

Abstract
The clinical use of human pluripotent stem cells and their derivatives is limited by the rejection of transplanted cells due to differences in their human leukocyte antigen (HLA) genes. This has led to the proposed use of histocompatible, patient-specific stem cells; however, the preparation of many different stem cell lines for clinical use is a daunting task. Here, we develop two distinct genetic engineering approaches that address this problem. First, we use a combination of gene targeting and mitotic recombination to derive HLA-homozygous embryonic stem cell (ESC) subclones from an HLA-heterozygous parental line. A small bank of HLA-homozygous stem cells with common haplotypes would match a significant proportion of the population. Second, we derive HLA class I–negative cells by targeted disruption of both alleles of the Beta-2 Microglobulin (B2M) gene in ESCs. Mixed leukocyte reactions and peptide-specific HLA-restricted CD8+ T cell responses were reduced in class I–negative cells that had undergone differentiation in embryoid bodies. These B2M−/− ESCs could act as universal donor cells in applications where the transplanted cells do not express HLA class II genes. Both approaches used adeno-associated virus (AAV) vectors for efficient gene targeting in the absence of potentially genotoxic nucleases, and produced pluripotent, transgene-free cell lines.
Introduction
If human pluripotent stem cells are to be used clinically, they must overcome the immunological barriers that limit the transplantation of allogeneic cells. A major immunologic barrier results from the cell surface expression of human leukocyte antigens (HLA), which are encoded by genes in the major histocompatibility complex on chromosome 6, and present self and foreign peptides to T cells. These polymorphic loci include the class I HLA-A, -B, and -C genes expressed on most nucleated cells in the body, and the class II HLA-DR, -DP, and -DQ genes expressed in specialized antigen-presenting cells such as dendritic cells and macrophages. Given that multiple alleles exist for each polymorphic HLA gene, the chance of any specific pair of HLA haplotypes being found in a potential transplant recipient is exceedingly small. Depending on the application, transplanted cells and organs can be rejected based on their HLA type, with hematopoietic stem cells requiring extensive matching of both class I and II alleles, and solid organs requiring less stringent matching of class I loci. Typically, prolonged treatment with immunosuppressive drugs is required to prevent the rejection of mismatched grafts, often with dangerous side effects.
One solution to the immunologic barrier imposed by HLA is to use autologous, induced pluripotent stem cells (iPSCs) derived from each patient. iPSCs resemble embryonic stem cells (ESCs) and can be derived from adult human somatic cells by introducing specific reprogramming factors.1,2 Although this approach ensures histocompatibility, it will be difficult to translate into clinical practice, due to the high cost for each patient, the prolonged cell culture period needed for reprogramming and differentiation into a therapeutic cell type, and the extensive validation and regulatory approval required of the final product. In addition, when treating genetic diseases, the responsible mutations must also be corrected before the cells are returned to the patient.
An alternative solution to this problem is to bank multiple stem cell lines with different HLA types, which allows therapeutic cell products derived from these lines to be prepared ahead of time. However, this would require large number of cell lines. The US bone marrow registry has >4,000,000 donors but accurately matches only 50–60% of the population at HLA-A and -B loci.3 One study estimated that 150 ESC lines derived from donors in the United Kingdom would produce a cell bank that matches <20% of the population at HLA-A, -B, and -DR loci.4 The use of HLA-homozygous cell lines would decrease the number required for matching. For example, 50 iPSC lines derived from HLA-homozygous individuals with common haplotypes could match ~73% of the relatively non-diverse Japanese population at HLA-A, -B, and -DRB1 loci, although it may still be difficult to identify donors homozygous for rare haplotypes.5 Any solution that requires the banking of multiple independent cell lines must also deal with the inherent variability of different pluripotent stem cell clones,6,7 and in the case of iPSCs, genetic and epigenetic variations may also occur during reprogramming that could influence the behavior of individual clones.8 This interclonal variation means that differentiation protocols must be optimized for each independent cell line, and that patients treated with distinct stem clones could have very different clinical outcomes.
Thus, there is a real need for developing pluripotent stem cell lines that are compatible with multiple allogeneic recipients, so that the number of cell lines required for clinical use can be reduced to a manageable level. Here, we develop two genetic engineering approaches that address this problem. First, we describe a method for deriving HLA-homozygous subclones from HLA-heterozygous ESC lines. A single HLA-homozygous line can be compatible with multiple recipients because only one haplotype requires matching. In the second approach, we develop HLA-negative stem cells that do not express any class I proteins on their cell surface by targeted disruption of the B2M gene. These B2M−/− ESCs could act as universal donor cells in applications where the transplanted cells do not express HLA class II genes.
Results
Creating HLA-homozygous cells
Figure 1a shows our two-step strategy for deriving HLA-homozygous cells from a heterozygous parental cell line. First, adeno-associated virus (AAV) gene targeting vectors are used to insert a HyTK fusion gene centromeric to the HLA locus on the short arm of chromosome 6, and targeted cells are selected with hygromycin. Then ganciclovir is used to select for loss of the TK component of the HyTK gene and isolate mitotic recombinants resulting from crossovers centromeric to HLA. Studies in mouse ESCs have shown that these recombination events typically produce homozygosity extending from the crossover point to the telomere,9 which includes the entire HLA locus. The crossover also removes the HyTK transgene from the cells.
Figure 1.

Derivation of HLA-homozygous ESCs. (a) Diagram showing the strategy for obtaining HLA-homozygous clones by targeting the HMGA1 gene centromeric to the HLA locus, then selecting for cells that had lost the HyTK gene by mitotic recombination with ganciclovir. The red and blue chromosomes represent the two copies of chromosome 6 in each cell, with the clusters of HLA class I, II, and III genes indicated. (b) Map of the AAV2-HMGA1-HyTKpA targeting vector and HMGA1 gene, showing the locations of the Southern blot probes and restriction enzyme sites (K, Kpn I). (c) Southern blot analysis of Kpn I–digested genomic DNA of parental H1 ESCs, two clones targeted at the HMGA1 gene (c4 and c5), and three subclones obtained by ganciclovir selection (c4A, c4B, and c5A), probed with HMGA1 and HyTK probes. Asterisks indicate the locations of the two HyTK-hybridizing fragments derived from the targeted allele. The faint HyTK-hybridizing band in clone c5A and H1 represents trace signal from the hygromycin-resistant mouse embryonic fibroblast feeder cells. Subclones c4A and c4B contain a novel HyTK-hybridizing fragment demonstrating a rearrangement of the HyTK gene that can account for ganciclovir resistance in these clones. (d) HLA typing results for parental H1 ESCs, gene-targeted clone c5, and ganciclovir-resistant subclone c5A. (e) Copy number analysis of SNP data. Loss of heterozygosity in clone c5A telomeric to the PRIM2A gene on chromosome 6 is shown by an increase in copy number of one allele and decrease of the other relative to the parental cell line (H1). GCV-R, ganciclovir-resistance; HLA, human leukocyte antigens.
