

Abstract
The HIV-1 coreceptor CCR5 is a validated target for HIV/AIDS therapy. The apparent elimination of HIV-1 in a patient treated with an allogeneic stem cell transplant homozygous for a naturally occurring CCR5 deletion mutation (CCR5Δ32/Δ32) supports the concept that a single dose of HIV-resistant hematopoietic stem cells can provide disease protection. Given the low frequency of naturally occurring CCR5Δ32/Δ32 donors, we reasoned that engineered autologous CD34+ hematopoietic stem/progenitor cells (HSPCs) could be used for AIDS therapy. We evaluated disruption of CCR5 gene expression in HSPCs isolated from granulocyte colony-stimulating factor (CSF)-mobilized adult blood using a recombinant adenoviral vector encoding a CCR5-specific pair of zinc finger nucleases (CCR5-ZFN). Our results demonstrate that CCR5-ZFN RNA and protein expression from the adenoviral vector is enhanced by pretreatment of HSPC with protein kinase C (PKC) activators resulting in >25% CCR5 gene disruption and that activation of the mitogen-activated protein kinase kinase (MEK)/extracellular signal-regulated kinase (ERK) signaling pathway is responsible for this activity. Importantly, using an optimized dose of PKC activator and adenoviral vector we could generate CCR5-modified HSPCs which engraft in a humanized mouse model (albeit at a reduced level) and support multilineage differentiation in vitro and in vivo. Together, these data establish the basis for improved approaches exploiting adenoviral vector delivery in the modification of HSPCs.
Introduction
HIV-1 continues to be a world-wide health problem with ≈50,000 new cases each year in the United States alone.1 The treatment of HIV-1 has improved through the use of combination antiretroviral therapy but antiviral treatment alone cannot eradicate HIV-1. Hütter et al. described a patient with acute myeloid leukemia, who was cured of AIDS following a bone marrow (BM) transplant from a donor who was homozygous for a 32-base pair deletion in the CCR5 gene (CCR5Δ32/Δ32).2 The engrafted donor phenotype appears to have conferred long-term control of HIV-1 replication as the patient has been off combination antiretroviral therapy for several years without HIV-1 being detected.3 However, the identification of human leukocyte antigen-matched, CCR5Δ32/Δ32 homozygous donors for transplantation presents a significant logistical barrier to the general application of this approach. An alternative approach to HIV-1 therapy is to use autologous hematopoietic stem and progenitor cells (HSPCs) to genetically engineer an HIV-1–resistant immune system that would prevent ongoing viral infection and/or replication.4
Several groups have performed clinical trials in which autologous peripheral blood T-cells or HSPCs were genetically modified to introduce anti-HIV moieties that are RNA or protein in nature.5 These trials have proven the safety and potential efficacy of a cell-based approach to immunotherapy for HIV-1 with observations of selective T-cell survival and reduced viral loads following infusion of gene-modified T-cells.6,7,8 Based on these results and our prior work in stem cell transplantation for HIV/AIDS-related lymphomas,9 we rationalized that transplantation of autologous HSPC that were genetically altered to prevent expression of CCR5 would result in an HIV-resistant immune system for patients without an allogeneic donor. We therefore sought to develop methods for engineering HSPC to eliminate expression of CCR5 at the genomic level so that the progeny of the HSPC inherit the resistance.
Zinc finger (ZF) proteins are sequence-specific DNA-binding proteins that when coupled to a DNA endonuclease for ZF nucleases (ZFNs) which can be engineered to cut DNA at a specific site.10 Moreover, the ZFN need to be expressed only for a short time to achieve permanent disruption of the target genomic sequence and thus are less likely to lead to antigen presentation and immune elimination as do other stably expressed transgenic proteins. Perez et al. have demonstrated inhibition of HIV-1 infection in T-cells modified to eliminate the CCR5 coreceptor expression using an adenoviral vector-encoded ZFN.11 Similarly, Holt et al. demonstrated that ZFN-mediated disruption of CCR5 gene sequences in fetal liver HSPC permitted engraftment of the modified cells in NOD/SCID/IL2rγnull (NSG) mice and conferred resistance to R5-tropic HIV-1 infection.12 Based on this result and our prior experience with stem cell gene therapy for HIV-1,13 we sought to create current good manufacturing practice compliant procedures for ZFN-mediated CCR5 disruption in HSPCs isolated from granulocyte colony-stimulating factor (CSF)-mobilized adults. We describe here optimized conditions for HSPC modification using cells derived from healthy donors and a clinical grade adenoviral vector encoding the previously described CCR5-specific ZFN. The goal of these studies is to develop reproducible cell-processing procedures that achieve maximal CCR5 disruption with minimal impact on the hematopoietic potential of autologous HSPC.
Results
Identification of optimal conditions for genomic modification of CCR5 in HSPC
We initially evaluated ZFN activity in adult CD34+ HSPCs using culture conditions that were previously used to support ex vivo genetic modification of HSPC for clinical use.13 HSPCs were isolated from granulocyte-CSF–mobilized peripheral blood using a CliniMACS cell selection device (Miltenyi Biotec, Auburn, CA) and routinely exceeded 95% purity. We employed an adenoviral vector expressing a CCR5-specific ZFN pair (CCR5-ZFN) that has been previously shown to effectively disrupt CCR5 genomic sequences in primary human T-cells.11 This vector uses the Ad5/F35 chimeric fiber protein to broaden the tropism to include hematopoietic stem cells.14,15 Freshly isolated HSPCs were prestimulated (12–24 hours) in 100 ng/ml each of stem cell factor (SCF) and fms-like tyrosine kinase 3 ligand (Flt-3L) and 10 ng/ml thrombopoietin (TPO) (referred collectively as SFT) and then transduced with CCR5-ZFN at multiplicity of infection (MOI) of 250–5,000. After overnight incubation (12–24 hours), HSPCs were washed and placed into liquid bulk culture with cytokines that promote hematopoietic cell growth and differentiation (SCF, Flt-3L, TPO, interleukin-3 (IL-3), IL-6, granulocyte macrophage-CSF, and erythropoietin) for 7–28 days. Cells were harvested on day 7 and analyzed for CCR5 gene disruption using the surveyor nuclease assay as previously described.16 Our results indicated that CCR5 gene disruption in adult HSPCs did not exceed 5% when ZFNs were delivered to adult HSPC via the CCR5-ZFN adenoviral vector under the described conditions (Supplementary Figure S1a). This was substantially lower than the 17% disruption we previously observed when ZFNs were introduced into cord blood HSPC through nucleofection of plasmid DNA.12 Extending the prestimulation time to 48 or 72 hours did not increase CCR5-ZFN activity (Supplementary Figure S1b). As a control for CCR5-ZFN function, we transduced K562 erythroleukemia cells (MOI 250) or activated primary human T-cells (MOI 500) and observed CCR5 genetic modification of 21 and 30.7%, respectively (Supplementary Figure S1c). We also used flow cytometric analysis to confirm the expression of the Ad5/F35 fiber receptor (CD46) on HSPC (Supplementary Figure S2a–c) and demonstrated transduction of HSPC using a similar adenoviral vector encoding green fluorescence protein (Ad5/F35-GFP) under the same conditions (Supplementary Figure S2d,e). CD34+ HSPC cells are readily transducible under standard culture conditions with cytokines (>65% of cells were GFP-positive). Together, these data indicated that the Ad5/F35 vector was functional and that adult HSPCs could be readily transduced with an Ad5/F35-pseudotyped adenoviral vector.
