Fischer MD, et al. Restoration of cone vision in the CNGA3-/- mouse model of congenital complete lack of cone photoreceptor function. Mol Ther. 2010;18:2057–2063. doi: 10.1038/mt.2010.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho LS, Xu J, Pearson RA, Smith AJ, Bainbridge JW, Morris LM, et al. Long-term and age-dependent restoration of visual function in a mouse model of CNGB3-associated achromatopsia following gene therapy. Hum Mol Genet. 2011;20:3161–3175. doi: 10.1093/hmg/ddr218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang JJ, Deng WT, Dai X, Lei B, Everhart D, Umino Y, et al. AAV-mediated cone rescue in a naturally occurring mouse model of CNGA3-achromatopsia. PLoS ONE. 2012;7:e35250. doi: 10.1371/journal.pone.0035250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons DL, Boye SL, Hauswirth WW, Wu SM. Gene therapy prevents photoreceptor death and preserves retinal function in a Bardet-Biedl syndrome mouse model. Proc Natl Acad Sci USA. 2011;108:6276–6281. doi: 10.1073/pnas.1019222108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boye SE, Boye SL, Pang J, Ryals R, Everhart D, Umino Y, et al. Functional and behavioral restoration of vision by gene therapy in the guanylate cyclase-1 (GC1) knockout mouse. PLoS ONE. 2010;5:e11306. doi: 10.1371/journal.pone.0011306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang JJ, Dai X, Boye SE, Barone I, Boye SL, Mao S, et al. Long-term retinal function and structure rescue using capsid mutant AAV8 vector in the rd10 mouse, a model of recessive retinitis pigmentosa. Mol Ther. 2011;19:234–242. doi: 10.1038/mt.2010.273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett J, Tanabe T, Sun D, Zeng Y, Kjeldbye H, Gouras P, et al. Photoreceptor cell rescue in retinal degeneration (rd) mice by in vivo gene therapy. Nat Med. 1996;2:649–654. doi: 10.1038/nm0696-649. [DOI] [PubMed] [Google Scholar]
- Ali RR, Sarra GM, Stephens C, Alwis MD, Bainbridge JW, Munro PM, et al. Restoration of photoreceptor ultrastructure and function in retinal degeneration slow mice by gene therapy. Nat Genet. 2000;25:306–310. doi: 10.1038/77068. [DOI] [PubMed] [Google Scholar]
- Sarra GM, Stephens C, de Alwis M, Bainbridge JW, Smith AJ, Thrasher AJ, et al. Gene replacement therapy in the retinal degeneration slow (rds) mouse: the effect on retinal degeneration following partial transduction of the retina. Hum Mol Genet. 2001;10:2353–2361. doi: 10.1093/hmg/10.21.2353. [DOI] [PubMed] [Google Scholar]
- Pang J, Boye SE, Lei B, Boye SL, Everhart D, Ryals R, et al. Self-complementary AAV-mediated gene therapy restores cone function and prevents cone degeneration in two models of Rpe65 deficiency. Gene Ther. 2010;17:815–826. doi: 10.1038/gt.2010.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong F, Li W, Li X, Zheng Q, Dai X, Zhou X, et al. Self-complementary AAV5 vector facilitates quicker transgene expression in photoreceptor and retinal pigment epithelial cells of normal mouse. Exp Eye Res. 2010;90:546–554. doi: 10.1016/j.exer.2010.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrs-Silva H, Dinculescu A, Li Q, Min SH, Chiodo V, Pang JJ, et al. High-efficiency transduction of the mouse retina by tyrosine-mutant AAV serotype vectors. Mol Ther. 2009;17:463–471. doi: 10.1038/mt.2008.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohl S, Varsanyi B, Antunes GA, Baumann B, Hoyng CB, Jägle H, et al. CNGB3 mutations account for 50% of all cases with autosomal recessive achromatopsia. Eur J Hum Genet. 2005;13:302–308. doi: 10.1038/sj.ejhg.5201269. [DOI] [PubMed] [Google Scholar]
- Sundin OH, Yang JM, Li Y, Zhu D, Hurd JN, Mitchell TN, et al. Genetic basis of total colourblindness among the Pingelapese islanders. Nat Genet. 2000;25:289–293. doi: 10.1038/77162. [DOI] [PubMed] [Google Scholar]
- Sidjanin DJ, Lowe JK, McElwee JL, Milne BS, Phippen TM, Sargan DR, et al. Canine CNGB3 mutations establish cone degeneration as orthologous to the human achromatopsia locus ACHM3. Hum Mol Genet. 2002;11:1823–1833. doi: 10.1093/hmg/11.16.1823. [DOI] [PubMed] [Google Scholar]
- Ding XQ, Harry CS, Umino Y, Matveev AV, Fliesler SJ, Barlow RB. Impaired cone function and cone degeneration resulting from CNGB3 deficiency: down-regulation of CNGA3 biosynthesis as a potential mechanism. Hum Mol Genet. 2009;18:4770–4780. doi: 10.1093/hmg/ddp440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen R, Tao W, Li Y, Sieving PA. CNTF and retina. Prog Retin Eye Res. 2012;31:136–151. doi: 10.1016/j.preteyeres.2011.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen R, Song Y, Kjellstrom S, Tanikawa A, Liu Y, Li Y, et al. Regulation of rod phototransduction machinery by ciliary neurotrophic factor. J Neurosci. 2006;26:13523–13530. doi: 10.1523/JNEUROSCI.4021-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher SK, Lewis GP, Linberg KA, Verardo MR. Cellular remodeling in mammalian retina: results from studies of experimental retinal detachment. Prog Retin Eye Res. 2005;24:395–431. doi: 10.1016/j.preteyeres.2004.10.004. [DOI] [PubMed] [Google Scholar]