We chose to target the HMGA1 gene because it is ~1 Mb centromeric to the HLA locus on chromosome 6 and expressed in pluripotent stem cells. The AAV2-HMGA1-HyTKpA vector is designed to insert the HyTK gene at exon 3 of HMGA1 and initiate translation from the HMGA1 start codon (Figure 1b). Initially, we transduced H1 ESCs10 and showed by Southern blots that the hygromycin-resistant clones were accurately targeted at the HMGA1 locus and did not contain random integrants (clones c4 and c5 in Figure 1c). We transduced the H7,10 BG01, BG02, and BG03 ESC lines11 with AAV2-HMGA1-HyTKpA as well, and 26 of the 27 hygromycin-resistant colonies screened were accurately targeted (Supplementary Table S1). Because the targeting step was so efficient, we also produced hygromycin-resistant polyclonal populations without picking individual colonies, most of which contained a majority of targeted cells (Supplementary Table S1).
Several of these HMGA1-targeted ESCs were then cultured in the presence of ganciclovir to select for loss of the HyTK transgene. The ganciclovir-resistant clones that underwent mitotic recombination should have removed the targeted HMGA1 allele and instead contain two wild-type alleles of HMGA1. In the case of H1 clone c5, we isolated ganciclovir-resistant subclone c5A that had lost the HyTK gene based on Southern blots (Figure 1c), and we confirmed by HLA typing that it contained a single HLA haplotype (Figure 1d). SNP chip analysis showed a loss of heterozygosity extending from the pericentromeric region to the telomere on chromosome 6 (Figure 1e), with the crossover occurring within the DNA Primase gene PRIM2 (data not shown). Thus, H1 subclone c5A had undergone the expected mitotic recombination event and become HLA-homozygous. These cells produced a trilineage teratoma when grown in immunodeficient mice, confirming that they remained pluripotent after both the gene targeting and mitotic recombination steps (Supplementary Figure S1). Several other ganciclovir selection experiments performed on other HMGA1-targeted ESCs failed to produce HLA-homozygous cell lines based on Southern blots and HLA typing, but instead produced resistant clones with HyTK gene rearrangements, mutations, or deletions (Supplementary Table S1). So, although subclone c5A clearly demonstrates that HLA-homozygous lines can be derived by mitotic recombination events, ganciclovir-resistant clones produced by other mechanisms are more commonly recovered. The unexpectedly low frequency of mitotic recombination events led us to investigate the derivation of HLA class I–negative cells as an alternative strategy.
Creating HLA class I–negative cells
The polymorphic HLA-A, -B, and -C class I proteins are expressed on the surface of nucleated cells where they present peptide antigens to the immune system, and matching at these loci is important for successful allogeneic transplantation. The Beta-2 Microglobulin (B2M) gene encodes a common subunit essential for cell surface expression of all the HLA class I antigen heterodimers (the other subunits are the heavy chains for HLA-A, -B, -C, -E, -F, or -G). Thus, deleting both B2M alleles should render ES cells class I deficient and represents an alternative approach for reducing their immunogenicity. Figure 2a shows our strategy for inactivating both copies of B2M in ESCs, which consists of two sequential rounds of gene targeting, followed by Cre-mediated excision of the selectable marker cassettes. The two AAV targeting vectors were designed to insert either a TKNeo or HyTK fusion gene encoding thymidine kinase linked to G418 or hygromycin resistance genes respectively into the B2M initiation codon located in exon 1. Both vectors were packaged in serotype 3 capsids to improve ESC transduction frequencies.12 H1 ESCs were transduced with the AAV3-B2M-ETKNpA vector and 30% of the G418-resistant cells were targeted at one B2M allele based on Southern blots (Supplementary Table S2). One of these clones was then transduced with the AAV3-B2M-EHyTKpA vector and 10% of hygromycin-resistant cells were targeted at B2M. None of the targeted clones analyzed contained random integrants.
Figure 2.

B2M targeting and transgene removal. (a) The structures of the AAV3-B2M-ETKNpA– and AAV3-EHyTKpA–targeting vectors, and the human B2M locus are shown before targeting, and after two rounds of targeting and transgene removal by Cre. The locations of Southern blot probes and enzymes (X, Xba I) are indicated. (b) Diagram of the wild-type, targeted, and transgene-deleted alleles of the B2M gene with the locations of PCR primers, and the results obtained from an analysis of 45 Cre-treated clones. The gel below shows a representative PCR analysis of Cre-transduced clones with the different primer combinations, and the resulting allele designations are shown. DNA from the parental H1 cells (+/+), and clones targeted in one (+/TKNeo) or both (HyTK/TKNeo) B2M alleles were used as controls. (c) Southern blot analysis of Xba I–digested genomic DNAs of parental H1 cells (+/+), one clone targeted at one B2M allele (+/TKNeo), one clone targeted at both B2M alleles (HyTK/TKNeo), and four clones obtained after removal of the transgenes with Cre (loxP/loxP c1–4), probed with B2M- or TK-specific probes. Positions of the different possible B2M fragments are shown at the left.
Cre-recombinase was then transiently expressed by transducing cells with a non-integrating foamy virus vector, which is a type of retroviral vector that efficiently infects human ESCs.13 Six of the 45 Cre-treated clones that were analyzed by allele-specific PCR had deleted both the HyTK and TKNeo transgenes (examples in Figure 2b), and deletion of the transgenes was confirmed by Southern blots (examples in Figure 2c). These transgene-deleted alleles contain a single residual loxP site in exon 1 of B2M, as well as stop codons in all three reading frames and a polyadenylation signal (Figure 2a) to ensure that no functional B2M protein can be synthesized. Two of these clones (B2MloxP/loxPc1 and c2) were also shown to have trilineage developmental potential by teratoma assays as well as normal karyotypes (example in Supplementary Figure S2).
B2M and HLA class I heavy chains were both detectable by flow cytometry on the surface of B2M+/+ H1 ESCs and at a lower level on cells with a single targeted B2M allele (B2M+/TKNeo), but were absent on cells with both alleles targeted (B2MTKNeo/HyTK) or targeted and treated with Cre to remove the transgenes (B2MloxP/loxP) (Figure 3a). B2M protein was also undetectable in B2MloxP/loxP cells by western blot, whereas class I heavy chain proteins were still present, albeit at lower levels in B2MloxP/loxP cells (Figure 3b). These results are consistent with the known requirement for B2M in the transport of class I heavy chains to the cell surface.14 Global gene expression analysis showed that B2M RNA was absent from both B2MloxP/loxP clones analyzed, but HLA class I heavy chain transcripts were present at wild-type levels (Figure 3c). The transcript levels of genes encoding other B2M-binding proteins, including CD1A-D, FCGRT, HFE, and MR1 were also unchanged in B2MloxP/loxP cells. Cluster analysis of the samples showed that the two B2M−/− cell lines did not cluster apart from the two parental H1 cell line duplicates (Figure 3d), demonstrating that the global gene expression pattern in these subclones had not diverged from the parental cell line. In addition, Pearson correlation coefficients for all crosswise comparisons were ≥0.98, suggesting that they had not undergone any major changes during their engineering, and that this approach could be used to generate cells for clinical use.