We postulated that the low CCR5 disruption obtained with the CCR5-ZFN vector in HSPCs may reflect a problem with viral entry, ZFN expression, stability and/or activation state of the HSPCs. Compounds known to mediate intracellular signaling, protein trafficking, histone acetylation, cycling and expansion of HSPC were tested in an attempt to identify conditions that supported increased ZFN activity (Table 1). Samples of CD34+ HSPC were prestimulated with SFT for 16–20 hours either with test compounds present during the entire prestimulation (pleoitrophin, interferon-α, IL-6, trichostatin A, valproic acid, bortezomib) or for 30 minutes before transduction (phorbol 12-myristate 13-acetate (PMA), bryostatin, chloroquine, MG132, and okadeic Acid). All pre-treated cells were washed, transduced with CCR5-ZFN at an MOI of 50 and allowed to incubate for 12–24 hours. Cells were then washed and placed in bulk liquid culture for 7 days as described above. Samples from each condition were harvested and analyzed for CCR5 disruption by surveyor nuclease assay. We observed little to no change over background in the level of CCR5 genetic modification in cells treated with most compounds tested. However, when we combined high MOI (150) with protein kinase C (PKC) activators (PMA or bryostatin), we observed poor cell viability but a significant increase in transduction efficiency (>99% GFP+ cells with greater than tenfold increase in mean fluorescent intensity when using Ad5/F35-GFP) (Supplementary Figure S2f). A similar increase in CCR5 disruption (26–31%) was observed in CCR5-ZFN–transduced HSPC when PKC activators (PMA or bryostatin) were included at the time of transduction (Figure 1). We subsequently performed titrations of PMA and bryostatin concentrations as well as vector MOI to identify optimal conditions for transduction of HSPCs (Supplementary Figure S3a,b). CCR5 disruption of >20% was observed using as little as 1 ng/ml PMA and CCR5-ZFN MOI of 150 or 10 nmol/l bryostatin and a vector MOI of 50. However, conditions that led to high levels of ZFN activity were also associated with low cell viability as determined by flow cytometric analysis using Annexin V/7-AAD staining. CCR5 disruption of ≈10% was observed when using vector MOI's of 5 and 10 following overnight prestimulation with SFT followed by a 30-minute incubation with 1 ng/ml PMA or 5 nmol/l bryostatin immediately preceding transduction (Supplementary Figure S3c,d). Cell viability under these latter conditions was >80%. Cells transduced with a control adenoviral vector (containing no ZFN sequences, Ad5/35 hIL2Rg10) in parallel with CCR5-ZFN at MOIs of 0, 10, and 50 showed similar vector dose-dependent toxicity (Supplementary Figure S4) suggesting that the loss of cell viability at high vector MOI was mediated by the adenoviral vector itself and not the ZFN.
Table 1. Effect of cytokines, chemokines, chemical compounds, and modulators on ZFN activity in HSPC CD34+ cells.

Figure 1.

Protein kinase C (PKC) activators (PMA and bryostatin) significantly enhanced CCR5-ZFN activity in CD34+ hematopoietic stem/progenitor cells (HSPC). Adult HSPCs were stimulated with SFT in SCGM medium overnight followed by PMA or bryostatin treatment for 30 minutes before CCR5-ZFN transduction (multiplicity of infection (MOI) 150). The percent CCR5 disruption was obtained by surveyor nuclease assay from transduced cells (MOI 150) without PKC activator treatment (left), with 20 ng/ml PMA treatment (middle), and 10 nmol/l bryostatin treatment (right). Percent of CCR5 disruption is indicated under each lane. PMA, phorbol 12-myristate 13-acetate; ZFN, zinc finger nuclease.
Evaluation of the effect of PKC activation and vector transduction on in vitro HSPC growth and differentiation
Having identified optimized conditions for CCR5 disruption, we wished to evaluate the hematopoietic potential of prestimulated, PKC-activated, CCR5-ZFN–transduced HSPC. HSPCs were prestimulated overnight in SFT and then activated with 1 ng/ml of PMA or 5 nmol/l bryostatin for 30 minutes. Cells were cultured in the absence of vector to serve as a control or transduced with CCR5-ZFN vector at MOIs of 5 or 10 for 16–24 hours. Cells were then washed, plated in methylcellulose media, and cultured for 2 weeks to determine colony-forming potential of each sample. No significant differences were seen in the number or types of colonies formed by the control and treatment group with MOI of 5 (Figure 2a). A modest but significant (P < 0.05) reduction in colony-forming potential was observed when using a vector MOI of 10.
Figure 2.

Effect of PMA and bryostatin treatment and vector transduction on hematopoietic stem/progenitor cell (HSPC) CD34+ cell growth and hematopoietic potential. (a) Colony-forming unit (CFU) assay. A total of 500 cells were plated in methylcellulose medium in triplicates for each condition and the average ± SD of CFU are shown. One way analysis of variance (ANOVA) with Dunnett's multiple comparison test was used for statistical analysis as compared with the untransduced control. *P < 0.05. (b) Growth of untransduced, PMA-, and bryostatin-treated, CCR5-ZFN–transduced cells cultured in bulk culture medium for 28 days. Average ± SD fold growth over starting cell number is shown, N = 3. (c) Phenotypic analysis of 2-week bulk culture and an one-way ANOVA performed with Bonferroni post-test. Error bars indicate average ± SEM of percent of total cells with each phenotype is shown, N = 3. ns, not significant; PMA, phorbol 12-myristate 13-acetate; Untd, untransduced; ZFN, zinc finger nuclease.
In order to more completely characterize the effect of transduction on lineage potential, transduced HSPC were also expanded in vitro in bulk liquid culture with weekly monitoring of cell proliferation and phenotypic analysis of myeloerythroid differentiation at 4 weeks. Cells from all cultures expanded over 200-fold, and there were no significant differences in total cell expansion between PMA- or bryostatin-treated and control cultures (Figure 2b). Flow cytometric analysis of bulk cultures was performed to assess lineage potential of the samples. Similar to the results from the colony-forming assays, no significant differences in the percentage of differentiated CD33+ progenitor cells, CD14+ monocytes, CD15+ granulocytes or glycophorin A+ erythrocytes were observed under these conditions as compared with control (Figure 2c).
CCR5-ZFN–induced CCR5 disruption is sustained and present in multiple lineages
To evaluate the stability of CCR5 disruption introduced by CCR5-ZFN in CD34+ HSPCs, 4-week liquid cultures of HSPC were analyzed weekly for CCR5 disruption by surveyor nuclease assay. These data demonstrate that CCR5-ZFN treatment results in persistent CCR5 gene disruption in the range of 12–24% on day 7 followed by a gradual decline to a range of 7–18% at day 28 for all conditions tested (Supplementary Figure S5a). In addition, we isolated differentiated CD14+ (monocytes), CD15+ (granulocytes), CD14−/CD15− (nonmyeloid), CD14+/CD15+ multipotent progenitors by flow cytometric cell sorting and analyzed for CCR5 disruption by surveyor nuclease assay. Although there were different levels of CCR5 genomic editing among the lineages analyzed, CCR5 disruption was detected in all the populations (Supplementary Figure S5b). In particular, there was a considerable level of CCR5 disruption (>10%) in the CD14+ monocyte population, a critical HIV-1 target cell type. We observed no difference in the number of granulocyte colonies derived from transduced cells when compared with untransduced cells in the colony-forming unit assays (data not shown). Thus, it is unlikely that the lower level of CCR5 disruption in the granulocyte population in this donor was due to an inefficient differentiation in this lineage after high expression levels of ZFN expression or high levels of adenovirus vector transduction.
Clonogenic analysis of CCR5 disruption
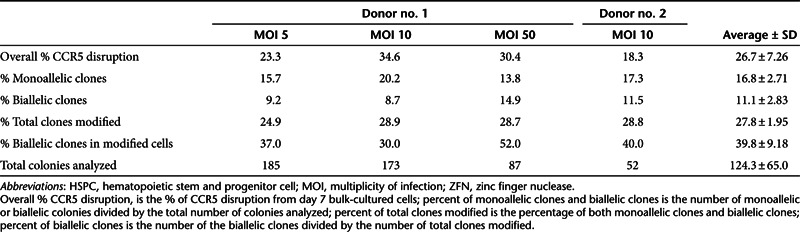
In order to establish the frequency of monoallelic and biallelic disruption of CCR5 gene, sequence analysis was performed on DNA from individual colony-forming units derived from PMA-treated CCR5-ZFN–transduced HSPCs. HSPCs from two different donors were transduced with vector MOIs of 5, 10, and 50 (donor 1) or MOI of 10 (donor 2) after treatment with 1 ng/ml of PMA for 30 minutes and cultured in methylcellulose medium for 2 weeks. Rapid hybridization-based screening was used to identify those clones with at least one disrupted CCR5 allele for further analysis. A total of 445 clones were analyzed from donor 1 and 52 clones from donor 2 (Table 2). An average of 27.8 ± 2% of the clones contained a detectable disruption of CCR5 with 16.8 ± 2.7% monoallelic and 11.1 ± 2.8% biallelic disruptions overall. Thus, ~40% of all CCR5-disrupted colonies contained biallelic disruptions and were thus “engineered” to be CCR5 homozygous deleted. In addition, the frequency of CCR5 disruption in the clones (% total clones modified) was similar to overall %CCR5 disruption in the bulk culture indicating that CCR5-disrupted cells had equal colony-forming potential as mock-treated cells. Our results reveal an overall biallelic disruption frequency of 10% of all clonogenic progenitors.
