

Abstract
Retroviral vectors (RVs) are powerful tools in clinical gene therapy. However, stable genomic integration of RVs can be oncogenic, as reported in several animal models and in clinical trials. Previously, we observed that T-cell receptor (TCR) polyclonal mature T cells are resistant to transformation after gammaretroviral transfer of (proto-)oncogenes, whereas TCR-oligoclonal T cells were transformable in the same setting. Here, we describe the induction of a mature T-cell lymphoma (MTCL) in TCR-oligoclonal OT-I transgenic T cells, transduced with an enhanced green fluorescent protein (EGFP)-encoding gammaretroviral vector. The tumor cells were of a mature T-cell phenotype and serially transplantable. Integration site analysis revealed a proviral hit in Janus kinase 1 (Jak1), which resulted in Jak1 overexpression and Jak/STAT-pathway activation, particularly via signal transducer and activator of transcription 3 (STAT3). Specific inhibition of Jak1 markedly delayed tumor growth. A systematic meta-analysis of available gene expression data on human mature T-cell lymphomas/leukemias confirmed the relevance of Jak/STAT overexpression in sporadic human T-cell tumorigenesis. To our knowledge, this is the first study to describe RV-associated insertional mutagenesis in mature T cells.
Introduction
Retroviral vectors (RVs) mediate stable integration and long-term expression of transgenes. Therefore, they are widely used in clinical gene therapy involving a broad range of different target cells and tissues. However, RVs have been associated with severe side effects. Gammaretroviruses are natural mutagens as they integrate semi-randomly into the host genome with regard to the target sequence, but have a preference for regions of active transcription and regulatory elements of transcriptionally active genes.1,2,3 Their integration can cause overexpression of adjacent genes or disruption of “target” gene expression. Other results of viral integration are modified transcripts and proteins through alternative or aberrant splicing or through premature termination of transcription.4
Induction of leukemia by the genotoxicity of a gammaretroviral vector in hematopoietic stem cells was initially described in a mouse model.5 Vector-related activation and subsequent overexpression of the oncogene ecotropic viral integration site-1 (Evi1) fostered clonal outgrowth and leukemogenesis. Furthermore, the genotoxicity of gammaretroviral vectors with RV-driven clonal dominance was observed in clinical gene therapy trials for X-linked severe combined immune deficiency,6,7 chronic granulomatous disease,8,9 and Wiskott-Aldrich syndrome.10 However, until now RV-mediated genotoxicity has not been described in clinical gene therapy trials involving adoptive transfer of RV-modified mature T cells.11,12,13 The lack of reported genotoxicity of gammaretroviral vectors in mature T cells is surprising, because T cells are long-lived and have a high capacity of self-renewal.14 In addition, murine retroviruses are known to transform T cells in mice by insertional mutagenesis, even though the majority of murine leukemia virus–induced T-cell malignancies present an immature phenotype.15
In a previous study, we compared the susceptibility with oncogenic transformation of mature T cells and hematopoietic stem cells.16 Our results demonstrated that T-cell receptor (TCR)-polyclonal mature T cells are far less prone to transformation after retroviral gene transfer in vivo than hematopoietic stem cells. Additional experiments revealed a rare event of T-cell immortalization in vitro following transduction of the T-cell proto-oncogene LMO2 and insertion-related activation of growth-promoting genes IL2RA and IL15RA.17 We have shown more recently that TCR-oligoclonal (OT-I and P14) mature T cells expressing the oncogenes NPM/ALK or ΔTrkA give rise to mature T-cell lymphomas (MTCLs).18
In this study, we transduced murine TCR-oligoclonal OT-I T cells with an enhanced green fluorescent protein (EGFP)-encoding RV and transplanted gene-modified T cells into RAG1−/− mice. After 16 months, including one round of serial transplantation, MTCLs emerged. Integration site analysis revealed a proviral insertion in the Janus kinase 1 (Jak1) gene. RV-driven overexpression of Jak1 was causally implicated in tumor growth promotion as specific inhibition of Jak1 significantly prolonged survival of mice transplanted with these Jak1-activated tumor cells. To our knowledge, although under very stringent experimental conditions, this is the first reported case of RV-induced insertional mutagenesis in mature T cells in vivo.
Results
Skewed TCR diversity fosters insertional mutagenesis in mature T cells
To evaluate if TCR-oligoclonal T cells are susceptible to RV-induced insertional mutagenesis, we transduced TCR-transgenic (tg) OT-I T cells with a replication-defective RV encoding EGFP (Figure 1a). RAG1−/− recipients were transplanted with 5 × 106 tg T cells (Figure 1b), which displayed a CD3+, CD8+, TCR+, and CD25+ phenotype. The transduction efficacy was 25% (data not shown). After 1 year, we killed one of the primary recipients and serially transplanted T cells from spleen and lymph nodes into three secondary RAG1−/− animals. Four months later, MTCLs developed. Symptomatic animals showed massive splenomegaly and lymphadenopathy as well as lymphomatous infiltrations of kidney, liver, and lung (Figure 1c). The tumor cells expressed the T-cell markers CD3, CD8, and TCR and the activation marker inducible co-stimulator (ICOS) as well as Sca-1 (Figure 1d).
Figure 1.

T-cell lymphomagenesis induced by transduction of an EGFP-encoding gammaretroviral control vector. (a) The gammaretroviral vector used in this study comprised long terminal repeats (LTRs) from myeloproliferative sarcoma virus (MPSV) and a leader region from the murine embryonic stem cell virus (MESV leader 71) including a splice donor and a splice acceptor site. A posttranscriptional regulatory element of the Woodchuck hepatitis virus (WPRE) was located 3′ of the transgene enhanced green fluorescent protein (EGFP). (b) T lymphocytes from OT-I donor mice were isolated and transduced with gammaretroviral particles encoding for EGFP and transplanted into RAG1−/−–recipient mice. One year after transplantation, splenocytes and lymphocytes were reisolated and serially transplanted into secondary RAG1−/−–recipient mice. Four months after serial transplantation, mature T-cell lymphoma emerged. (c) Representative histological sections (HE stained) from lymph node, spleen, kidney, liver, and lung of a diseased animal (magnification 200× scale bar = 100 µm). (d) Cell surface characterization of control vector–induced malignancy. After single cell suspension of tumor samples, cells from massively enlarged spleens were stained for the α chain of the OT-I TCR (Vα2), CD3, CD8, and ICOS (inducible co-stimulator). Staining for CD4 was negative. Representative blots (gated on EGFP-positive cells) from the primary tumor are depicted. Cell surface marking was comparable in all secondary and tertiary recipients. Staining of lymph node samples showed the same results.