Figure 3.
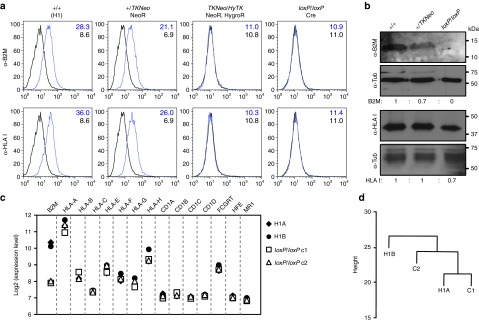
Gene expression in B2M-targeted clones. (a) Flow cytometry analysis showing surface expression of B2M and HLA class I heavy chains (antibody W6/32 binds HLA-A, -B, and -C) on undifferentiated parental H1 ESCs (+/+), ESCs targeted at one B2M allele (+/TKNeo), both B2M alleles (HyTK/TKNeo), or after Cre-mediated transgene removal (loxP/loxP). Mean fluorescence intensity is indicated in each case for the specific antibody (blue numbers) and isotype controls (black numbers). (b) Western blot analysis of total cellular protein extracts from parental H1 ESCs (+/+), ESCs targeted at one B2M allele (+/TKNeo), or both B2M alleles after Cre-mediated transgene excision (loxP/loxP) performed with the indicated antibodies. Signal ratios for B2M or HLA class I heavy chains are shown after normalization to the wild-type (+/+) sample. (c) Transcriptional array analysis. The relative mRNA expression levels for B2M, HLA class I heavy chains, and other B2M-binding protein genes (CD1A-D, FCGRT, HFE, and MR1) are shown for two B2M−/− clones (loxP/loxP c1 and c2) and two independent cultures of parental H1 ESCs (+/+). Values are expressed as log2, after normalization and background subtraction. (d) Sample relation between two duplicate cultures of parental H1 cells (H1A and H1B) and two B2M−/− clones (loxP/loxP c1 and c2) based on a global expression analysis of 18,174 genes with SD/mean >0.1. HLA, human leukocyte antigens.
Human immune cell responses to class I–deficient cells
The lack of HLA class I surface expression on B2MloxP/loxP cells should prevent their recognition by CD8+ T cells. We first examined this with Mixed Leukocyte Reaction (MLR) assays in which allogeneic peripheral blood mononuclear cells (PBMCs) from normal blood donors were mixed with undifferentiated, irradiated B2M+/+ or B2MloxP/loxP ESCs. In both cases, we did not observe proliferation responses as evidenced by the lack of 3H-thymidine uptake (Figure 4a, left panel). These findings confirm prior results showing a lack of MLR responses to undifferentiated ESCs,15 and presumably reflect the absence of costimulatory molecules and/or the presence of inhibitors on pluripotent cells.16 To elicit a more robust MLR response, we differentiated ESCs into embryoid bodies (EBs) for 15 days, which allows for the development of cells entering the endothelial/hematopoietic lineages that might be competent for antigen presentation.17 At this timepoint, 12–15% of cells were CD34+ by flow cytometry (Figure 4b), but HLA class II expression remained low (<1.2% of cells), allowing us to focus on class I–dependent responses. Wild-type B2M+/+ EB cells were capable of stimulating PBMC proliferation in an MLR assay, albeit at 14–28% the level of allogeneic PBMCs (Figure 4a, right panel), perhaps due to the smaller proportion of EB cells capable of antigen presentation. In contrast, the MLR assay using B2MloxP/loxP EB cells was similar to that of unstimulated PBMCs. This suggested that class I–deficient cells produce a decreased allogeneic response, at least when HLA class II expression is limited. However, these MLR results should be interpreted cautiously, because human ESCs can also inhibit MLR responses of PBMCs against allogeneic dendritic cells, and ESC-derived EB cells did not induce significant PBMC proliferation in a prior study.15
Figure 4.

Immune responses to B2M-targeted cells. (a) MLR results showing 3[H]-thymidine incorporation by PBMC responder cells mixed with irradiated, undifferentiated B2M+/+ or B2MloxP/loxP ESCs (left panel) or with day 15 EB-derived cells (right panel) at a ratio of 1:1. Two independent PBMC responder cell preparations were used when analyzing EB cells. Controls included unstimulated PBMCs, and PBMCs stimulated by allogeneic PBMCs. (b) Flow cytometry analysis of day 15 B2M+/+ or B2MloxP/loxP EB cells showing the surface expression of CD34, HLA class I (HLA-A, -B, and -C), and HLA class II (HLA-DR), with isotype controls. All preparations were labeled with 7-amino-actinomycin D (7AAD) to improve gating and remove dead cells. (c) Intracellular cytokine staining of HLA-A*0201/NLV-CMVpp65-specific T cells stimulated with an equal number of B2M+/+ or B2MloxP/loxP EB cells, HLA class I–deficient K562 cells, or HLA-A*0201–expressing K562-A2 cells, with or without prior pulsing with NLVPMVATV (NLV) peptide from the CMV pp65 protein. HLA, human leukocyte antigens.
To demonstrate more directly that HLA class I–restricted T cell responses were not elicited by B2MloxP/loxP cells, we looked for interferon-γ (IFN-γ) expression in an HLA-A*0201–restricted human CD8+ T cell clone that recognizes the specific peptide NLVPMVATV derived from cytomegalovirus. K562 cells served as a negative control lacking HLA class I antigens, and K562-A2 cells transfected with the HLA-A*0201 gene and pulsed with the peptide served as a positive control. Exposure to wild-type B2M+/+ EB cells (HLA-A*0201+) induced significant IFN-γ expression in these CD8+ T cells, which was comparable with that induced by K562-A2 cells, but only after loading with the peptide, demonstrating the specificity of the response (Figure 4c). In contrast, IFN-γ expression was significantly reduced in T cells exposed to B2MloxP/loxP EB cells (2.11% as compared with 17.8% for B2M+/+ cells). The basis for this low-level residual IFN-γ expression is unclear, but possible explanations include peptide uptake and presentation by class I–expressing T cells exposed to EB cell membranes, and/or the effects of free HLA class I heavy chains present in EB cells.
Class I–negative hematopoietic cells may be lysed by natural killer (NK) cells,18 so this is a potential concern for our approach. We incubated human NK cells with either B2M+/+ or B2M−/− EB cells and did not observe an increase in surface expression of CD107a, which is an indicator of NK cell degranulation and cytotoxicity (Figure 5). HLA class I–deficient K562 control cells did induce significant degranulation. Thus, there was no NK-mediated cytotoxicity against B2M−/−cells, at least at the EB stage of differentiation.
Figure 5.

NK cells do not lyse B2M−/− EB cells. Flow cytometry analysis of CD107a expression on CD56+ NK effector cells when incubated with B2M+/+ or B2MloxP/loxP day 15 EB cells at a ratio of 1:1. Controls included unstimulated NK cells, and NK cells incubated with class I–negative K562 cells. NK, natural killer.