Table 2. CCR5-ZFN generated both monoallelic and biallelic disruptions in HSPC CD34+ cells.
Off-target analysis
In order to understand the specificity of the CCR5-ZFN's activities, deep-sequencing analysis of the top 23 off-target sites (predicted by in silico modeling, ref. 11 and integration site analysis, ref. 17) was performed on PMA/CCR5-ZFN–treated cells (Supplementary Table S1). A low but detectable level of off-target modification was detected at CCR2, about 1 log lower than at the intended (CCR5) site. In addition, very low levels of modification were also observed at three additional sites. Importantly, the three additional sites where low levels of modification was detected occurred in noncoding (either at intergenic or intronic) sequences in the genome. The remaining 19 sites analyzed by deep sequencing showed no modification above background levels (0.01%). A list of top eight genes in which off-target cleavage is predicted to occur is presented in Supplementary Table S2.
Engraftment of ZFN-modified CD34+ HSPCs in NSG mice
The use of immunodeficient mice as a model for hematopoietic potential of candidate HSPC has been extensively documented.18,19 We used the NOD.Cg-Prkdcscid IL2rgtm1Wjl/SzJ (NSG) mouse model to evaluate the hematopoietic potential of ZFN-modified HSPC cells. NSG mice were transplanted with prestimulated HSPC, prestimulated HSPC treated with PMA or bryostatin alone, or prestimulated HSPC treated with PMA or bryostatin plus CCR5-ZFN at MOI's of 5 or 10. When non-transduced HSPC, or cells pre-treated with 1 ng/ml of PMA were tested, 100% of the animals engrafted with at least 1% human (CD45+) cells in the spleen and BM at 26–28 weeks. The combination of PMA (1 ng/ml) plus CCR5-ZFN transduction reduced the frequency of engraftment to between 80 and 91% (as measured in spleen and BM, respectively) at MOI of 5, to 50–68% at MOI of 10 (Table 3).
Table 3. Engraftment of NSG mice after transplantation of PMA-, bryostatin-treated, and CCR5-ZFN–transduced HSPC.
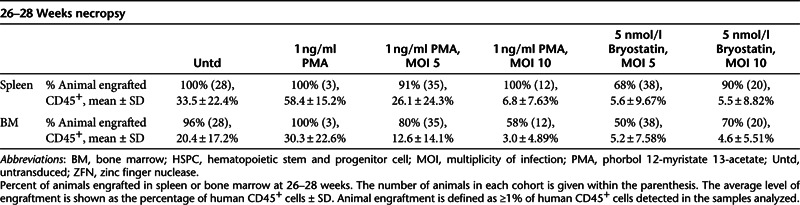
Although evaluation of human cell engraftment (%CD45+ cells) in the spleen and BM showed no statistically significant reduction at 26–28 weeks in the PMA alone group, or for PMA plus CCR5-ZFN at MOI 5 when compared with untransduced cells (P > 0.05), a reduction of about twofold was evident for the PMA plus CCR5-ZFN at MOI 10 treated group (P < 0.001 in spleen and P < 0.01 in BM). A significant reduction in the level of engraftment in spleen and BM was also observed when cells were treated with bryostatin plus CCR5-ZFN at either MOI of 5 or 10 (P < 0.001 in all cases). Despite any effects on absolute levels of engraftment, no significant differences were observed in the percentage of specific hematopoietic lineages (CD4+ and CD8+ T-cells, CD19+ B-cells, monocytes) for any treatment group at this 26–28 weeks necropsy in either spleen (Supplementary Figure S6a) or BM (Supplementary Figure S6b).
Multiple lineage CCR5 disruption in transplanted, HIV-1–challenged NSG mice
At 28 weeks of age, five animals in a transplant cohort of 10 were necropsied and human cell (CD45+) engraftment in the BM and spleen was determined. The remaining five mice were challenged with HIV-1BaL and monitored for an additional 11 weeks. Of these five mice, the mock- (2) or PMA alone-treated (1) animals displayed sustained HIV-1 infections throughout the period examined, with viral kinetics likely reflecting differences in human CD4+ T cell levels in the mice (data not shown). Of the mice engrafted with PMA/CCR5-ZFN (MOI 10)-treated HSPC, one animal became infected with HIV-1 with the same kinetics as the mock- and PMA-treated mice, but then underwent a marked reduction in HIV-1 levels between 8 and 10 weeks post-infection and at necropsy. This animal had the highest levels of human CD45+ cells, and CD4+ T cells, which is consistent with protective effects of CCR5-ZFN modification in the face of a HIV-1 infection (data not shown). Surveyor nuclease analysis was performed on organs (BM and spleen) from all animals at necropsy to determine the extent of CCR5 disruption of human cells (Supplementary Figure S7). In the cohort of animals analyzed before HIV-1 infection, the CCR5 disruption rates were ~3% in the BM of the PMA/CCR5-ZFN animals. Following 11 weeks of HIV-1 infection, the frequency of CCR5 disruption in the two remaining PMA/CCR5-ZFN–treated mice showed a modest increase to ~5% in the BM and 7% in the spleen (Supplementary Figure S7a). However, upon sorting of splenocytes in the HIV-infected mouse into CD4+, CD8+, and CD19+ (B-cell) population (Supplementary Figure S7b), evidence for selective advantage was observed in both CD4+ and CD8+ populations exhibiting an increase of 20% in CCR5 disruption, (Supplementary Figure S7c). Previously, Holt et al. noted that infections with HIV-1BaL in humanized NSG mice ultimately selects for CCR5-negative CD8+ cells as well as CD4+ cells due to selection occurring at the double-positive stage within the thymus following CCR5 upregulation in this organ,12 and likely accounts for the increase observed in CCR5 disruption in both T-cell subsets.
Mechanism of PKC activation in enhancing ZFN activity
We observed significant enhancement of adenovirally delivered ZFN activity upon treatment with PMA and bryostatin. PKC is a serine/threonine kinase and is known to activate multiple intracellular signaling pathways in cells of the hematopoietic lineage.20 In order to investigate the mechanism of PKC-mediated enhancement of CCR5 gene disruption in HSPC, we analyzed the downstream activation of several intracellular signaling pathways known to be regulated by serine or threonine phosphorylation status. HSPCs from four donors were prestimulated overnight with cytokines (SFT) and treated the next day with (or without) 1 ng/ml PMA. Both treated and untreated cells were fixed, permeabilized, and labeled with antibodies to pSTAT3, pSTAT5, pp38 mitogen-activated protein kinase, pAkt, p-β-Catenin, pERK1/2, and p-NFκBp65. Samples were washed and analyzed by flow cytometry for the phosphorylation levels of each signaling epitope (Figure 3a). Among the phospho-signaling molecules evaluated, we only detected statistically significant (P < 0.001) induction of extracellular signal-regulated kinase (ERK)1/2 phosphorylation which peaked between 15 and 20 minutes following PMA addition. In a separate experiment, we confirmed that 5 nmol/l bryostatin also induced ERK1/2 phosphorylation to similar levels (Figure 3b). Although NFκBp65 signaling is known to be responsive to PMA activation, we did not observe phosphorylation of NFκBp65 or its cognate inhibitor IκB (data not shown).
Figure 3.