Retrovirus-mediated T-cell lymphomagenesis is associated with Jak1 overexpression and activation of its target signal transducer and activator of transcription 3
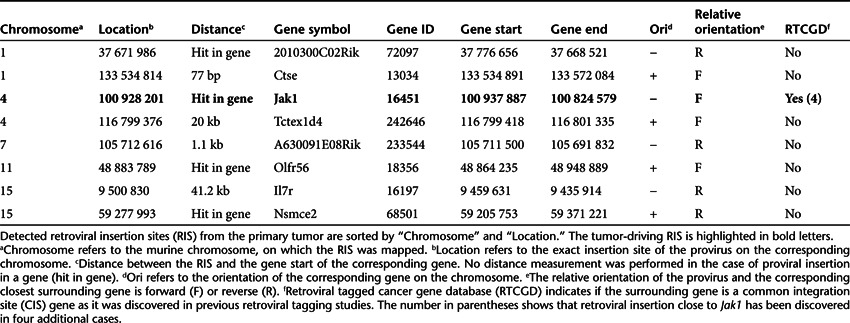
By conventional culturing methods, we were not able to establish stable in vitro lines from these lymphoma cells. However, tumor cells were transplantable into secondary and tertiary hosts (Figure 2a). These animals developed malignancies that were histologically and immunophenotypically identical to the primary tumor (data not shown). To reveal a potential oncogenic target, we performed retroviral integration site (RIS) analysis by ligation-mediated polymerase chain reaction. In total, we identified eight unique RIS (Table 1); only one RIS was detectable in all animals that succumbed to lymphoma after two rounds of transplantation (Figure 2b). The gene surrounding this RIS is enlisted in the retroviral tagged cancer gene database19 as a common integration site. The intriguing proviral insertion was located on chromosome 4 in sense within Jak1 between exons 1 and 2 (Figure 2c). The RIS was already detectable 54 days after initial transplantation, analyzed retrospectively by integration site-specific PCR of peripheral blood samples from the primary recipient (data not shown).
Figure 2.

Serial transplantation of primary tumor cells reveals outgrowth of a T-cell clone with a retroviral insertion site in Jak1. (a) Tumor cells from the primary mature T-cell tumor (1st generation) were serially transplantable into secondary (2nd generation) and tertiary (3rd generation) recipients and established histological and immunophenotypical identical malignancies. 5 × 106 tumor cells from spleen and lymph node were injected intravenously into secondary and tertiary RAG1−/− recipients. (b) Retroviral integration site (RIS) analysis revealed eight unique proviral insertions in the primary tumor, whereof one RIS was detectable in all animals of the three generations. The RIS was amplified via integration site-specific PCR and verified by sequencing. Primary, secondary, and tertiary tumor samples are depicted by 1, 2, and 3, respectively. Negative control (N), untransduced OT-I cells. M, base pair marker. (c) Using ligation-mediated PCR (LM-PCR) and integration site-specific PCR, we identified the proviral insertion in Jak1, that was common to all animals, which succumbed to mature T-cell lymphoma. The EGFP-encoding provirus integrated between exon 1 and exon 2 of the Jak1 gene on chromosome 4. The start codon of the 25 exons spanning the Jak1 gene is depicted on exon 2. Proviral sequence was orientated in sense with Jak1.
Table 1. Retroviral insertion sites obtained from primary tumor sample.
We next studied potential mechanisms of Jak1 activation. A fusion product of Jak1 and EGFP was excluded at the level of mRNA and protein expression (Figure 3a). Interestingly, we detected an aberrant transcript which comprised the proviral LTR and Jak1 exons located downstream of the RIS. This transcript was lost in cells of tertiary recipients although the RIS remained detectable (Figure 3b). We performed methylation profiling of the proviral LTR to investigate the loss of the aberrant Jak1 transcript. Interestingly, no significant methylation in the proviral promoter and enhancer elements was identified (Figure 4). Therefore, we reasoned that the LTR enhancer was still active and could influence the nearby Jak1 promoter.
Figure 3.

Exclusion of an EGFP/Jak1 fusion product and detection of an aberrant transcript. (a) Detection of EGFP by western blot to exclude a fusion-protein of Jak1 and EGFP. EGFP was solely detectable at a molecular weight (MW) of 27 kDa in all diseased animals. This result was confirmed by PCR on cDNA. Primary, secondary, and tertiary tumor samples are depicted by 1, 2, and 3, respectively. Negative control (N), untransduced OT-I T cells. (b) Insertion site and orientation of EGFP-encoding provirus and schematic overview of different detectable Jak1 transcripts in tumor samples. Retroviral insertion site was located on chromosome 4 in the intron between exons 1 and 2 of Jak1 upstream of the start codon, which is depicted on exon 2. Black boxes represent the three 5′ exons of Jak1 (25 exons in total, whereof 24 are coding). Filled arrowheads schematically show the location of the proviral promoter in the 5′ LTR and 3′ LTR, the splice donor (SD), and the splice acceptor (SA) in the leader region (MESV-L), respectively. The presence of different transcripts was investigated by PCR on cDNA. In all recipients, the Jak1 nascent chain transcript (transcript 1) and the transgene transcript (transcript 3) could be detected. In tumor cells from the second generation, we found an aberrant transcript comprising of proviral LTR, the MESV leader region until the SD site and the 3′ located Jak1 exons (transcript 2). Secondary and tertiary tumor samples are depicted by 2 and 3, respectively. Negative control (N), untransduced OT-I T cells. Only vector-related transcripts are shown. Nascent chain transcript was detectable in all investigated samples including controls. M, base pair marker.
Figure 4.

Methylation analysis of CpG islands within the proviral LTR in Jak1. DNA methylation analysis was carried out using sodium bisulfite sequencing to determine methylation status of CpG islands within the long terminal repeat (LTR) of the proviral integration in Jak1. The enhancer is located in the U3-region; promoter is spanning the U3-, R-, and U5-region. Filled boxes resemble methylated CpG islands; open boxes resemble non-methylated CpG islands.