- Beltran WA, Wen R, Acland GM, Aguirre GD. Intravitreal injection of ciliary neurotrophic factor (CNTF) causes peripheral remodeling and does not prevent photoreceptor loss in canine RPGR mutant retina. Exp Eye Res. 2007;84:753–771. doi: 10.1016/j.exer.2006.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Tao W, Luo L, Huang D, Kauper K, Stabila P, et al. CNTF induces regeneration of cone outer segments in a rat model of retinal degeneration. PLoS ONE. 2010;5:e9495. doi: 10.1371/journal.pone.0009495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mowat FM, Petersen-Jones SM, Williamson H, Williams DL, Luthert PJ, Ali RR, et al. Topographical characterization of cone photoreceptors and the area centralis of the canine retina. Mol Vis. 2008;14:2518–2527. [PMC free article] [PubMed] [Google Scholar]
- Gropp KE, Szél A, Huang JC, Acland GM, Farber DB, Aguirre GD. Selective absence of cone outer segment beta 3-transducin immunoreactivity in hereditary cone degeneration (cd). Exp Eye Res. 1996;63:285–296. doi: 10.1006/exer.1996.0117. [DOI] [PubMed] [Google Scholar]
- Komáromy AM, Alexander JJ, Cooper AE, Chiodo VA, Glushakova LG, Acland GM, et al. Targeting gene expression to cones with human cone opsin promoters in recombinant AAV. Gene Ther. 2008;15:1049–1055. doi: 10.1038/gt.2008.32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akhmedov NB, Piriev NI, Pearce-Kelling S, Acland GM, Aguirre GD, Farber DB. Canine cone transducin-gamma gene and cone degeneration in the cd dog. Invest Ophthalmol Vis Sci. 1998;39:1775–1781. [PubMed] [Google Scholar]
- Berta ÁI, Boesze-Battaglia K, Genini S, Goldstein O, O'Brien PJ, Szél Á, et al. Photoreceptor cell death, proliferation and formation of hybrid rod/S-cone photoreceptors in the degenerating STK38L mutant retina. PLoS ONE. 2011;6:e24074. doi: 10.1371/journal.pone.0024074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltran WA, Hammond P, Acland GM, Aguirre GD. A frameshift mutation in RPGR exon ORF15 causes photoreceptor degeneration and inner retina remodeling in a model of X-linked retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2006;47:1669–1681. doi: 10.1167/iovs.05-0845. [DOI] [PubMed] [Google Scholar]
- Beltran WA, Rohrer H, Aguirre GD. Immunolocalization of ciliary neurotrophic factor receptor alpha (CNTFRalpha) in mammalian photoreceptor cells. Mol Vis. 2005;11:232–244. [PubMed] [Google Scholar]
- Acland GM, Aguirre GD. Retinal degenerations in the dog: IV. Early retinal degeneration (erd) in Norwegian elkhounds. Exp Eye Res. 1987;44:491–521. doi: 10.1016/s0014-4835(87)80160-4. [DOI] [PubMed] [Google Scholar]
- Matulef K, Zagotta WN. Cyclic nucleotide-gated ion channels. Annu Rev Cell Dev Biol. 2003;19:23–44. doi: 10.1146/annurev.cellbio.19.110701.154854. [DOI] [PubMed] [Google Scholar]
- Thapa A, Morris L, Xu J, Ma H, Michalakis S, Biel M, et al. Endoplasmic reticulum stress-associated cone photoreceptor degeneration in cyclic nucleotide-gated channel deficiency. J Biol Chem. 2012;287:18018–18029. doi: 10.1074/jbc.M112.342220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curcio CA, Allen KA, Sloan KR, Lerea CL, Hurley JB, Klock IB, et al. Distribution and morphology of human cone photoreceptors stained with anti-blue opsin. J Comp Neurol. 1991;312:610–624. doi: 10.1002/cne.903120411. [DOI] [PubMed] [Google Scholar]
- Kohl S, Marx T, Giddings I, Jägle H, Jacobson SG, Apfelstedt-Sylla E, et al. Total colourblindness is caused by mutations in the gene encoding the alpha-subunit of the cone photoreceptor cGMP-gated cation channel. Nat Genet. 1998;19:257–259. doi: 10.1038/935. [DOI] [PubMed] [Google Scholar]
- Genead MA, Fishman GA, Rha J, Dubis AM, Bonci DM, Dubra A, et al. Photoreceptor structure and function in patients with congenital achromatopsia. Invest Ophthalmol Vis Sci. 2011;52:7298–7308. doi: 10.1167/iovs.11-7762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buch PK, MacLaren RE, Durán Y, Balaggan KS, MacNeil A, Schlichtenbrede FC, et al. In contrast to AAV-mediated Cntf expression, AAV-mediated Gdnf expression enhances gene replacement therapy in rodent models of retinal degeneration. Mol Ther. 2006;14:700–709. doi: 10.1016/j.ymthe.2006.05.019. [DOI] [PubMed] [Google Scholar]
- Tao W, Wen R, Goddard MB, Sherman SD, O'Rourke PJ, Stabila PF, et al. Encapsulated cell-based delivery of CNTF reduces photoreceptor degeneration in animal models of retinitis pigmentosa. Invest Ophthalmol Vis Sci. 2002;43:3292–3298. [PubMed] [Google Scholar]
- Beltran WA, Boye SL, Boye SE, Chiodo VA, Lewin AS, Hauswirth WW, et al. rAAV2/5 gene-targeting to rods:dose-dependent efficiency and complications associated with different promoters. Gene Ther. 2010;17:1162–1174. doi: 10.1038/gt.2010.56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buyukmihci N, Aguirre GD. Rod disc turnover in the dog. Invest Ophthalmol. 1976;15:579–584. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.