Discussion
In this report, we describe two gene targeting approaches for engineering the HLA locus in human pluripotent stem cells to overcome immunogenicity and potentially enhance their therapeutic utility. AAV vectors were used in both cases, which allowed us to efficiently isolate targeted clones from a small number of antibiotic-resistant colonies. Over 95% of resistant clones were targeted by the HMGA1 initiation codon trap vector, and 10–30% of resistant clones were targeted when inserting a functional promoter-transgene cassette at the B2M locus. Both approaches produced transgene-free cells. There was minimal genotoxicity from these manipulations, because site-specific endonucleases were not used to stimulate homologous recombination, the vector uses host cell enzymes for integration, and infection with AAV vectors does not lead to increased mutation frequencies.19 This was largely confirmed by the normal karyotypes we obtained in both of the B2MloxP/loxP clones that were analyzed and is consistent with our prior cytogenetic studies of ESCs and iPSCs that underwent AAV-mediated gene targeting.20 The microarray data also showed a consistency in gene expression patterns, because the B2M−/− clones could not be distinguished from the parental H1 cells except for their lack of B2M transcripts. One potential caveat in this regard was our use of Cre-recombinase to remove the antibiotic resistance genes in the B2M targeting approach, because this may have introduced unwanted recombination events at pseudo-loxP sites that were difficult to detect.21 Given the importance of maintaining genomic integrity in cells destined for clinical use, it may be preferable to eliminate this step in the future. This could be accomplished by using a retargeting strategy, in which the antibiotic resistance genes are removed instead by an additional gene targeting step.
The two approaches have different advantages and potential applications. HLA-homozygous cells could be derived from heterozygous ESCs to generate a stem cell bank comprised of relatively few cell lines that matches a significant percentage of the population. A bank of 30 HLA-homozygous ESC lines with common haplotypes would match 38–59% of the US population at HLA-A, -B, and -DR, and a single line with the most common haplotype would match 9% of Caucasian Americans.3 Our strategy overcomes some of the limitations of other approaches for creating HLA-homozygous stem cells. For example, ESCs could be derived directly from HLA-homozygous embryos, but this might require the ethically questionable use of HLA-typed sperm and egg donors specifically for ESC derivation. Parthenogenesis could be used to generate HLA-homozygous ESCs from heterozygous oocytes; however, this results in genome-wide homozygosity that could unmask recessive mutations. A more realistic alternative may be to derive a bank of iPSCs from HLA-homozygous individuals,5 although iPSCs may not be identical to ESCs in their therapeutic potential.22 If iPSCs are ever used clinically, our method would allow the derivation of histocompatible, HLA-homozygous iPSCs from the family members of patients that share one HLA haplotype, which could provide a source of matched, genetically normal cells for the treatment of genetic diseases.
One technical difficulty in our experiments was the low frequency of recovering HLA-homozygous cells by ganciclovir selection as compared to other types of TK mutations. This is consistent with recent results showing that mitotic recombination with loss of heterozygosity accounts for <1% of ganciclovir-resistant colonies in human pluripotent stem cells,23 in contrast to what one would expect based on mouse ESC experiments.9 Although this will hinder the routine application of our approach in regenerative medicine, there may be ways to improve the recovery of extended loss of heterozygosity events by inducing mitotic recombination or improving selection stringency by introducing a second TK gene telomeric to the HLA locus on chromosome 6.
The major advantage of B2M knockout cells is that they could serve as universal donors when transplanting cells that express HLA class I but not class II, including most solid organ cells. This would produce substantial savings in time and money, because a single cell line could be differentiated into therapeutic cell products, characterized, certified by regulatory agencies, and used in all recipients. Universal donor cells also avoid many of the potential problems of HLA-typed cell banks and patient-specific stem cells, including variability among different stem cell lines in gene expression patterns,24 culture characteristics,25 differentiation potential,6,7 and genetic changes acquired during culture.26 Unlike patient-derived iPSCs, universal donor cells can be prepared ahead of time and are ideal for treating genetic diseases, because they do not harbor disease-causing mutations. Additional benefits of HLA class I–deficient cells are that they will not present autoantigens that might otherwise prevent the successful treatment of autoimmune diseases, such as peptide antigens derived from GAD65 or insulin that can cause the destruction of β cells in diabetes,27 and immune responses against therapeutic gene products can be avoided when treating genetic diseases, such as antidystrophin responses in muscular dystrophy.28
Significant evidence supports the concept that HLA class I–negative cells will function normally and avoid allogeneic rejection after transplantation. First, rare individuals who are class I–negative due to mutations in the TAP1 or TAP2 genes encoding the transporter associated with antigen processing are relatively healthy, with a spectrum of manifestations ranging from the absence of symptoms to chronic upper respiratory infections and skin inflammation,29 showing that HLA class I–deficient human cells can form all essential organs. Second, B2m−/− mice have been extensively studied and they are relatively healthy except for a notable lack of CD4–CD8+ T cells.30 Importantly, transplantation experiments have shown that organs or cells from B2m−/− mouse donors survive longer than allogeneic cells in B2m+/+ recipients (sometimes persisting indefinitely), including liver cells,31 kidneys,32 hearts,33 pancreatic islets,34 and dendritic cells.35 These mouse experiments suggest that B2m−/− human cells may also survive longer than allogeneic cells in many of the clinical settings being considered for pluripotent stem cells. In support of this, our data show that in comparison with wild-type cells, B2M−/− human cells induce less of an immune response in allogeneic human PBMCs and HLA-A*0201–restricted CD8+ T cells as determined by MLR and IFN-γ expression assays respectively. Although the lack of B2M could also affect the function of non-HLA, B2M-binding proteins such as HFE, which is associated with hepatic iron overload,36 this would not be expected to impact transplant recipients with B2M+/+ host cells.
The complete lack of HLA class I expression raises potential concerns. In mice, host NK cells have been shown to eliminate B2m−/− hematopoietic donor cells,37 which occurs in the absence of specific inhibitory interactions between donor cell class I antigens and NK cell Ly49 receptors.38 An analogous inhibition of human NK cells is mediated through interactions of HLA-C, -E, and -G with several NK cell receptors.39,40 Although this is clearly an issue for hematopoietic cells, it is not known if this “missing self” response will be observed after transplanting non-hematopoietic cell types, because the same phenomena was not observed when solid organs from B2m−/− mice were transplanted in B2m+/+ recipients.31,32,33,34 We did not detect NK-mediated lysis in day 15 B2M−/− EB cells, so further differentiation down the hematopoietic lineage may be necessary to demonstrate the “missing self” phenomenon in ESC-derived cells. A second concern is the potential for tumor formation, because HLA expression is frequently lost or reduced in cancer cells.41 However, neither HLA class I–deficient mice30 nor humans29 are tumor-prone. A third concern is viral infection of B2M−/− cells, which may escape class I–restricted immune responses. However, B2M−/− mice are able to survive infection with influenza42 or Sendai virus,43 and this would be mitigated when transplanting B2M−/− human cells to anatomically sequestered locations in individuals with fully functional immune systems.