Protein kinase C activators induce phosphorylation of ERK1/2 in hematopoietic stem/progenitor cell (HSPC) CD34+ cells and result in enhancement of CCR5-ZFN activity. (a) CD34+ HSPCs from four healthy donors were stimulated with 1 ng/ml PMA and stained with anti-phospho-signal protein antibodies as indicated. Mean fluorescence intensity (MFI) of each phospho-signal protein was displayed. Two-way analysis of variance with Bonferroni post-test was used for statistical analysis, ***P < 0.001. (b) CD34+ HSPC from four donors were either untreated or treated in parallel with 1 ng/ml PMA or 5 nmol/l bryostatin. (c,d) CD34+ HSPCs from three donors were either mock-treated or treated with bryostatin (Bryo) and MEK1/2 inhibitor GSK1120212 (Bryo + 5 or 50 nmol/l GSK) at the concentration indicated 1 hour before bryostatin stimulation, followed by CCR5-ZFN vector transduction at multiplicity of infection 10 (all samples). (c) MFI of pERK and (d) % CCR5 disruption of the same samples as in c were measured. The relative fold change of the parameters listed was normalized to samples treated with bryostatin and vector only. Average ± SEM is shown for (c) pERK MFI and (d) CCR5 disruption. ERK, extracellular signal-regulated kinase; MAPK, mitogen-activated protein kinase; NF-κB, nuclear factor-κB; PMA, phorbol 12-myristate 13-acetate; ZFN, zinc finger nuclease.
In order to determine whether the activation of mitogen-activated protein kinase kinase (MEK)/ERK pathway was required for increased ZFN activity, a specific inhibitor of MEK1/2 GSK1120212 was used to prevent phosphorylation of ERK and the effect on CCR5 disruption in PMA or bryostatin-activated HSPC was measured. HSPCs from three independent donors were prestimulated with three cytokines (SFT) as described above and then exposed to 5 or 50 nmol/l GSK1120212 for 1 hour before PKC activation with bryostatin and CCR5-ZFN transduction. Our data demonstrate a dose-dependent reduction of ERK phosphorylation in the HSPC using this inhibitor (Figure 3c). In the same experiment, we performed analysis of CCR5 disruption on the aforementioned samples using the surveyor nuclease assay. The bryostatin-induced ZFN-mediated CCR5 disruption was also reduced in a dose-dependent manner by the MEK inhibitor (Figure 3d). Similar results were obtained when PMA was used as the PKC activator (data not shown).
We then investigated how mitogen-activated protein kinase activation leads to increased ZFN activity. First, ZFN RNA expression was analyzed by reverse transcription-PCR analysis using primers specific for Fok1 endonuclease domain sequences of the ZFN. We compared untreated, CCR5-ZFN–transduced, and PMA-activated/ZFN-transduced CD34+ HSPCs from five healthy donors for Fok-1 RNA expression. Our data demonstrated a 50-fold increase in the Fok1 RNA expression upon PMA stimulation relative to cells transduced with vector alone (Figure 4a). In addition, western blot analysis was performed on samples treated with PMA or bryostatin and demonstrated that low levels of CCR5-ZFN protein was expressed when the cells were prestimulated with SFT and transduced with vector alone. However, a substantial dose-dependent increase in the expression of ZFN protein was observed upon treatement of HSPC with either PMA or bryostatin (Figure 4b). Pretreatment of HSPC with MEK inhibitor before transduction resulted in dose-dependent reduction of CCR5-ZFN protein expression (Figure 4c). Thus, in this adenoviral delivery system, the activation of the MEK/ERK pathway is critical for enhancing ZFN activity primarily through increased viral entry and cytomegalovirus (CMV) promoter-driven transcription of ZFN RNA leading to enhanced protein expression.
Figure 4.

Protein kinase C (PKC) activation increases CCR5-ZFN RNA expression leading to enhanced protein production. (a) Reverse transcription-PCR analysis of CCR5-ZFN mRNA using CCR5-ZFN-Fok1 endonuclease-specific primers. Untreated, CCR5-ZFN transduced (multiplicity of infection (MOI) 50) without PMA treatment (0PMA-50), and 1 ng/ml PMA-treated and CCR5-ZFN–transduced (1PMA-50) CD34+ cells from five donors were analyzed. (b) Western blot analysis of Fok1 endonuclease protein expression in hematopoietic stem/progenitor cells treated with PKC activators at the concentrations indicated and transduced with CCR5-ZFN vector (MOI 50). (c) Dose-dependent inhibition of the Fok1 endonuclease protein by MEK inhibitor GSK1120212 at indicated concentrations before bryostatin treatment (5 nmol/l) and CCR5-ZFN transduction (MOI 50). PMA, phorbol 12-myristate 13-acetate; ZFN, zinc finger nuclease.
Discussion
Allogeneic BM transplantation with HIV-resistant (CCR5Δ32/Δ32) hematopoietic stem cells has been shown to provide long-term control of HIV-1 viremia and a sterilizing cure of the infection. However, the identification of a suitable human leukocyte antigen-matched BM donor with a CCR5Δ32/Δ32 (or equivalent) genotype is exceedingly difficult and not practical for widespread application of this approach. Our previous studies (and those of others) have demonstrated that transplantation of gene-modified autologous HSPCs is a feasible approach to HIV-1 gene therapy. Several groups have demonstrated the utility of ZFN technology in disruption of the CCR5 gene-reading frame resulting in effective inhibition of CCR5 expression, and this technology is currently under clinical investigation in T-cells.11 In our prior animal studies, we used electroporation to deliver ZFN-encoding sequences to cord blood HSPC and were able to observe CCR5 disruption of ≈17% without the use of any additional stimulation agents.12 In our current report, we evaluated an adenoviral vector-based CCR5/ZFN construct to determine whether we could generate similar levels of CCR5 disruption in adult HPSC. Among the advantages of using adenoviral vectors are the availability of good manufacturing practice methods for production of safe, high-titer stocks and the ability to transduce both dividing and nondividing cells without integration into genome, minimizing the potential risk of vector-mediated oncogenesis. Surprisingly, we were unable to achieve similar levels of CCR5 disruption using adult HSPC with this vector. We demonstrate that this is at least partially due to suboptimal transduction and low level of transcription of the ZFN protein from the CMV promoter in HSPC, but may also involve other mechanisms related to DNA repair in the nonhomologous end joining (NHEJ) pathway.
We explored ways to increase the activity of ZFNs in HSPC by assessing treatments that could improve adenoviral vector transduction through alteration of HSPC activation (cytokines, pleoitrophin, interferon-α, PMA, bryostatin), chromatin condensation (trichostatin A, valproic Acid), intracellular trafficking (okadeic Acid, chloroquin) or ZFN expression and stability (PMA, bryostatin, bortezomib, MG132). We found that only short-term exposure to low concentrations of the PKC activators, PMA or bryostatin resulted in increased CCR5-ZFN activity (CCR5 disruption). Our results using Ad/GFP vector demonstrated that pretreatment of HSPC with PKC activators demonstrated an increase in the percentage of GFP+ cells from 66 to >99% and greater than tenfold increase in GFP expression levels. This data supports a mechanism that includes both higher levels of viral entry (% transduced cells) and CMV-driven gene expression (GFP). We confirmed that PKC activation results in activation of the MEK/ERK pathway and verified the role of this pathway using MEK inhibitors and measuring ZFN RNA and protein expression and CCR5 disruption. This mechanism is consistent with previous reports that enhancement of CMV promoter-driven gene expression results in the recruitment of c-fos and c-jun (AP-1) transcriptional complexes to AP-1–binding sites on the CMV promoter and enhances its transcriptional activity.21,22 It is also possible that PKC activation results in changes to the alternate NHEJ DNA repair pathway making it more prone to errors and leading to a higher rate of CCR5 disruption. However, reports by several other groups have shown that PKC activation actually inhibits poly(adenosine diphosphate-ribose) polymerase-1 (PARP-1) activity which is required for alternative NHEJ activity leading to inaccurate DNA repair.23,24 Thus, based on our Ad5/GFP and ZFN RNA and protein expression analysis, we believe the most likely explanation for increased activity is a combination of enhanced viral transduction and increased ZFN gene expression, although we cannot formally rule out additional mechanisms.