To solidify the specific RV-mediated activation of Jak1, replicating retroviruses were excluded by gag-based PCR analysis (data not shown). In addition, derepression of an endogenous murine retrovirus was dismissed as an alternative, because the proviral sequence in the Jak1 intron contained gammaretroviral vector–derived EGFP (data not shown). Nonetheless, a selective overexpression of Jak1 in the murine tumor samples could be demonstrated by western blot analysis and quantitative PCR (qPCR), also after serial transplantation of lymphoma cells (Figure 5a,b). Next, we analyzed the phosphorylation state of the signaling molecules signal transducer and activator of transcription 3 (STAT3) and STAT5, which are known to act as major targets downstream of Jak1. STAT-phosphorylation in tumor cells of this RV-induced murine MTCL was restricted to STAT3 (Figure 5c).
Figure 5.

Provirus-induced activation of the Jak/STAT-pathway. (a) Western blot analysis showing highly elevated levels of Jak1 in tumor tissue derived from spleen and lymph node carrying the insertion site in Jak1. Primary, secondary, and tertiary tumor samples are depicted by 1, 2, and 3, respectively. Untransduced OT-I cells isolated from RAG1−/− mice were used as negative control N1; ex vivo obtained EGFP-transduced OT-I T cells w/o RIS in Jak1 as negative control N2. (b) Elevated Jak1 RNA expression determined by real-time quantitative PCR. Measurements were performed in duplicates. Relative quantification was based on normalization on the housekeeping gene 18S rRNA. As negative controls, untransduced OT-I cells (N1) isolated from RAG1−/− mice and ex vivo obtained EGFP-transduced OT-I cells (N2) w/o RIS in Jak1 were used. RNA from the primary mature T-cell lymphoma was not isolated. (c) Western blot analysis showing the selective phosphorylation of STAT3 in tumor tissue obtained from spleen and lymph node carrying the RIS in Jak1. Phosphorylation of STAT5 could not be detected. Primary, secondary, and tertiary tumor samples are depicted by 1, 2, and 3, respectively. Untransduced OT-I T cells served as negative control (N).
Selective inhibition of Jak1 improves overall survival of T-cell tumor-bearing mice
To further corroborate the concept of a retroviral insertion in Jak1 to be an initiating event and of relevance in the sustenance of this experimental T-cell lymphoma, we selectively inhibited Jak1 kinase activity pharmacologically. We serially transplanted equal numbers of tumor cells (8 × 106 cells/animal) from secondary hosts into RAG1−/− recipients (n = 16). Half of the cohort was treated with the Jak1/2-inhibitor INCB018424 at a dose of 45 mg/kg and the other half received equimolar vehicle control20 (0.5% methylcellulose) by daily intraperitoneal injections. INCB018424-treated mice transplanted with tg T cells bearing the retroviral integration in Jak1 demonstrated a highly significant overall survival advantage (P = 0.0001) over animals of the vehicle-treated control group (Figure 6). ΔTrkA-induced T-cell lymphoma cells18 were transplanted as a negative control (8 × 106 cells/animal). Coactivation of STAT-signaling pathways by ΔTrkA was excluded in previous studies.21 INCB018424 had no influence on the survival of ΔTrkA-transplanted animals compared with the vehicle-treated group (n = 8).
Figure 6.

Reduced tumor growth kinetics in vivo after specific Jak1 inhibition by INCB018424. Survival of animals transplanted with either EGFP control vector–transformed tumor cells, carrying the genetic lesion in Jak1 (n = 16), or ΔTrkA-induced tumor cells (n = 8) under treatment of Jak1/2 inhibitor INCB018424 or vehicle only. Treated as well as untreated animals transplanted with ΔTrkA-induced tumor cells developed hematopoietic malignancies within 8 days. Tumor development in recipients transplanted with T cells bearing the RIS in Jak1 was significantly delayed by the inhibitor INCB018424 (***P = 0.0001, log-rank).
Elevated expression of Jak1 and STAT3 in primary samples of human MTCL
To provide assurance of relevance of these findings by data from human tumorigenesis, we performed a comprehensive in silico meta-analysis of published whole-genome gene expression profiles across a large spectrum of human MTCL.22,23,24,25,26,27,28,29,30,31 We selected Jak and STAT family members for the specific subanalysis. Overall, among the Jak family members, Jak1 showed a markedly elevated expression (particularly over Jak2 and Jak3) in most MTCL entities. Furthermore, there was a strong association of expression of Jak1 with STAT3 in an unsupervised clustering analysis as both genes show the closest resemblance of expression profiles across MTCL (Figure 7).
Figure 7.

Gene expression levels of Jak and STAT family members in human primary mature T-cell lymphomas. Array-based mRNA expression levels of the Jak and STAT family genes were retrieved from publically available primary in silico datasets of whole-genome profilings and summarized in a heatmap showing upregulation (red) and downregulation (yellow) across various human MTCL entities and subsets of normal T cells. On the X-axis, the first digit(s) indicate(s) the number of samples per entity per dataset; the second field abbreviates the entity followed by the primary data reference. Unsupervised clustering reveals the association of Jak1 with STAT3 by showing the closest resemblance of expression profiles across MTCL and normal T-cell isolates. The predominantly high expression of Jak1 and to a lesser degree of STAT3 in most MTCL except for some anaplastic large-cell lymphomas and normal T cells, emphasizes the relevance of these molecules in spontaneous mature T-cell tumorigenesis. Another Jak family member highly expressed in the analyzed MTCLs is Tyk2, which shows a closer resemblance with the STAT1 pattern. AB HSTL, α-β hepatosplenic T-cell lymphoma; AITL, Angioimmunoblastic T-cell lymphoma; ALCL-ALK−, Anaplastic large-cell lymphoma ALK negative; ALCL-ALK+, Anaplastic large-cell lymphoma ALK positive; ATLL, Adult T-cell lymphoma; cALCL, primary cutaneous anaplastic large cell lymphoma; CTCL, Cutaneous T-cell lymphoma; EATL, Enteropathy-associated T-cell lymphoma; HSTL, hepatosplenic T-cell lymphoma; PTCL-NOS, Peripheral T-cell lymphoma not otherwise specified; T-cells DR+, T-cells DR−, DR: HLA-DR, human leukocyte antigen receptor DR positive/negative; T-PLL, T-cell prolymphocytic leukemia; T-PLL no inv(14), T-cell prolymphocytic leukemia without inv(14)/t(14;14); T-PLL inv (14), T-cell prolymphocytic leukemia with inv(14)/t(14;14); T-cells CD8A, activated CD8 T cells; T-cells CD8R, resting CD8 T cells; T-cells CD4A, activated CD4 T cells; T-cells CD4R, resting CD4 T cells.