Some of these concerns could be addressed by further engineering of B2M−/− stem cells, including the introduction of single chain fusion constructs encoding non-polymorphic HLA class I proteins fused to B2M. Both HLA-G and HLA-E have been shown to interact with inhibitory NK cell receptors and prevent NK cell–mediated lysis.39,40 The potential tumor risk could be addressed by the introduction of an inducible suicide gene that could be activated if aberrant cell proliferation occurred.44 The additional disruption of HLA class II genes in B2M−/− cells could also be beneficial in some settings, because solid organ cells may be induced to express class II genes under various stress responses45 and this would allow the derivation of universal donor hematopoietic cell products that might otherwise be rejected due to class II antigen presentation.
Materials and Methods
Cell culture. Undifferentiated human ESCs were cultured as described.20 Differentiation of ESCs into embryoid bodies was performed as described.46 Standard G-banding karyotypes were carried out in the Cytogenetics Laboratory of the Department of Pathology, University of Washington (Seattle, WA). Teratoma assays were performed as described.20 All animal experiments (teratoma assays) were approved by the University of Washington Institutional Animal Care and Use Committee.
CD8+ T cell clone 6G1-82, specific for the HLA-A*0201 presenting the peptide NLVPMVATV from the CMV pp65 protein,47 was obtained and expanded as described.48 K562 human chronic myeloid leukemia cells K562-A2 cells expressing HLA-A*0201 were previously described.49 All cells were grown at 37 °C in a 5% CO2 atmosphere.
Viral vectors. The AAV2-HMGA1-HyTKpA vector was described previously.20 The AAV3-B2M-ETKNpA and AAV3-B2M-EHyTKpA vectors contain genomic DNA from the B2M locus (chromosome 15: nucleotides 45,002,685–45,004,747; and nucleotides 45,002,938–45,004,747, respectively, February 2009 assembly); with an EF1α (eukaryotic translation elongation factor 1α) promoter; TKNeo gene (herpes simplex virus thymidine kinase—neomycin phosphotransferase fusion gene) or HyTK gene (hygromycin phosphotransferase—herpes simplex virus thymidine kinase fusion gene) respectively, flanking loxP sites, stop codons in all three frames and a polyadenylation signal at the 3′end. The TKNeo and HyTK cassettes were inserted at the initiation codon in exon 1 of B2M. AAV vector stocks were produced and titered as described.12 The non-integrating foamy virus vector NIFV-EokCreW is identical to the NIFV-ECreW vector described previously,50 but with an optimized Kozak sequence in the Cre gene.
Gene targeting and ganciclovir selection. AAV-mediated gene targeting experiments were performed as described previously. For HMGA1 targeting, 5 × 104 H1 ESCs were plated per well of a 24-well plate on day 0, and on the next day cells were infected with the AAV2-HMGA1-HyTKpA vector at a multiplicity of infection of 2 × 104 genome-containing vector particles per cell. On day 4, hygromycin B (20 µg/ml) was added to the culture and selection was continued for 16 days. To isolate HLA-homozygous clones, HMGA1-targeted cells were selected with 3 µmol/l ganciclovir for 9 days.
For B2M targeting, 7.5 × 105 H1 ESCs were plated per 10 cm dish on day 0 and infected on day 1 with the AAV-B2M-ETKNpA vector at a multiplicity of infection of 20,000 genome-containing vector particles per cell. On day 4, the cells were cultured in 50 µg/ml G418 and selection was continued for 22–36 days. When targeting the second B2M allele, 2.5 × 105 ESCs targeted at the first allele with AAV-B2M-ETKNpA were plated per 6 cm dish on day 0 and infected on day 1 with the AAV-B2M-EHyTKpA vector at a multiplicity of infection of 2,000 genome-containing vector particles per cell. On day 4, the cells were cultured in 20 µg/ml hygromycin and selection was continued for 26–28 days. In each case, a portion of cells was also plated without selection to determine the percentage of transduced colony-forming units.
Cre-mediated transgene excision. 2.5 × 104 ESC targeted at both B2M alleles were plated per well of a 48-well plate on day 0. On day 1, the cells were infected with the non-integrating Foamy vector NIFV-EokCreW at a multiplicity of infection of 50,000 genome-containing vector particles per cell. On day 15, these cells were disaggregated into single cells with accutase (Stemgent, San Diego, CA) counted, and plated at densities of 106 or 0.5 × 106 cells per 10 cm dish. On day 28, individual colonies were picked and expanded for further analysis.
DNA isolation and analysis. Genomic DNA was isolated by using the Puregene DNA purification system (Gentra Systems, Minneapolis, MN). Southern blot analysis, plasmid preparation, and restriction digestion were performed according to standard protocols. Radiolabeled probes were synthesized by random priming using Rediprime II (GE Healthcare, Piscataway, NJ). For PCR analysis, cells were lysed in 50 mmol/l Tris pH 8, 1 mmol/l EDTA, 0.2 mol/l sodium chloride, treated with 200 µg/ml Proteinase K (Invitrogen, Carlsbad, CA) at 55 °C for 1 hour, and heated at 95 °C for 5 minutes to inactivate Proteinase K. The lysate was clarified by centrifugation at 16,000g for 10 minutes and the supernatant was used as template for the PCR. Primers used in the PCR were: (A) forward primer: CGCCGATGTACAGACAGCAAA; (B) reverse primer: TATCGACGGATCCCACACAA; (C) reverse primer: ACCGTCTATATAAACCCGCAGT; and (D) reverse primer: GCCAAAGGTCTCCCCTGCTCC. For the PCR, GoTaq polymerase (Promega, Madison, WI) was used, and the conditions were as follows: 150 ng of the template genomic DNA, 0.5 µmol/l each primer, 0.25 mmol/l dNTPs, and 2 mmol/l MgCl2. The annealing temperatures were 56 °C (primers A + C and A + D) or 58 °C (primers A + B) and amplification was run for 35 cycles in a PTC-200 Thermo Cycler (Biorad, MJ Research, Hercules, CA). Short extension times (40 seconds) were used for primers A + D to amplify only the loxP or the wild-type alleles.
Western blots. Protein extracts were obtained by lysing cells in 50 mmol/l Tris pH 8, 150 mmol/l NaCl, 1 mmol/l EDTA, 1% (v/v) NP40, and Complete Protease Inhibitor Cocktail (Roche Diagnostics, Mannheim, Germany), and clarified by centrifugation at 16,000g for 30 minutes at 4 °C. Samples were analyzed in precast SDS-PAGE gels (Biorad), under non-reducing conditions for the anti-HLA antibody or reducing conditions for the anti-B2M antibody; and electroblotted to nitrocellulose membranes (Biorad). After blocking with 5% milk in phosphate-buffered saline, the membranes were probed with the corresponding primary and secondary antibodies (see below), developed with the enhanced luminescence (ECL, Pierce, Thermo Scientific, Rockford, IL), and exposed to X-ray films. Anti-HLA-ABC antibody clone W6/32 was obtained from Sigma-Aldrich (St Louis, MO). Anti-B2M BBM.1 antibody was a gift from D Geraghty. Antitubulin antibody was from Abcam (Cambridge, MA). Secondary antibodies and antimouse horseradish peroxidase were obtained from Santa Cruz Biotechnology (Santa Cruz, CA).