It is well established that prolonged activation of PKC can lead to terminal differentiation of hematopoietic cells along the erythromyeloid lineages or even promote cellular transformation.20,21,25,26 In order to assess the effect of brief exposure to PKC activation on the hematopoietic potential of HSPCs, we used a series of in vitro and in vivo assays to evaluate cell proliferation, colony formation, and lineage-specific differentiation of treated versus untreated HSPC. Using brief activation of PKC and an Ad5/F35 ZFN MOI of 5, HSPCs maintain high viability and show no evidence of disruption of normal hematopoietic differentiation in vitro. When PKC-activated HSPC were used to transplant immunodeficient NSG mice, no reduction of engraftment was observed. However, when we combined PKC activation and adenoviral transduction, we noted a reduction in the level of human cell engraftment that correlated with the MOI of the adenoviral vector used but observed no changes in the distribution of B, T, and myeloid cells in the spleen and BMs of engrafted mice. Thus, inhibition of engraftment was associated with viral transduction and not PKC activation alone. HIV-1 challenge of mice successfully reconstituted with CCR5-ZFN–treated HSPC resulted in enrichment in the level of CCR5 disruption in both CD4+ T-cells and CD8+ T-cells. We postulate that the increase in gene-modified CD8 cells (not normally a target of HIV-1 infection) is either due to viral (HIV-1) depletion of CD4+/CD8+ immature T-cells during T-lymphopoiesis or is an indirect outcome of HIV-1 bystander effect.27,28 Irrespective of the mechanism, there is a resultant enrichment in both mature subsets consistent with our previous findings using neonatal HSPC.12 Thus, the data presented here support the concept that gene-modified HSPC will produce modified progeny in vivo that are resistant to HIV-1 infection.
The methods developed here are intended to support eventual clinical trials on CCR5-ZFN–modified stem cell gene therapy for HIV-1 and may have important implications for other applications of ZFN or stem cell technology. It is unknown whether the reduction observed in the NSG mice transplanted with PMA CCR5-ZFN–treated cells is a quantitative predictor of what would happen in the context of an autologous human transplant. Although we have found that the reduction in human cell engraftment is correctable by the use of an increase in cell dosage, we are also currently exploring alternative approaches to delivering ZFN including DNA or RNA electroporation and non-integrating lentiviral vectors. These studies will permit the selection of the ideal delivery mode and CCR5 disruption level for clinical translation.
Materials and Methods
CD34+ cells. CD34+ HSPC were isolated from granulocyte-CSF–mobilized peripheral blood from healthy donors and purchased from vendors using previously described methods.13 Briefly, the washed and concentrated mobilized peripheral blood were labeled with CliniMACS CD34 microbeads (Miltenyi Biotec) and enriched with the CliniMACS Cell Separation System (Miltenyi Biotec). The purity of cells was usually >98%. Enriched CD34+ cells were either used freshly for transduction or cryopreserved using a controlled rate freezer and stored in the vapor phase of a liquid nitrogen dewar for later use.
Adenoviral vector. The Ad5/F35-pseudotyped CCR5-ZFN (CCR5-ZFN) and Ad5/F35-pseudotyped GFP (Ad-GFP) used in this study were provided by Sangamo BioSciences (Richmond, CA) and described previously (Perez et al.11 and Nilsson, M. et al.29). Briefly, CCR5-ZFN and Ad-GFP adenoviral vectors were generated on an E1/E3-deleted backbone. The pair of CCR5 ZFNs were linked via a 2A-peptide sequence and cloned into the pAdEasy-1/F35 vector under control of the CMV TetO promoter (24). The Ad5/F35 virus for each construct was generated using TREx 293T cells as described.29 The vector was tested negative for the presence of replication-competent adenovirus before use in the study.
Adenoviral transduction of HSPCs and in vitro culture. Freshly isolated or frozen CD34+ HSPC cells (1 × 106 cells/ml) were prestimulated in CellGro SCGM (CellGenix, Freiburg, Germany) supplemented with three cytokines (SFT): 100 ng/ml recombinant human SCF, 100 ng/ml recombinant human Flt-3L (Invitroge, Carlsbad, CA), and 10 ng/ml recombinant human TPO (CellGenix) 12–24 hours before transduction. After overnight prestimulation, cells were treated with PMA (Sigma-Aldrich, St Louis, MO) or bryostatin 1 (bryostatin) (Sigma-Aldrich) at indicated concentrations for 30 minutes and washed before transduction. Treated cells were suspended to a density of 1 × 106 cells/ml. Vectors were added to the cells at MOI of 0–5,000 for 12–24 hours and incubated at 37 °C in 5% CO2. For myeloid hematopoietic potential, cells were incubated in a bulk culture medium (Iscove's modified Dulbecco's medium, 20% fetal bovine serum, 1% L-glutamine, 100 ng/ml of SCF, 100 ng/ml Flt-3L, 10 ng/ml of TPO, 40 ng/ml of IL-6, 20 ng/ml of IL-3, 3 IU/ml erythropoietin, and 100 ng/ml granulocyte macrophage-CSF) for up to 4 weeks.13 Samples were collected at the time points indicated for cell counts using Guava PCA-96 (EMD Millipore, Danvers, MA) and performance of other assays as listed below.
Colony-forming unit assay. Mock-transduced and PMA-treated, CCR5-ZFN–transduced CD34+ HSPCs were harvested 16–24 hours after transduction. A total of 500 cells were plated in triplicate in MethoCult H4435-enriched methylcellulose medium according to the manufacturer's instruction (Stem Cell Technologies, Vancouver, British Columbia, Canada); 12–14 days after incubation, total colonies were enumerated under inverted microscope.
Cell viability-apoptosis analysis by annexin V/7-AAD staining. Aliquots of control and transduced cells were stained with Annexin V FITC Apoptosis Detection Kit (BD Pharmingen, San Jose, CA) in Annexin-binding buffer (BD Pharmingen) as instructed by the manufacturer and phenotypic data was collected on an Cytomics FC500 flow cytometer (Beckman Coulter, Brea, CA) and analyzed with the FCS Express Version 3 software (DeNovo Software, Los Angeles, CA). Viable cells are the population that is negative for both annexin V and 7-AAD staining and are compared with the unstained control.
Surveyor nuclease assay. The percentage of CCR5 alleles disrupted by ZFN treatment was measured by surveyor nuclease assay. Briefly, genomic DNA was extracted using the MasterPure Complete Purification kit (Epicentre, Madison, WI) according to the manufacturer's instructions. The purified genomic DNA was used as a template to amplify a fragment of the CCR5 gene using the primers C5_Cel_160_F1: AAGATGGATTATCAAGTGTCAAGTCC; and C5_Cel_160_R1: CAAAGTCCCACTGGGCG in the presence of a 32P-dATP and dCTP. The PCR products were then heated, allowed to re-anneal followed by treatment with the mismatch-sensitive surveyor nuclease (Transgenomic, Omaha, NE) as described in order to detect insertions and deletions caused by NHEJ. The resulting radiolabeled products digested with the surveyor nuclease were resolved by polyacrylamide gel electrophoresis, and the ratio of cleaved to uncleaved products was calculated to give a measure of the frequency of gene disruption. The assay is sensitive enough to detect single-nucleotide changes.
Flow cytometric analysis. Aliquots of cells from in vitro culture were taken weekly for phenotypic analysis using antibodies to lineage-specific cell surface antigens. Antibodies CD33-PE, CD15-FITC, GlyA-PC5, (BD Biosciences, San Jose, CA), and CD14-APC-Alexa750 (Invitrogen, Carlsbad, CA) were used to identify for progenitors, granulocyte, erythyrocyte, and monocyte subpopulations, respectively. Phenotypic data were collected on a Gallios flow cytometer (Beckman Coulter) and analyzed with FCS Express Version 3 software (DeNovo Software). Lineage-specific subpopulations, CD14+, CD15+, CD14+/CD15+, and CD14−/CD15− cells from PMA-treated and CCR5-ZFN–transduced cells were sorted by fluorescence-activated cell sorting after 4 weeks culture in the bulk culture media that promotes myeloid-erythroid lineage differentiation.
Genotyping for single and biallele CCR5 knockout. Mock-transduced and CCR5-ZFN–transduced CD34+ HSPC cells were harvested 16–24 hours after transduction and plated in triplicate in MethoCult H4435-enriched methylcellulose medium as described above in colony-forming unit assay. Individual colonies were isolated and analyzed for single and biallele CCR5 knockout by first screening for unmodified colonies using either the surveyor nuclease assay or a second PCR-based assay capable of detecting mutations at the CCR5-ZFN cleavage site using primers designed to specifically amplify unmodified wild-type sequences but not sequences with deletions, as previously described.30 These assays identified 160 methylcellulose colonies for further evaluation. The CCR5 locus was PCR-amplified from these cells, cloned into a bacterial plasmid, and then genotyped by directly sequencing the plasmid inserts.