Discussion
Although, RV-mediated insertional mutagenesis has not been described for adoptive transfer of gene-modified mature T cells, the risk of genotoxicity by gammaretroviral vectors in T-cell–based gene therapy remains poorly elucidated. In the present study, we investigated the susceptibility of mature T cells to be transformed by mechanisms of insertional mutagenesis after gammaretroviral transduction. We found that in the setting of a skewed TCR repertoire mature T-cell lymphomagenesis can take place after provirus-driven activation of the growth-promoting gene Jak1. We show that this specific Jak1 activation is transmitted to its target STAT3 and that pharmacologic Jak1 inhibition reduces RV-induced tumor growth in vivo.
In previous work, we could show that TCR-polyclonal mature T cells, as opposed to hematopoietic progenitor cells, are less vulnerable to transformation after RV-mediated ectopic expression of known T-cell (proto-)oncogenes.16 This finding reflects the current perception of a high safety of clinical trials dealing with RV-based gene transfer in mature T lymphocytes. So far, clonal imbalances and oncogenesis has never been observed in clinical trials with gene-modified T cells.11,12,13,32–34 Nevertheless, in previous in vitro experiments, we demonstrated that insertional activation can contribute to immortalization of a mature T-cell clone.17 Moreover, extended experiments revealed that certain conditions revoke the resistance of mature T cells to transformation. In those studies, we proposed that the transformation-resistance of TCR-polyclonal mature T cells is mainly mediated by a broad TCR repertoire and the competition for MHC-self-peptide stimulatory niches.18 This hypothesis was corroborated by our findings of induction of MTCL by RV-mediated transfer of certain T-cell oncogenes into TCR-oligoclonal T-cell populations (OT-I and P14).18 Moreover, we could inhibit the outgrowth of preleukemic clones by cotransplantation of non-modified TCR-polyclonal T cells. From this, we concluded that a skewed TCR repertoire predisposes mature T cells to oncogenic transformation.
To address questions on the potential mechanisms of T-cell tumorigenesis by retroviral gene transfer, we capitalized on an observation of the outgrowth of a mature T-cell tumor after experimental T-cell targeting by a RV that carried no encoding oncogene. This enabled us to investigate the susceptibility of TCR-oligoclonal mature T cells to insertional mutagenesis after gammaretroviral gene transfer. We transduced TCR-transgenic OT-I T cells with an empty EGFP-encoding RV and transplanted gene-modified T cells into RAG1−/− recipients. Remarkably, MTCL developed with a latency of 16 months. Serial transplantation of tumor cells over three generations resulted in histologically and immunophenotypically identical malignancies. RIS analysis revealed provirus localization in the Jak1 gene. Although a second (aberrant) transcript was lost after serial transplantation, we were still able to detect overexpression of Jak1 in later generations of tumors. As we could not identify any significant methylation in the proviral promoter and enhancer elements, we conclude that Jak1 transcription was not only activated by expression from the LTR promoter and splicing into exon 2 of Jak1, but also by the retroviral enhancer that influences the nearby Jak1 promoter elements.
Our additional experiments revealed the activation of the Jak/STAT-pathway as an important event in this particular case of T-cell lymphomagenesis. Constitutive activation of the Jak/STAT-pathway has been linked to T-cell malignancies before. Human T-cell lymphotropic virus I transformed T cells develop IL-2 independence by constitutive activation of the Jak/STAT-pathway via Jak1/3 and STAT3/5.35 Lymphocyte-specific protein tyrosine kinase (Lck)–mediated oncogenesis in the murine T-cell lymphoma line LSTRA was accompanied by constitutively activated Jak1 and Jak2, as well as STAT3 and STAT5.36 Recently, a somatic Jak1 mutation was reported among adults with T-cell acute lymphoblastic leukemia, which could be associated with a poor therapy response and an inferior overall survival.37 Furthermore, enhanced STAT3 activity has been linked to anaplastic lymphoma kinase (ALK)-positive anaplastic large-cell lymphoma.38 Moreover, the activation of STAT3 in NPM/ALK-transduced T cells was documented in our previous study,18 as well as in clinical investigations of anaplastic large-cell lymphoma and cutaneous T-cell lymphoma.39,40 In addition, in a comprehensive in-silico meta-analysis, we found here a positively correlated gene expression pattern between Jak1 and STAT3 across a large spectrum of (spontaneous) human MTCL. Taken together, these findings support the importance of Jak/STAT deregulation during T-cell tumorigenesis and justify the exploration of inhibitors of the Jak/STAT-pathway for clinical applications.
Given the long latency of lymphoma development in our stringent experimental setup, the RV-mediated activation of Jak1 and subsequent activation of the Jak/STAT-pathway is most likely not the only component of relevance in sustenance of this overt murine lymphoma. Besides accumulation of specific Jak1-inhibitor resistance-promoting mutations, such a multistep tumorigenesis and driver-gene shifts explain why the survival-prolonging antitumor effects of INCB018424 in those hosts with proliferations of RV-mediated Jak1 activation was of limited magnitude.
Our data strongly suggest that RV-mediated activation of Jak1 played a pivotal role in lymphomagenesis. In our hands, longer observation times of EGFP-transduced animals, which were generally used as controls in our previous experiments, were not necessary as recipients transplanted with NPM/ALK- or ΔTrkA-transduced OT-I T cells succumbed to lymphoma within very short latencies.18 RV-induced clonal imbalance has never been observed in a TCR-polyclonal wild-type T-cell population despite stable transgene expression for up to 510 days (n = 9).16 So far, we are not able to predict the frequency of RV-induced T-cell malignancies in a TCR-oligoclonal T-cell population, but long-term follow-up of recipients is currently under investigation.