Flow cytometry. Undifferentiated ESC suspensions were washed with flow buffer (phosphate-buffered saline containing 2% fetal bovine serum, 0.05% [v/v] sodium azide, and 2 mmol/l EDTA), then incubated with the appropriate antibodies for 30 minutes in the dark on ice. Embryoid body cells were obtained by trypsinization and preincubated with Fc blocking reagent (Miltenyi Biotec, Auburn, CA) for 10 minutes at 4 °C before staining with the antibodies. 7-amino-actinomycin D from Via-Probe (BD Biosciences, Bedford, MA) was added last to exclude dead cells from the analysis. Samples were analyzed on a FACscan or Calibur (BD Biosciences) instrument and the data were analyzed using FlowJo software (TreeStar, Ashland, OR). The antibodies used for flow cytometry were: anti-B2M PE clone B2M-01 (Santa Cruz Biotechnology), anti-HLA-ABC clone W6/32 FITC (eBioscience, San Diego, CA), anti-HLA-DR FITC (Miltenyi Biotec), anti-CD34 FITC (BD Pharmingen, San Diego, CA), anti-CD45 FITC (BD Pharmingen), anti-CD8 APC (BD Pharmingen), and anti-IFN-γ FITC (BD Pharmingen). Isotype control antibodies were obtained from Miltenyi Biotec or BD Biosciences.
Mixed lymphocyte reactions. PBMCs were obtained from healthy donors at the Puget Sound Blood Center (Seattle, WA) by centrifugation in Ficoll (LSM, MP Biomedicals, Santa Ana, CA) gradients as previously described. MLRs were done following established protocols. Briefly, stimulator cells (hESCs, day 15 EB cells, or PBMCs for the allogeneic positive control) were irradiated with 4,000 rad, and 105 cells were incubated with the responder PBMCs at a ratio of 1:1 in a 96-well round-bottom plate in RPMI-1640 supplemented with glutamine, 10% heat-inactivated human AB serum (GemCell, Sacramento, CA), and 50 µmol/l β-mercaptoethanol. After 6 days, these mixtures were pulsed with 3[H]-thymidine (Perkin Elmer, Santa Clara, CA) for 16–18 hours and harvested onto glass fiber filters (Filtermat A; Perkin Elmer) using a Filtermate Harvester. The amount of radioactivity incorporated was determined with an LS6500 counter (Beckman, Indianapolis IN).
Intracellular cytokine staining. Cells derived from trypsinized embryoid bodies on day 15–18 of the differentiation protocol, K562 cells, or K562-A2 cells were pulsed with 5 µg/ml of peptide NLVPMVATV (>90% purity; GeneScript, Piscataway, NJ) at 37 °C for 2 hours. After the pulse, cells were washed four times then mixed with the responder CD8+ T cell clone 6G1-82 at a ratio 1:1, and incubated for 4 hours at 37 °C, with brefeldin A (Sigma-Aldrich) being added to the cells after 1 hour. The cells were then fixed and permeabilized with Fix and Perm kit (BD Biosciences), costained with FITC-conjugated anti-IFN-γ antibody (BD Biosciences), and APC-conjugated anti-CD8 antibody (BD Biosciences), and analyzed by flow cytometry.
CD107a expression. NK effector cells were isolated from blood samples by positive selection using Miltenyi's magnetic beads and the CliniMacs purification system. The sorted cells were >94% CD56+ by flow cytometry. Effector (NK) and target cells (K562 or EB-derived cells) were incubated at a ratio of 1:1 in the presence of anti-CD107a-PeCy5 (BD Pharmingen) at 37 °C in 5% CO2. After 1 hour, 6 µg/ml monensin (Golgi-Stop, BD Biosciences) was added to the culture and the cells were incubated for an additional 4 hours. The cells were then stained with anti-CD56 PE (Invitrogen) and the samples were analyzed by flow cytometry.
Microarray analysis. RNA was isolated from hESCs cultured without feeder cells and hybridized to the HumanHT 12 v4 BeadChip (Illumina, San Diego, CA) by the Fred Hutchinson Cancer Research Center Genomics Shared Resource. The gene expression levels of all RefSeq genes were quantile normalized. Cell comparisons were conducted using the Bioconductor package “limma” (http://bioconductor.org/) by calculating the 75th percentile of negative controls for each array and using this as a minimum signal intensity threshold value to be applied to each array. For probe sets considered detectable, microarray data normalized to the median and log2 ratios were calculated. Microarray data have been deposited under Gene Expression Omnibus accession number (GSE42289).
For the analysis of the of SNPs copy number, genomic DNA was isolated from parental H1 hESCs and GCV-resistant clone c5A, and hybridized to the Affymetrix GeneChip Human Mapping 250K Nsp Array at the University of Washington Center for Array Technology. Data were analyzed using the GeneChip Genotyping Analysis Software (GTYPE; Affymetrix, Santa Clara, CA) and Copy Number Analysis Tool.
SUPPLEMENTARY MATERIAL Figure S1. Trilineage teratoma formation in HLA-homozygous ESC clone c5A. Figure S2. Trilineage teratoma formation and normal karyotype of B2MloxP/loxP cells. Table S1. HMGA1 targeting and GCV-resistant clone analysis. Table S2. B2M targeting experiments.
Acknowledgments
We thank Daniel Geraghty (Fred Hutchinson Cancer Research Center, Seattle, WA) for providing the anti-B2M BBM-1 antibody, Li B Li for help with array data analysis, and Raisa Stolitenko for technical assistance. This work was supported by US National Institutes of Health grants DK55759, HL53750, and GM086497 to D.W.R. and AI053193 to S.R.R. D.W.R. is on the Advisory Board of Horizon Discovery. The other authors declare no conflict of interest.