Off-target site analysis. CD34+ cells were prestimulated in SCGM medium with SCF, Flt-3L, and TPO at 37 °C in 5% CO2 overnight. On the next day, cells were treated with 1 ng/ml PMA for 30 minutes. Cells were washed and transduced with Ad5/35 CCR5-ZFN at MOI of 10, and then cultured for 7 days before harvesting and isolating genomic DNA for initial PCR for the target loci followed by illumina deep sequencing. Briefly, initial PCR products were purified, treated with Klenow fragment (3′-5′ exo-; New England BioLabs, Ipswich, MA), ligated to adaptors, gel-purified, and then PCR-amplified (14 cycles) with illumina primers. Deep sequencing was performed on the top 23 off-target sites. The original top 15 potential off-target sites identified by a Systematic Evolution of Ligands by Exponential Enrichment (SELEX) protocol-guided analysis of the human genome; and eight additional sites obtained by an unbiased approach to off-target sites identification via mapping the locations of integrase-defective lentiviral integration in CCR5-ZFN–treated cells by linear amplification-mediated PCR.17
Phosphoprotein analysis. The CD34+ cells were stained with phosphoprotein-specific monoclonal antibody as previously described.31 Briefly, 1 × 106 CD34+ cells cultured in SCGM medium with three cytokines at 37 °C in 5% CO2 were harvested after 15 minutes of treatment with either PMA or bryostatin or mock-treated in a suitable amount of dimethyl sulfoxide containing medium. Cells were fixed with an equal volume of BD PhosFlow Fix Buffer I (BD Biosciences) for 10 minutes at 37 °C to arrest signaling activity and washed with staining buffer (phosphate-buffered saline supplemented with 0.5% bovine serum albumin) followed by permeabilization in 90% cold methanol at 4 °C for 30 minutes, washed twice with staining buffer and split evenly for parallel staining with anti-STAT3 (pY705)-PE, anti-ERK1/2 (pT202/pY204)-PE, anti-STAT5 (pY694)-PE, anti-β-catenin (pS45)-PE, anti-Akt (pS473)–Alexa Fluor 647, anti-p38 (pT180/pY182)-PE, anti-NF-κBp65(pS529)-PE, and isotype controls (BD Biosciences) for 30 minutes at room temperature. After incubation with antibody, cells were washed with staining buffer, pelleted, and resuspended in phosphate-buffered saline buffer containing 0.1% bovine serum albumin for flow cytometric analysis. At least 50,000 events from each sample were collected using Cytomics FC500 flow cytometer (Beckman Coulter). Cells were gated on live population (clearly demarcated in forward/side scatter plots) for analysis of geometric mean of fluorescence intensity using FCS Express 3.0 software (Denovo Software).
RNA preparation and quantitative reverse transcription-PCR. The effect of PMA on the expression of ZFN (Fok1) mRNA was examined using reverse transcription-PCR. CD34+ cells were seeded in six-well plates and supplemented with SCGM containing SCF, TPO, and Flt-3L as described above. One day after seeding, cells were stimulated with or without PMA (1 ng/ml) for 30 minutes. Then cells were washed, transduced with CCR5-ZFN (MOI 50), and cultured in SCGM with three cytokines for an additional 3 hours. Total RNA was extracted from untreated control; CCR5-ZFN–transduced; and PMA-treated, CCR5-ZFN–transduced CD34+ cells, with RNeasy Mini kit (Qiagen, Valencia, CA) according to the manufacturer's manual. Approximately 1 μg RNA was used for cDNA synthesis using the RT2 First strand kit (Qiagen) following the manufacturer's instruction. The following primers were used for amplification; Fok1 forward 5′-CCTGACGGCGCCATCTAT-3′ and reverse 5′-CGATCACGCCGTAATCGAT-3′, GAPDH forward 5′-GAAGGTGAAGGTCGGAGTC-3′ and reverse 5′-GAAGATGGTGATGGGATTTC-3′. Quantitative PCR measurements were performed on five biological replicates, PCR-amplified as three technical replicates and plotted as fold increase of ZFN-Fok1 RNA expression. Changes in the cycle threshold (ΔCT) values for Fok1 gene expression were obtained by subtracting the mean threshold cycle (CT) of the housekeeping gene (GAPDH) from the threshold cycle value of the Fok1 gene. Changes in the ΔCT values for Fok1 gene were obtained by subtracting the mean CT of the housekeeping gene (GAPDH) from the CT value of the Fok1 gene. The ΔΔCT value for five experiments was calculated using formula ΔΔCT = ΔCT (treatment) − ΔCT (untransduced control). The fold upregulation of Fok1 transcription was calculated as follows: 2(−ΔΔCT).
Western blot analysis. CD34+ cells (1 × 106 cells/ml) were treated with bryostatin or PMA in presence or absence of GSK1120212 inhibitor (Selleck Chemicals, Houston, TX) for 30 minutes followed by transduction of CCR5-ZFN at MOI of 50 overnight (12–24 hours). Total cell lysates were prepared in a RIPA Lysis Buffer system (Santa Cruz Biotechnology, Santa Cruz, CA). The protein content of the samples was measured using the BCA protein assay reagent (Thermo Scientific, Pierce Protein Research Products, Rockford, IL). A total of 20 or 50 μg protein samples were then subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis using 10% Tris-HCl gels and blot transferred onto a polyvinylidene difluotide membrane, immunoblotted with anti-Fok1 antibody (Sangamo BioSciences) anti-ERK1/2, phospho-ERK1/2, IkBa, phospho-IkBa (Cell Signaling Technology, Danvers, MA) or β-actin (Sigma-Aldrich) antibodies. The bands were detected using the SuperSignal West Pico chemiluminescent detection system (Thermo Scientific, Pierce Protein Research Products).
NSG mouse transplantation and engraftment analysis. NOD.Cg-Prkdc scid Il2rg tm1Wj/SzJ (NSG) mice were originally obtained from Jackson Laboratories (Bar Harbor, ME). Adult mice within 7–10 weeks of age received 250 cGy radiation, then either immediately or 2–4 hours later mice were retro-orbitally injected with 1 × 106 or 2 × 106 CCR-ZFN–modified or mock-treated human CD34+ HSPCs in 50 μl phosphate-buffered saline containing 1% heparin. Peripheral blood samples were collected every month starting at 4 weeks postengraftment using retro-orbital sampling. Whole blood was blocked in fetal bovine serum (Mediatech, Manassas, VA) for 30 minutes. The RBCs were lysed using Pharmlyse solution (BD Biosciences), and cells were washed with phosphate-buffered saline. Mice were necropsied between 8 and 28 weeks post-transplantation and tissues (spleen, BM) were collected and processed either immediately for cell isolation and fluorescence-activated cell sorting analysis as previously described by Holt and colleagues12 or spun down and frozen for later DNA extraction and surveyor nuclease analysis.
Fluorescence-activated cell sorting analysis of human cells was performed using a FACS Cyan instrument (BD Biosciences) with FlowJo software version 7.6.5 for PC (TreeStar). The initial gating strategy performed was based on forward scatter versus side scatter gate to exclude debris, followed by a human CD45 gate. Specific lineages (B cell, CD4+ T cell, CD8+ T cell, and monocytes) were determined using specific antibodies, with monocytes defined as CD4dim. All antibodies (BD Biosciences) were fluorochrome-conjugated and human-specific. The clones used in the studies are: CD45PerCP Cy5.5, clone 2D1; CD19APC, clone HIB19; CD4FITC, clone SK3; CD8PE, clone HIT8a.
HIV-1 challenge and CCR5 disruption analysis of NSG mice. NSG mice were transplanted/engrafted with either untreated or PMA/CCR5-ZFN–modified HSPCs as described in the text. At 26 weeks of transplant, half of the cohort was necropsied and analyzed for % CD45 and CCR5 disruption in the BM and spleen. Of the remaining animals, five mice were infected with a cell-free virus stock of HIV-1BaL that was propagated and administered as previously described.12 Eleven weeks post-infection, tissue samples were collected at necropsy and processed immediately for cell isolation and fluorescence-activated cell sorting analysis, or kept in freezing media for later analysis and DNA extraction. CCR5 disruption were analyzed using the Surveyor nuclease assay on bulk unsorted spleen cells and CD4+, CD8+, and CD19+ cells.