Overall, our findings echo the reported safety of RV-modified T cells in clinical application, as determined in >500 patient-years of follow-up.11,12,13 Nevertheless, we support the importance of long-term monitoring of genetically altered T cells in vivo, as the present study demonstrates that a skewed TCR repertoire potentially enables clonal outgrowth of T lymphocytes transduced with a RV encoding a non-carcinogenic transgene.
Materials and Methods
Mice (OT-I and RAG1−/−), RVs, RV-production, retroviral transduction and transplantation of mature T cells (OT-I), histopathological analysis, and retroviral insertion site analysis are described elsewhere.16,18,41,42
Flow cytometry analysis. Flow cytometry analyses were carried out on single cell suspensions of spleen and lymph nodes. The following antimouse monoclonal antibodies were used for staining: rat antimouse monoclonal antibodies (Invitrogen, Carlsbad, CA): phycoerythrin (PE)-conjugated CD8a (CT-CD8a), phycoerythrin Cy5.5 (PE-Cy5.5)-conjugated CD4 (RM4-5), phycoerythrin Cy5 (PE-Cy5)-conjugated CD3 (145-2C11); (BD, Franklin Lakes, NJ); (PE)-conjugated Sca-1 (E13-161.7) (eBioscience, San Diego, CA); allophycocyanin (APC)-conjugated Vα2 TCR (B20.1), (PE-Cy5)-conjugated CD278/ICOS (15F9). To prevent non-specific binding to Fc receptors, samples were incubated with mouse seroblock FcR (Serotec, Düsseldorf, Germany). After staining lymph node and spleen, samples were treated with Cal-lysis solution (Invitrogen) to remove red blood cells. Analyses were performed on FACS Canto II and MACSQuant using FACSDiva software (BD) and MACSQuantify software (Miltenyi Biotech, Bergisch Gladbach, Germany).
Western blotting. For western blot analysis, we lysed 1 × 106 lymphoma cells after single cell suspension. As negative control, we prepared lysates of equal cell numbers from untransduced OT-I T cells and OT-I T cells transduced with EGFP (w/o RIS in Jak1), isolated from RAG1−/− recipients 3 months after transplantation. Western blotting was performed according to the manufacturer's instructions. As primary antibodies, we used monoclonal rat anti-Jak1 antibody (clone 413104) in a 1:500 dilution (R&D systems, Minneapolis, MN), rabbit antiphospho-STAT3 (Tyr705) in a 1:1,000 dilution (Cell Signaling, Danvers, MA), rabbit anti-STAT3 (C-20) in a 1:1,000 dilution (Santa Cruz Biotechnology, Santa Cruz, CA), rabbit antiphospho-STAT5 (Tyr694) in a 1:1,000 dilution (Cell Signaling), rabbit anti-STAT5 (3H7) in a 1:1,000 dilution (Cell Signaling), mouse anti-GFP in a 1:500 dilution (Abcam, Cambridge, UK), and as loading control monoclonal mouse anti-β-actin antibody in a 1:40,000 dilution (Sigma-Aldrich, St Louis, MO). The primary antibodies were detected with horseradish peroxidase–conjugated secondary antibodies: goat antirat horseradish peroxidase–conjugated in a 1:5,000 dilution (Dianova, Hamburg, Germany), goat antirabbit horseradish peroxidase–conjugated in a 1:2,000 dilution (Dako, Glostrup, Denmark), and rabbit antimouse horseradish peroxidase–conjugated in a 1:2,000 dilution (Dako).
Quantitative real-time PCR. Small tissue sections from inguinal and mesenteric lymph nodes of diseased and healthy control animals were stored in RNAlater RNA Stabilization Reagent (Qiagen, Hilden, Germany) until purification of RNA using the RNeasy Mini Kit (Qiagen). Subsequently, RNA was reverse transcribed using the High Capacity RNA-to-cDNA Kit (Applied Biosystems, Carlsbad, CA). For qPCR procedure, the following oligonucleotides and probe were used: Jak1_qfor: 5′-GCA CGG AAC CAA TGA CAA CGA ACA-3′ Jak1_qrev: 5′-ACT CCA GTG AAC TGG CAT CAA GGA-3′ Jak1_qprobe: 5′-(LC610) TGG CGA CAT TCT CCA AAG AAG CAG A (BHQ2)-3′. As reference, the housekeeping gene 18S rRNA was chosen and oligonucleotides and probe had the following sequence: rRNA18S_qfor: 5′-TCA ACT TTC GAT GGT AGT CGC CGT-3′ rRNA18S_qrev: 5′-TCC TTG GAT GTG GTA GCC GTT TCT-3′ rRNA18S_qprobe: 5′-(LC610) AAT CAG GGT TCG ATT CCG GAG AGG GA (BHQ2)-3′. Real-time PCR was performed with the TaqMan Gene Expression Master Mix (Applied Biosystems) using a LightCycler 480 Real-Time PCR System (Roche, Basel, Switzerland).
Serial transplantation. All in vivo experiments were performed in accordance with the local animal experimentation guidelines and approved by the regional council (Regierungspräsidium, Darmstadt, Germany). One year after transplantation of EGFP-transduced OT-I T cells, RAG1−/− recipient was killed and 5 × 106 lymphocytes/splenocytes were serially transplanted into a new RAG1−/− recipients. Four months after serial transplantation, MTCL emerged (primary tumor). Tumor cells (5 × 106) were intravenously transplantable after single cell suspension into RAG1−/− recipients (secondary and tertiary tumor).
Methylation profiling. Genomic DNA from tumor samples was isolated using the DNeasy Blood and Tissue Kit (Qiagen). Subsequent DNA conversion with sodium bisulfite was performed using the Epitect Kit (Qiagen). Bisulfite specific primer were designed using the MethPrimer software43 and had the following sequences: F1_Meth: 5′-TAT AGA TGG TAA GTT TTT TTG ATG TGG T-3′ F2_Meth: 5′-GAT GTA TTA GTT GGT ATT AAA GAA ATG TTT-3′ and R1_Meth: 5′-CAA TCA ATC AAT CTA AAA AAA CCC T-3′. Amplification of proviral LTR elements was performed with a seminested PCR approach. PCR products were subcloned with TOPO TA Cloning Kit (Invitrogen) followed by sequencing.