Supplementary Material
References
- Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, et al. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007;131:861–872. doi: 10.1016/j.cell.2007.11.019. [DOI] [PubMed] [Google Scholar]
- Yu J, Vodyanik MA, Smuga-Otto K, Antosiewicz-Bourget J, Frane JL, Tian S, et al. Induced pluripotent stem cell lines derived from human somatic cells. Science. 2007;318:1917–1920. doi: 10.1126/science.1151526. [DOI] [PubMed] [Google Scholar]
- Faden RR, Dawson L, Bateman-House AS, Agnew DM, Bok H, Brock DW, et al. Public stem cell banks: considerations of justice in stem cell research and therapy. Hastings Cent Rep. 2003;33:13–27. [PubMed] [Google Scholar]
- Taylor CJ, Bolton EM, Pocock S, Sharples LD, Pedersen RA, Bradley JA. Banking on human embryonic stem cells: estimating the number of donor cell lines needed for HLA matching. Lancet. 2005;366:2019–2025. doi: 10.1016/S0140-6736(05)67813-0. [DOI] [PubMed] [Google Scholar]
- Okita K, Matsumura Y, Sato Y, Okada A, Morizane A, Okamoto S, et al. A more efficient method to generate integration-free human iPS cells. Nat Methods. 2011;8:409–412. doi: 10.1038/nmeth.1591. [DOI] [PubMed] [Google Scholar]
- Chang KH, Nelson AM, Fields PA, Hesson JL, Ulyanova T, Cao H, et al. Diverse hematopoietic potentials of five human embryonic stem cell lines. Exp Cell Res. 2008;314:2930–2940. doi: 10.1016/j.yexcr.2008.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pekkanen-Mattila M, Kerkelä E, Tanskanen JM, Pietilä M, Pelto-Huikko M, Hyttinen J, et al. Substantial variation in the cardiac differentiation of human embryonic stem cell lines derived and propagated under the same conditions–a comparison of multiple cell lines. Ann Med. 2009;41:360–370. doi: 10.1080/07853890802609542. [DOI] [PubMed] [Google Scholar]
- Mayshar Y, Ben-David U, Lavon N, Biancotti JC, Yakir B, Clark AT, et al. Identification and classification of chromosomal aberrations in human induced pluripotent stem cells. Cell Stem Cell. 2010;7:521–531. doi: 10.1016/j.stem.2010.07.017. [DOI] [PubMed] [Google Scholar]
- Lefebvre L, Dionne N, Karaskova J, Squire JA, Nagy A. Selection for transgene homozygosity in embryonic stem cells results in extensive loss of heterozygosity. Nat Genet. 2001;27:257–258. doi: 10.1038/85808. [DOI] [PubMed] [Google Scholar]
- Thomson JA, Itskovitz-Eldor J, Shapiro SS, Waknitz MA, Swiergiel JJ, Marshall VS, et al. Embryonic stem cell lines derived from human blastocysts. Science. 1998;282:1145–1147. doi: 10.1126/science.282.5391.1145. [DOI] [PubMed] [Google Scholar]
- Mitalipova M, Calhoun J, Shin S, Wininger D, Schulz T, Noggle S, et al. Human embryonic stem cell lines derived from discarded embryos. Stem Cells. 2003;21:521–526. doi: 10.1634/stemcells.21-5-521. [DOI] [PubMed] [Google Scholar]
- Khan IF, Hirata RK, Russell DW. AAV-mediated gene targeting methods for human cells. Nat Protoc. 2011;6:482–501. doi: 10.1038/nprot.2011.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gharwan H, Hirata RK, Wang P, Richard RE, Wang L, Olson E, et al. Transduction of human embryonic stem cells by foamy virus vectors. Mol Ther. 2007;15:1827–1833. doi: 10.1038/sj.mt.6300244. [DOI] [PubMed] [Google Scholar]
- Arce-Gomez B, Jones EA, Barnstable CJ, Solomon E, Bodmer WF. The genetic control of HLA-A and B antigens in somatic cell hybrids: requirement for beta2 microglobulin. Tissue Antigens. 1978;11:96–112. doi: 10.1111/j.1399-0039.1978.tb01233.x. [DOI] [PubMed] [Google Scholar]
- Li L, Baroja ML, Majumdar A, Chadwick K, Rouleau A, Gallacher L, et al. Human embryonic stem cells possess immune-privileged properties. Stem Cells. 2004;22:448–456. doi: 10.1634/stemcells.22-4-448. [DOI] [PubMed] [Google Scholar]
- Drukker M, Katz G, Urbach A, Schuldiner M, Markel G, Itskovitz-Eldor J, et al. Characterization of the expression of MHC proteins in human embryonic stem cells. Proc Natl Acad Sci USA. 2002;99:9864–9869. doi: 10.1073/pnas.142298299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan X, Dravid G, Ye Z, Hammond H, Shamblott M, Gearhart J, et al. Functional antigen-presenting leucocytes derived from human embryonic stem cells in vitro. Lancet. 2004;364:163–171. doi: 10.1016/S0140-6736(04)16629-4. [DOI] [PubMed] [Google Scholar]
- Ljunggren HG, Kärre K. In search of the ‘missing self': MHC molecules and NK cell recognition. Immunol Today. 1990;11:237–244. doi: 10.1016/0167-5699(90)90097-s. [DOI] [PubMed] [Google Scholar]
- Miller DG, Petek LM, Russell DW. Adeno-associated virus vectors integrate at chromosome breakage sites. Nat Genet. 2004;36:767–773. doi: 10.1038/ng1380. [DOI] [PubMed] [Google Scholar]
- Khan IF, Hirata RK, Wang PR, Li Y, Kho J, Nelson A, et al. Engineering of human pluripotent stem cells by AAV-mediated gene targeting. Mol Ther. 2010;18:1192–1199. doi: 10.1038/mt.2010.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thyagarajan B, Guimarães MJ, Groth AC, Calos MP. Mammalian genomes contain active recombinase recognition sites. Gene. 2000;244:47–54. doi: 10.1016/s0378-1119(00)00008-1. [DOI] [PubMed] [Google Scholar]
- Kim K, Doi A, Wen B, Ng K, Zhao R, Cahan P, et al. Epigenetic memory in induced pluripotent stem cells. Nature. 2010;467:285–290. doi: 10.1038/nature09342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li LB, Chang KH, Wang PR, Hirata RK, Papayannopoulou T, Russell DW. Trisomy correction in down syndrome induced pluripotent stem cells. Cell Stem Cell. 2012;11:615–619. doi: 10.1016/j.stem.2012.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosler ES, Fisk GJ, Ares X, Irving J, Miura T, Rao MS, et al. Long-term culture of human embryonic stem cells in feeder-free conditions. Dev Dyn. 2004;229:259–274. doi: 10.1002/dvdy.10430. [DOI] [PubMed] [Google Scholar]
- Ware CB, Nelson AM, Blau CA. A comparison of NIH-approved human ESC lines. Stem Cells. 2006;24:2677–2684. doi: 10.1634/stemcells.2005-0452. [DOI] [PubMed] [Google Scholar]
- Närvä E, Autio R, Rahkonen N, Kong L, Harrison N, Kitsberg D, et al. High-resolution DNA analysis of human embryonic stem cell lines reveals culture-induced copy number changes and loss of heterozygosity. Nat Biotechnol. 2010;28:371–377. doi: 10.1038/nbt.1615. [DOI] [PubMed] [Google Scholar]