SUPPLEMENTARY MATERIAL Figure S1. CCR5-ZFN transduction resulted in low level CCR5 disruption in HSPC CD34+ cells under standard conditions. Figure S2. CD46 is expressed on HSPCs permitting transduction with an Ad5/F35-based adenovirus vector expressing GFP. Figure S3. PMA, bryostatin dose response, and adenoviral vector MOI on CCR5 disruption and viability of HSPC CD34+ cells. Figure S4. Viability of PMA-treated HSPC following Ad5/F35 CCR5-ZFN or Ad5/F35 hIL2Rg10 transduction. Figure S5. CCR5-ZFN–induced CCR5 disruption is sustained and present in multiple lineages. Figure S6. Multiple lineage engraftment of PKC activator-treated and CCR5-ZFN–transduced HSPCs in the spleen and bone marrow of transplanted NSG mice. Figure S7. CCR5 disruption was present and enhanced in multiple lineages in long-term transplanted, HIV-1–challenged NSG mice. Table S1. Off-site target analysis of PMA-treated and Ad/35ZFN-transduced cells. Table S2. List of genes in which off-target cleavage may occur.
Acknowledgments
This work was performed in Duarte, CA; Richmond, CA; and Los Angeles, CA. The research was supported by a grant to D.L.D., P.M.C., and J.A.Z. from the California Institute for Regenerative Medicine (CIRM) (grant no. DR1-01490). The contents of this publication are solely the responsibility of the authors and do not necessarily represent the official views of CIRM or any other agency of the State of California. U.H. was supported by a fellowship from the Swiss National Science Foundation, PBZHP3-133452. We thank David Ann (City of Hope) for critical reading of the manuscript. J.W., K.K., P.-Q.L., M.C.H., and P.D.G. are full time employees of Sangamo BioSciences, Inc. The others authors declared no conflict of interest.
Supplementary Material
References
- Prejean J, Song R, Hernandez A, Ziebell R, Green T, Walker F, HIV Incidence Surveillance Group et al. Estimated HIV incidence in the United States, 2006-2009. PLoS ONE. 2011;6:e17502. doi: 10.1371/journal.pone.0017502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hütter G, Nowak D, Mossner M, Ganepola S, Müssig A, Allers K, et al. Long-term control of HIV by CCR5 Delta32/Delta32 stem-cell transplantation. N Engl J Med. 2009;360:692–698. doi: 10.1056/NEJMoa0802905. [DOI] [PubMed] [Google Scholar]
- Allers K, Hütter G, Hofmann J, Loddenkemper C, Rieger K, Thiel E, et al. Evidence for the cure of HIV infection by CCR5?32/?32 stem cell transplantation. Blood. 2011;117:2791–2799. doi: 10.1182/blood-2010-09-309591. [DOI] [PubMed] [Google Scholar]
- Baltimore D. Gene therapy. Intracellular immunization. Nature. 1988;335:395–396. doi: 10.1038/335395a0. [DOI] [PubMed] [Google Scholar]
- Hoxie JA, June CH. Novel cell and gene therapies for HIV. Cold Spring Harb Perspect Med. 2012;2:pii: a007179. doi: 10.1101/cshperspect.a007179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podsakoff GM, Engel BC, Carbonaro DA, Choi C, Smogorzewska EM, Bauer G, et al. Selective survival of peripheral blood lymphocytes in children with HIV-1 following delivery of an anti-HIV gene to bone marrow CD34(+) cells. Mol Ther. 2005;12:77–86. doi: 10.1016/j.ymthe.2005.02.024. [DOI] [PubMed] [Google Scholar]
- Levine BL, Humeau LM, Boyer J, MacGregor RR, Rebello T, Lu X, et al. Gene transfer in humans using a conditionally replicating lentiviral vector. Proc Natl Acad Sci USA. 2006;103:17372–17377. doi: 10.1073/pnas.0608138103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsuyasu RT, Merigan TC, Carr A, Zack JA, Winters MA, Workman C, et al. Phase 2 gene therapy trial of an anti-HIV ribozyme in autologous CD34+ cells. Nat Med. 2009;15:285–292. doi: 10.1038/nm.1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnan A, Molina A, Zaia J, Smith D, Vasquez D, Kogut N, et al. Durable remissions with autologous stem cell transplantation for high-risk HIV-associated lymphomas. Blood. 2005;105:874–878. doi: 10.1182/blood-2004-04-1532. [DOI] [PubMed] [Google Scholar]
- Santiago Y, Chan E, Liu PQ, Orlando S, Zhang L, Urnov FD, et al. Targeted gene knockout in mammalian cells by using engineered zinc-finger nucleases. Proc Natl Acad Sci USA. 2008;105:5809–5814. doi: 10.1073/pnas.0800940105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez EE, Wang J, Miller JC, Jouvenot Y, Kim KA, Liu O, et al. Establishment of HIV-1 resistance in CD4+ T cells by genome editing using zinc-finger nucleases. Nat Biotechnol. 2008;26:808–816. doi: 10.1038/nbt1410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt N, Wang J, Kim K, Friedman G, Wang X, Taupin V, et al. Human hematopoietic stem/progenitor cells modified by zinc-finger nucleases targeted to CCR5 control HIV-1 in vivo. Nat Biotechnol. 2010;28:839–847. doi: 10.1038/nbt.1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran CA, Torres-Coronado M, Gardner A, Gu A, Vu H, Rao A, et al. Optimized processing of growth factor mobilized peripheral blood CD34+ products by counterflow centrifugal elutriation. Stem Cells Transl Med . 2012;1 (5:422–429. doi: 10.5966/sctm.2011-0062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knaän-Shanzer S, Van Der Velde I, Havenga MJ, Lemckert AA, De Vries AA, Valerio D. Highly efficient targeted transduction of undifferentiated human hematopoietic cells by adenoviral vectors displaying fiber knobs of subgroup B. Hum Gene Ther. 2001;12:1989–2005. doi: 10.1089/104303401753204562. [DOI] [PubMed] [Google Scholar]
- Nilsson M, Karlsson S, Fan X. Functionally distinct subpopulations of cord blood CD34+ cells are transduced by adenoviral vectors with serotype 5 or 35 tropism. Mol Ther. 2004;9:377–388. doi: 10.1016/j.ymthe.2003.12.014. [DOI] [PubMed] [Google Scholar]
- Guschin DY, Waite AJ, Katibah GE, Miller JC, Holmes MC, Rebar EJ. A rapid and general assay for monitoring endogenous gene modification. Methods Mol Biol. 2010;649:247–256. doi: 10.1007/978-1-60761-753-2_15. [DOI] [PubMed] [Google Scholar]
- Gabriel R, Lombardo A, Arens A, Miller JC, Genovese P, Kaeppel C, et al. An unbiased genome-wide analysis of zinc-finger nuclease specificity. Nat Biotechnol. 2011;29:816–823. doi: 10.1038/nbt.1948. [DOI] [PubMed] [Google Scholar]
- Shultz LD, Ishikawa F, Greiner DL. Humanized mice in translational biomedical research. Nat Rev Immunol. 2007;7:118–130. doi: 10.1038/nri2017. [DOI] [PubMed] [Google Scholar]
- Ishikawa F, Yasukawa M, Lyons B, Yoshida S, Miyamoto T, Yoshimoto G, et al. Development of functional human blood and immune systems in NOD/SCID/IL2 receptor {gamma} chain(null) mice. Blood. 2005;106:1565–1573. doi: 10.1182/blood-2005-02-0516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redig AJ, Platanias LC. The protein kinase C (PKC) family of proteins in cytokine signaling in hematopoiesis. J Interferon Cytokine Res. 2007;27:623–636. doi: 10.1089/jir.2007.0007. [DOI] [PubMed] [Google Scholar]