Jak1 inhibition. Jak1 specific inhibition experiments were carried out in 6-week-old, female RAG1−/− recipients, transplanted either with 8 × 106 EGFP-transduced OT-I T cells (n = 16), bearing the proviral insertion in Jak1, or with 8 × 106 ΔTrkA-transduced OT-I T cells (n = 8). Each of the two cohorts was split in two, whereof half of each group was treated intraperitoneally once a day with the Jak1/2 specific inhibitor INCB018424 (Novartis, Basel, Switzerland/Incyte, Wilmington, DE) or its vehicle 0.5% methylcellulose. Treatment with INCB018424 and vehicle started 24 hours after cell inoculation (daily administration dose of INCB018424 was 45 mg/kg). Cumulative survival analysis was performed using the log-rank (Mantel–Cox) test.
Integration site-specific PCR and detection of aberrant transcript. Integration site-specific PCR was performed on genomic DNA extracted with the DNeasy Blood and Tissue Kit (Qiagen). To identify the proviral insertion in Jak1, we performed a nested PCR using the following primers: Int1_Jak1: 5′-GAT GGT AAG TCC TCT TGA TGT G-3′ Int2_Jak1: 5′-CAC TAG CTG GCA CTA AAG AAA TG-3′ A2RV: 5′-GCC CTT GAT CTG AAC TTC TC-3′ and A3RV: 5′-CCA TGC CTT GCA AAA TGG C-3′. Amplified sequences were subcloned with TOPO TA Cloning Kit (Invitrogen) followed by sequencing and BLAST verification (NCBI37/mm9).
For the detection of aberrant transcripts, we isolated RNA using RNeasy Mini Kit (Qiagen). For reverse transcription of RNA, we used the High Capacity RNA-to-cDNA Kit (Applied Biosystems). The following primers were used for identification of aberrant transcripts by a nested PCR approach: U5_for_int1: 5′-TGG ACT CGC TGA TCC TTG GG-3′ (binding the proviral LTR), U5_for_int2: 5′-ATC CTT GGG AGG GTC TCC TC-3′ (binding the proviral LTR), Jak1_for_int1: 5′-CAG CCT CGC TCG TCC TTT C-3′ (binding Jak1 exon 1), Jak1_for_int2: 5′-GCT GAG TTG CCT GCC AGA C-3′ (binding Jak1 exon 1), Jak1_rev_int1: 5′-GAA GCT CCT CAT TTT AGC ACA GAA C-3′ (binding Jak1 exon 3), and Jak1_rev_int2: 5′-GCC ATG GCA TTG CAG TCC TC-3′ (binding Jak1 exon 3).
In silico gene expression analysis. Out of 38 reports on array-based gene expression profiling of mature T-cell leukemias/lymphomas and normal T cells, we identified 10 batches with available primary data from standardized oligonucleotide arrays (7 on Affymetrix HG-U133 Plus 2.0 and 3 on older U133A arrays with less than half of the probe sets).22,23,24,25,26,27,28,29,30,31 All datasets were separately background-corrected and preannotated using the BioConductor packages “affy” in R. Replicates were combined by mean. The dataset was quantile normalized and the genes were re-annotated via Ensembl ID using “biomaRt.” The resulting ambiguous probe sets assigned to a gene were also reduced by calculating their mean. The 11 genes of the Jak/STAT family were then retrieved from the datasets. Expression values from all datasets were again together quantile normalized to account for variability in platform specifications and noise instead of working on gene overlap.
Acknowledgments
The authors thank Margot Landersz, Sabine Albrecht, Ekaterini Hadzoglou, and Susanne Hansen for excellent technical assistance (Margot Landersz from Georg-Speyer-Haus, Institute for Biomedical Research, Frankfurt am Main, Germany as well as Sabine Albrecht, Ekaterini Hadzoglou, and Susanne Hansen from Senckenberg Institute of Pathology, Goethe-University Hospital, Frankfurt am Main, Germany). We acknowledge the helpful advice by Christian Brendel, Stephan Schultze-Strasser, and Stefan Stein (all Georg-Speyer-Haus, Institute for Biomedical Research, Frankfurt am Main, Germany). We thank Thomas Radimerski, Jordan Fridman, and Heidrun Bartels (Novartis Pharma AG/Incyte Corporation) for supplying the Jak1/2 inhibitor INCB018424. We thank "Alfons und Gertrud Kassel-Stiftung" and the "Research Support Foundation, Vaduz" for funding of the MACSQuant cytometer. This work was supported by the Deutsche Forschungsgemeinschaft (LA1135/9-1) as a part of the SPP1230 (to D.v.L.) as well as by the Deutsche Krebshilfe (Max-Eder Program) and the BMBF (Federal Ministry of Education and Research, Germany; grant number 01 KI 0771; both to M.H.). The authors declare no conflict of interest.