- Di Lorenzo TP, Peakman M, Roep BO. Translational mini-review series on type 1 diabetes: Systematic analysis of T cell epitopes in autoimmune diabetes. Clin Exp Immunol. 2007;148:1–16. doi: 10.1111/j.1365-2249.2006.03244.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendell JR, Campbell K, Rodino-Klapac L, Sahenk Z, Shilling C, Lewis S, et al. Dystrophin immunity in Duchenne's muscular dystrophy. N Engl J Med. 2010;363:1429–1437. doi: 10.1056/NEJMoa1000228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmer J, Andrès E, Donato L, Hanau D, Hentges F, de la Salle H. Clinical and immunological aspects of HLA class I deficiency. QJM. 2005;98:719–727. doi: 10.1093/qjmed/hci112. [DOI] [PubMed] [Google Scholar]
- Zijlstra M, Bix M, Simister NE, Loring JM, Raulet DH, Jaenisch R. Beta 2-microglobulin deficient mice lack CD4-8+ cytolytic T cells. 1990. J Immunol. 2010;184:4587–4591. [PubMed] [Google Scholar]
- Li X, Faustman D. Use of donor beta 2-microglobulin-deficient transgenic mouse liver cells for isografts, allografts, and xenografts. Transplantation. 1993;55:940–946. doi: 10.1097/00007890-199304000-00046. [DOI] [PubMed] [Google Scholar]
- Coffman T, Geier S, Ibrahim S, Griffiths R, Spurney R, Smithies O, et al. Improved renal function in mouse kidney allografts lacking MHC class I antigens. J Immunol. 1993;151:425–435. [PubMed] [Google Scholar]
- Qian S, Fu F, Li Y, Lu L, Rao AS, Starzl TE, et al. Impact of donor MHC class I or class II antigen deficiency on first- and second-set rejection of mouse heart or liver allografts. Immunology. 1996;88:124–129. doi: 10.1046/j.1365-2567.1996.d01-633.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prange S, Zucker P, Jevnikar AM, Singh B. Transplanted MHC class I-deficient nonobese diabetic mouse islets are protected from autoimmune injury in diabetic nonobese recipients. Transplantation. 2001;71:982–985. doi: 10.1097/00007890-200104150-00025. [DOI] [PubMed] [Google Scholar]
- Matsunaga Y, Fukuma D, Hirata S, Fukushima S, Haruta M, Ikeda T, et al. Activation of antigen-specific cytotoxic T lymphocytes by beta 2-microglobulin or TAP1 gene disruption and the introduction of recipient-matched MHC class I gene in allogeneic embryonic stem cell-derived dendritic cells. J Immunol. 2008;181:6635–6643. doi: 10.4049/jimmunol.181.9.6635. [DOI] [PubMed] [Google Scholar]
- Feder JN, Gnirke A, Thomas W, Tsuchihashi Z, Ruddy DA, Basava A, et al. A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis. Nat Genet. 1996;13:399–408. doi: 10.1038/ng0896-399. [DOI] [PubMed] [Google Scholar]
- Bix M, Liao NS, Zijlstra M, Loring J, Jaenisch R, Raulet D. Rejection of class I MHC-deficient haemopoietic cells by irradiated MHC-matched mice. Nature. 1991;349:329–331. doi: 10.1038/349329a0. [DOI] [PubMed] [Google Scholar]
- Karlhofer FM, Ribaudo RK, Yokoyama WM. MHC class I alloantigen specificity of Ly-49+ IL-2-activated natural killer cells. Nature. 1992;358:66–70. doi: 10.1038/358066a0. [DOI] [PubMed] [Google Scholar]
- Pazmany L, Mandelboim O, Valés-Gómez M, Davis DM, Reyburn HT, Strominger JL. Protection from natural killer cell-mediated lysis by HLA-G expression on target cells. Science. 1996;274:792–795. doi: 10.1126/science.274.5288.792. [DOI] [PubMed] [Google Scholar]
- Lee N, Llano M, Carretero M, Ishitani A, Navarro F, López-Botet M, et al. HLA-E is a major ligand for the natural killer inhibitory receptor CD94/NKG2A. Proc Natl Acad Sci USA. 1998;95:5199–5204. doi: 10.1073/pnas.95.9.5199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hicklin DJ, Wang Z, Arienti F, Rivoltini L, Parmiani G, Ferrone S. beta2-Microglobulin mutations, HLA class I antigen loss, and tumor progression in melanoma. J Clin Invest. 1998;101:2720–2729. doi: 10.1172/JCI498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichelberger M, Allan W, Zijlstra M, Jaenisch R, Doherty PC. Clearance of influenza virus respiratory infection in mice lacking class I major histocompatibility complex-restricted CD8+ T cells. J Exp Med. 1991;174:875–880. doi: 10.1084/jem.174.4.875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hou S, Doherty PC, Zijlstra M, Jaenisch R, Katz JM. Delayed clearance of Sendai virus in mice lacking class I MHC-restricted CD8+ T cells. J Immunol. 1992;149:1319–1325. [PubMed] [Google Scholar]
- Di Stasi A, Tey SK, Dotti G, Fujita Y, Kennedy-Nasser A, Martinez C, et al. Inducible apoptosis as a safety switch for adoptive cell therapy. N Engl J Med. 2011;365:1673–1683. doi: 10.1056/NEJMoa1106152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yousem SA, Curley JM, Dauber J, Paradis I, Rabinowich H, Zeevi A, et al. HLA-class II antigen expression in human heart-lung allografts. Transplantation. 1990;49:991–995. doi: 10.1097/00007890-199005000-00030. [DOI] [PubMed] [Google Scholar]
- Silk KM, Tseng SY, Nishimoto KP, Lebkowski J, Reddy A, Fairchild PJ. Differentiation of dendritic cells from human embryonic stem cells. Methods Mol Biol. 2011;767:449–461. doi: 10.1007/978-1-61779-201-4_33. [DOI] [PubMed] [Google Scholar]
- Wills MR, Carmichael AJ, Mynard K, Jin X, Weekes MP, Plachter B, et al. The human cytotoxic T-lymphocyte (CTL) response to cytomegalovirus is dominated by structural protein pp65: frequency, specificity, and T-cell receptor usage of pp65-specific CTL. J Virol. 1996;70:7569–7579. doi: 10.1128/jvi.70.11.7569-7579.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manley TJ, Luy L, Jones T, Boeckh M, Mutimer H, Riddell SR. Immune evasion proteins of human cytomegalovirus do not prevent a diverse CD8+ cytotoxic T-cell response in natural infection. Blood. 2004;104:1075–1082. doi: 10.1182/blood-2003-06-1937. [DOI] [PubMed] [Google Scholar]
- Terakura S, Yamamoto TN, Gardner RA, Turtle CJ, Jensen MC, Riddell SR. Generation of CD19-chimeric antigen receptor modified CD8+ T cells derived from virus-specific central memory T cells. Blood. 2012;119:72–82. doi: 10.1182/blood-2011-07-366419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deyle DR, Khan IF, Ren G, Wang PR, Kho J, Schwarze U, et al. Normal collagen and bone production by gene-targeted human osteogenesis imperfecta iPSCs. Mol Ther. 2012;20:204–213. doi: 10.1038/mt.2011.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.