- Lebkowski JS, McNally MA, Okarma TB, Lerch LB. Inducible gene expression from multiple promoters by the tumor-promoting agent, PMA. Nucleic Acids Res. 1987;15:9043–9055. doi: 10.1093/nar/15.21.9043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isern E, Gustems M, Messerle M, Borst E, Ghazal P, Angulo A. The activator protein 1 binding motifs within the human cytomegalovirus major immediate-early enhancer are functionally redundant and act in a cooperative manner with the NF-{kappa}B sites during acute infection. J Virol. 2011;85:1732–1746. doi: 10.1128/JVI.01713-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegedus C, Lakatos P, Oláh G, Tóth BI, Gergely S, Szabó E, et al. Protein kinase C protects from DNA damage-induced necrotic cell death by inhibiting poly(ADP-ribose) polymerase-1. FEBS Lett. 2008;582:1672–1678. doi: 10.1016/j.febslet.2008.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaheen M, Allen C, Nickoloff JA, Hromas R. Synthetic lethality: exploiting the addiction of cancer to DNA repair. Blood. 2011;117:6074–6082. doi: 10.1182/blood-2011-01-313734. [DOI] [PubMed] [Google Scholar]
- Myklebust JH, Smeland EB, Josefsen D, Sioud M. Protein kinase C-alpha isoform is involved in erythropoietin-induced erythroid differentiation of CD34(+) progenitor cells from human bone marrow. Blood. 2000;95:510–518. [PubMed] [Google Scholar]
- Davis TA, Saini AA, Blair PJ, Levine BL, Craighead N, Harlan DM, et al. Phorbol esters induce differentiation of human CD34+ hemopoietic progenitors to dendritic cells: evidence for protein kinase C-mediated signaling. J Immunol. 1998;160:3689–3697. [PubMed] [Google Scholar]
- Moon HS, Yang JS. Role of HIV Vpr as a regulator of apoptosis and an effector on bystander cells. Mol Cells. 2006;21:7–20. [PubMed] [Google Scholar]
- Muthumani K, Choo AY, Hwang DS, Premkumar A, Dayes NS, Harris C, et al. HIV-1 Nef-induced FasL induction and bystander killing requires p38 MAPK activation. Blood. 2005;106:2059–2068. doi: 10.1182/blood-2005-03-0932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson M, Ljungberg J, Richter J, Kiefer T, Magnusson M, Lieber A, et al. Development of an adenoviral vector system with adenovirus serotype 35 tropism; efficient transient gene transfer into primary malignant hematopoietic cells. J Gene Med. 2004;6:631–641. doi: 10.1002/jgm.543. [DOI] [PubMed] [Google Scholar]
- Liu PQ, Chan EM, Cost GJ, Zhang L, Wang J, Miller JC, et al. Generation of a triple-gene knockout mammalian cell line using engineered zinc-finger nucleases. Biotechnol Bioeng. 2010;106:97–105. doi: 10.1002/bit.22654. [DOI] [PubMed] [Google Scholar]
- Perez OD, Nolan GP. Simultaneous measurement of multiple active kinase states using polychromatic flow cytometry. Nat Biotechnol. 2002;20:155–162. doi: 10.1038/nbt0202-155. [DOI] [PubMed] [Google Scholar]
- Zielske SP, Gerson SL. Cytokines, including stem cell factor alone, enhance lentiviral transduction in nondividing human LTCIC and NOD/SCID repopulating cells. Mol Ther. 2003;7:325–333. doi: 10.1016/s1525-0016(03)00005-4. [DOI] [PubMed] [Google Scholar]
- Himburg HA, Muramoto GG, Daher P, Meadows SK, Russell JL, Doan P, et al. Pleiotrophin regulates the expansion and regeneration of hematopoietic stem cells. Nat Med. 2010;16:475–482. doi: 10.1038/nm.2119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikelis C, Koutsioumpa M, Papadimitriou E. Pleiotrophin as a possible new target for angiogenesis-related diseases and cancer. Recent Pat Anticancer Drug Discov. 2007;2:175–186. doi: 10.2174/157489207780832405. [DOI] [PubMed] [Google Scholar]
- Essers MA, Offner S, Blanco-Bose WE, Waibler Z, Kalinke U, Duchosal MA, et al. IFNalpha activates dormant haematopoietic stem cells in vivo. Nature. 2009;458:904–908. doi: 10.1038/nature07815. [DOI] [PubMed] [Google Scholar]
- Passegué E, Ernst P. IFN-alpha wakes up sleeping hematopoietic stem cells. Nat Med. 2009;15:612–613. doi: 10.1038/nm0609-612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friebe A, Volk HD. Stability of tumor necrosis factor alpha, interleukin 6, and interleukin 8 in blood samples of patients with systemic immune activation. Arch Pathol Lab Med. 2008;132:1802–1806. doi: 10.5858/132.11.1802. [DOI] [PubMed] [Google Scholar]
- Gschwend JE, Fair WR, Powell CT. Bryostatin 1 induces prolonged activation of extracellular regulated protein kinases in and apoptosis of LNCaP human prostate cancer cells overexpressing protein kinase calpha. Mol Pharmacol. 2000;57:1224–1234. [PubMed] [Google Scholar]
- May WS, Sharkis SJ, Esa AH, Gebbia V, Kraft AS, Pettit GR, et al. Antineoplastic bryostatins are multipotential stimulators of human hematopoietic progenitor cells. Proc Natl Acad Sci USA. 1987;84:8483–8487. doi: 10.1073/pnas.84.23.8483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zen K, Biwersi J, Periasamy N, Verkman AS. Second messengers regulate endosomal acidification in Swiss 3T3 fibroblasts. J Cell Biol. 1992;119:99–110. doi: 10.1083/jcb.119.1.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brehm A, Miska EA, McCance DJ, Reid JL, Bannister AJ, Kouzarides T. Retinoblastoma protein recruits histone deacetylase to repress transcription. Nature. 1998;391:597–601. doi: 10.1038/35404. [DOI] [PubMed] [Google Scholar]
- Bartels M, Geest CR, Bierings M, Buitenhuis M, Coffer P. JHistone deacetylase inhibition modulates cell fate decisions during myeloid differentiation. Haematologica. 2010;95:1052–1060. doi: 10.3324/haematol.2009.008870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai S, Maurin M, Smith MA, Bolick SC, Dessureault S, Tao J, et al. PRDM1 is required for mantle cell lymphoma response to bortezomib. Mol Cancer Res. 2010;8:907–918. doi: 10.1158/1541-7786.MCR-10-0131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavelin I, Beer A, Kam Z, Rotter V, Oren M, Navon A, et al. Discovery of novel proteasome inhibitors using a high-content cell-based screening system. PLoS ONE. 2009;4:e8503. doi: 10.1371/journal.pone.0008503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiraishi T, Nielsen PE. Enhanced delivery of cell-penetrating peptide-peptide nucleic acid conjugates by endosomal disruption. Nat Protoc. 2006;1:633–636. doi: 10.1038/nprot.2006.92. [DOI] [PubMed] [Google Scholar]
- Fredericksen BL, Wei BL, Yao J, Luo T, Garcia JV. Inhibition of endosomal/lysosomal degradation increases the infectivity of human immunodeficiency virus. J Virol. 2002;76:11440–11446. doi: 10.1128/JVI.76.22.11440-11446.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaghen M, Bouhallaoui A, Taleb H, Idrissi H, Tagmouti F, Talbi M, et al. Okadaic acid and its interaction with sodium, potassium, magnesium and calcium ions: complex formation and transport across a liquid membrane. Toxicon. 1997;35:843–847. doi: 10.1016/s0041-0101(96)00199-7. [DOI] [PubMed] [Google Scholar]
- Namboodiripad AN, Jennings ML. Permeability characteristics of erythrocyte membrane to okadaic acid and calyculin A. Am J Physiol. 1996;270 2 Pt 1:C449–C456. doi: 10.1152/ajpcell.1996.270.2.C449. [DOI] [PubMed] [Google Scholar]
- Pruett-Miller SM, Reading DW, Porter SN, Porteus MH. Attenuation of zinc finger nuclease toxicity by small-molecule regulation of protein levels. PLoS Genet. 2009;5:e1000376. doi: 10.1371/journal.pgen.1000376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shayakhmetov DM, Li ZY, Ternovoi V, Gaggar A, Gharwan H, Lieber A. The interaction between the fiber knob domain and the cellular attachment receptor determines the intracellular trafficking route of adenoviruses. J Virol. 2003;77:3712–3723. doi: 10.1128/JVI.77.6.3712-3723.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.