References
- Wu X, Li Y, Crise B, Burgess SM. Transcription start regions in the human genome are favored targets for MLV integration. Science. 2003;300:1749–1751. doi: 10.1126/science.1083413. [DOI] [PubMed] [Google Scholar]
- Laufs S, Nagy KZ, Giordano FA, Hotz-Wagenblatt A, Zeller WJ, Fruehauf S. Insertion of retroviral vectors in NOD/SCID repopulating human peripheral blood progenitor cells occurs preferentially in the vicinity of transcription start regions and in introns. Mol Ther. 2004;10:874–881. doi: 10.1016/j.ymthe.2004.08.001. [DOI] [PubMed] [Google Scholar]
- Bushman F, Lewinski M, Ciuffi A, Barr S, Leipzig J, Hannenhalli S, et al. Genome-wide analysis of retroviral DNA integration. Nat Rev Microbiol. 2005;3:848–858. doi: 10.1038/nrmicro1263. [DOI] [PubMed] [Google Scholar]
- Staal FJ, Pike-Overzet K, Ng YY, van Dongen JJ. Sola dosis facit venenum. Leukemia in gene therapy trials: a question of vectors, inserts and dosage. Leukemia. 2008;22:1849–1852. doi: 10.1038/leu.2008.219. [DOI] [PubMed] [Google Scholar]
- Li Z, Düllmann J, Schiedlmeier B, Schmidt M, von Kalle C, Meyer J, et al. Murine leukemia induced by retroviral gene marking. Science. 2002;296:497. doi: 10.1126/science.1068893. [DOI] [PubMed] [Google Scholar]
- Hacein-Bey-Abina S, Garrigue A, Wang GP, Soulier J, Lim A, Morillon E, et al. Insertional oncogenesis in 4 patients after retrovirus-mediated gene therapy of SCID-X1. J Clin Invest. 2008;118:3132–3142. doi: 10.1172/JCI35700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe SJ, Mansour MR, Schwarzwaelder K, Bartholomae C, Hubank M, Kempski H, et al. Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients. J Clin Invest. 2008;118:3143–3150. doi: 10.1172/JCI35798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ott MG, Schmidt M, Schwarzwaelder K, Stein S, Siler U, Koehl U, et al. Correction of X-linked chronic granulomatous disease by gene therapy, augmented by insertional activation of MDS1-EVI1, PRDM16 or SETBP1. Nat Med. 2006;12:401–409. doi: 10.1038/nm1393. [DOI] [PubMed] [Google Scholar]
- Stein S, Ott MG, Schultze-Strasser S, Jauch A, Burwinkel B, Kinner A, et al. Genomic instability and myelodysplasia with monosomy 7 consequent to EVI1 activation after gene therapy for chronic granulomatous disease. Nat Med. 2010;16:198–204. doi: 10.1038/nm.2088. [DOI] [PubMed] [Google Scholar]
- Avedillo Díez I, Zychlinski D, Coci EG, Galla M, Modlich U, Dewey RA, et al. Development of novel efficient SIN vectors with improved safety features for Wiskott-Aldrich syndrome stem cell based gene therapy. Mol Pharm. 2011;8:1525–1537. doi: 10.1021/mp200132u. [DOI] [PubMed] [Google Scholar]
- Bonini C, Grez M, Traversari C, Ciceri F, Marktel S, Ferrari G, et al. Safety of retroviral gene marking with a truncated NGF receptor. Nat Med. 2003;9:367–369. doi: 10.1038/nm0403-367. [DOI] [PubMed] [Google Scholar]
- Scholler J, Brady TL, Binder-Scholl G, Hwang WT, Plesa G, Hege KM, et al. Decade-long safety and function of retroviral-modified chimeric antigen receptor T cells. Sci Transl Med. 2012;4:132ra53. doi: 10.1126/scitranslmed.3003761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Recchia A, Bonini C, Magnani Z, Urbinati F, Sartori D, Muraro S, et al. Retroviral vector integration deregulates gene expression but has no consequence on the biology and function of transplanted T cells. Proc Natl Acad Sci USA. 2006;103:1457–1462. doi: 10.1073/pnas.0507496103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surh CD, Sprent J. Regulation of mature T cell homeostasis. Semin Immunol. 2005;17:183–191. doi: 10.1016/j.smim.2005.02.007. [DOI] [PubMed] [Google Scholar]
- Hays EF, Bristol GC, McDougall S, Klotz JL, Kronenberg M. Development of lymphoma in the thymus of AKR mice treated with the lymphomagenic virus SL 3-3. Cancer Res. 1989;49:4225–4230. [PubMed] [Google Scholar]
- Newrzela S, Cornils K, Li Z, Baum C, Brugman MH, Hartmann M, et al. Resistance of mature T cells to oncogene transformation. Blood. 2008;112:2278–2286. doi: 10.1182/blood-2007-12-128751. [DOI] [PubMed] [Google Scholar]
- Newrzela S, Cornils K, Heinrich T, Schläger J, Yi JH, Lysenko O, et al. Retroviral insertional mutagenesis can contribute to immortalization of mature T lymphocytes. Mol Med. 2011;17:1223–1232. doi: 10.2119/molmed.2010.00193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newrzela S, Al-Ghaili N, Heinrich T, Petkova M, Hartmann S, Rengstl B, et al. T-cell receptor diversity prevents T-cell lymphoma development. Leukemia. 2012;26:2499–2507. doi: 10.1038/leu.2012.142. [DOI] [PubMed] [Google Scholar]
- Akagi K, Suzuki T, Stephens RM, Jenkins NA, Copeland NG. RTCGD: retroviral tagged cancer gene database. Nucleic Acids Res. 2004;32 Database issue:D523–D527. doi: 10.1093/nar/gkh013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quintás-Cardama A, Vaddi K, Liu P, Manshouri T, Li J, Scherle PA, et al. Preclinical characterization of the selective JAK1/2 inhibitor INCB018424: therapeutic implications for the treatment of myeloproliferative neoplasms. Blood. 2010;115:3109–3117. doi: 10.1182/blood-2009-04-214957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer J, Rhein M, Schiedlmeier B, Kustikova O, Rudolph C, Kamino K, et al. Remarkable leukemogenic potency and quality of a constitutively active neurotrophin receptor, deltaTrkA. Leukemia. 2007;21:2171–2180. doi: 10.1038/sj.leu.2404882. [DOI] [PubMed] [Google Scholar]
- Iqbal J, Weisenburger DD, Chowdhury A, Tsai MY, Srivastava G, Greiner TC, International Peripheral T-cell Lymphoma Project et al. Natural killer cell lymphoma shares strikingly similar molecular features with a group of non-hepatosplenic ?d T-cell lymphoma and is highly sensitive to a novel aurora kinase A inhibitor in vitro. Leukemia. 2011;25:348–358. doi: 10.1038/leu.2010.255. [DOI] [PubMed] [Google Scholar]
- Iqbal J, Weisenburger DD, Greiner TC, Vose JM, McKeithan T, Kucuk C, International Peripheral T-Cell Lymphoma Project et al. Molecular signatures to improve diagnosis in peripheral T-cell lymphoma and prognostication in angioimmunoblastic T-cell lymphoma. Blood. 2010;115:1026–1036. doi: 10.1182/blood-2009-06-227579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccaluga PP, Agostinelli C, Califano A, Rossi M, Basso K, Zupo S, et al. Gene expression analysis of peripheral T cell lymphoma, unspecified, reveals distinct profiles and new potential therapeutic targets. J Clin Invest. 2007;117:823–834. doi: 10.1172/JCI26833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Travert M, Huang Y, de Leval L, Martin-Garcia N, Delfau-Larue MH, Berger F, et al. Molecular features of hepatosplenic T-cell lymphoma unravels potential novel therapeutic targets. Blood. 2012;119:5795–5806. doi: 10.1182/blood-2011-12-396150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Doorn R, van Kester MS, Dijkman R, Vermeer MH, Mulder AA, Szuhai K, et al. Oncogenomic analysis of mycosis fungoides reveals major differences with Sezary syndrome. Blood. 2009;113:127–136. doi: 10.1182/blood-2008-04-153031. [DOI] [PubMed] [Google Scholar]
- de Leval L, Rickman DS, Thielen C, Reynies Ad, Huang YL, Delsol G, et al. The gene expression profile of nodal peripheral T-cell lymphoma demonstrates a molecular link between angioimmunoblastic T-cell lymphoma (AITL) and follicular helper T (TFH) cells. Blood. 2007;109:4952–4963. doi: 10.1182/blood-2006-10-055145. [DOI] [PubMed] [Google Scholar]
- Dürig J, Bug S, Klein-Hitpass L, Boes T, Jöns T, Martin-Subero JI, et al. Combined single nucleotide polymorphism-based genomic mapping and global gene expression profiling identifies novel chromosomal imbalances, mechanisms and candidate genes important in the pathogenesis of T-cell prolymphocytic leukemia with inv(14)(q11q32). Leukemia. 2007;21:2153–2163. doi: 10.1038/sj.leu.2404877. [DOI] [PubMed] [Google Scholar]
- Shin J, Monti S, Aires DJ, Duvic M, Golub T, Jones DA, et al. Lesional gene expression profiling in cutaneous T-cell lymphoma reveals natural clusters associated with disease outcome. Blood. 2007;110:3015–3027. doi: 10.1182/blood-2006-12-061507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckerle S, Brune V, Döring C, Tiacci E, Bohle V, Sundström C, et al. Gene expression profiling of isolated tumour cells from anaplastic large cell lymphomas: insights into its cellular origin, pathogenesis and relation to Hodgkin lymphoma. Leukemia. 2009;23:2129–2138. doi: 10.1038/leu.2009.161. [DOI] [PubMed] [Google Scholar]
- Lamant L, de Reyniès A, Duplantier MM, Rickman DS, Sabourdy F, Giuriato S, et al. Gene-expression profiling of systemic anaplastic large-cell lymphoma reveals differences based on ALK status and two distinct morphologic ALK+ subtypes. Blood. 2007;109:2156–2164. doi: 10.1182/blood-2006-06-028969. [DOI] [PubMed] [Google Scholar]
- Deeks SG, Wagner B, Anton PA, Mitsuyasu RT, Scadden DT, Huang C, et al. A phase II randomized study of HIV-specific T-cell gene therapy in subjects with undetectable plasma viremia on combination antiretroviral therapy. Mol Ther. 2002;5:788–797. doi: 10.1006/mthe.2002.0611. [DOI] [PubMed] [Google Scholar]
- Mitsuyasu RT, Anton PA, Deeks SG, Scadden DT, Connick E, Downs MT, et al. Prolonged survival and tissue trafficking following adoptive transfer of CD4zeta gene-modified autologous CD4(+) and CD8(+) T cells in human immunodeficiency virus-infected subjects. Blood. 2000;96:785–793. [PubMed] [Google Scholar]
- Walker RE, Bechtel CM, Natarajan V, Baseler M, Hege KM, Metcalf JA, et al. Long-term in vivo survival of receptor-modified syngeneic T cells in patients with human immunodeficiency virus infection. Blood. 2000;96:467–474. [PubMed] [Google Scholar]
- Migone TS, Lin JX, Cereseto A, Mulloy JC, O'Shea JJ, Franchini G, et al. Constitutively activated Jak-STAT pathway in T cells transformed with HTLV-I. Science. 1995;269:79–81. doi: 10.1126/science.7604283. [DOI] [PubMed] [Google Scholar]
- Yu CL, Jove R, Burakoff SJ. Constitutive activation of the Janus kinase-STAT pathway in T lymphoma overexpressing the Lck protein tyrosine kinase. J Immunol. 1997;159:5206–5210. [PubMed] [Google Scholar]
- Flex E, Petrangeli V, Stella L, Chiaretti S, Hornakova T, Knoops L, et al. Somatically acquired JAK1 mutations in adult acute lymphoblastic leukemia. J Exp Med. 2008;205:751–758. doi: 10.1084/jem.20072182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Q, Raghunath PN, Xue L, Majewski M, Carpentieri DF, Odum N, et al. Multilevel dysregulation of STAT3 activation in anaplastic lymphoma kinase-positive T/null-cell lymphoma. J Immunol. 2002;168:466–474. doi: 10.4049/jimmunol.168.1.466. [DOI] [PubMed] [Google Scholar]
- Khoury JD, Medeiros LJ, Rassidakis GZ, Yared MA, Tsioli P, Leventaki V, et al. Differential expression and clinical significance of tyrosine-phosphorylated STAT3 in ALK+ and ALK- anaplastic large cell lymphoma. Clin Cancer Res. 2003;9 10 Pt 1:3692–3699. [PubMed] [Google Scholar]
- Sommer VH, Clemmensen OJ, Nielsen O, Wasik M, Lovato P, Brender C, et al. In vivo activation of STAT3 in cutaneous T-cell lymphoma. Evidence for an antiapoptotic function of STAT3. Leukemia. 2004;18:1288–1295. doi: 10.1038/sj.leu.2403385. [DOI] [PubMed] [Google Scholar]
- Schambach A, Wodrich H, Hildinger M, Bohne J, Kräusslich HG, Baum C. Context dependence of different modules for posttranscriptional enhancement of gene expression from retroviral vectors. Mol Ther. 2000;2:435–445. doi: 10.1006/mthe.2000.0191. [DOI] [PubMed] [Google Scholar]
- Newrzela S, Gunda B, von Laer D. T cell culture for gammaretroviral transfer. Methods Mol Biol. 2009;506:71–82. doi: 10.1007/978-1-59745-409-4_6. [DOI] [PubMed] [Google Scholar]
- Li LC, Dahiya R. MethPrimer: designing primers for methylation PCRs. Bioinformatics. 2002;18:1427–1431. doi: 10.1093/bioinformatics/18.11.1427. [DOI] [PubMed] [Google Scholar]