

Abstract
Filamentous inclusions made of hyperphosphorylated tau are characteristic of numerous human neurodegenerative diseases, including Alzheimer’s disease, tangle-only dementia, Pick disease, argyrophilic grain disease (AGD), progressive supranuclear palsy, and corticobasal degeneration. In Alzheimer’s disease and AGD, it has been shown that filamentous tau appears to spread in a stereotypic manner as the disease progresses. We previously demonstrated that the injection of brain extracts from human mutant P301S tau-expressing transgenic mice into the brains of mice transgenic for wild-type human tau (line ALZ17) resulted in the assembly of wild-type human tau into filaments and the spreading of tau inclusions from the injection sites to anatomically connected brain regions. Here we injected brain extracts from humans who had died with various tauopathies into the hippocampus and cerebral cortex of ALZ17 mice. Argyrophilic tau inclusions formed in all cases and following the injection of the corresponding brain extracts, we recapitulated the hallmark lesions of AGD, PSP and CBD. Similar inclusions also formed after intracerebral injection of brain homogenates from human tauopathies into nontransgenic mice. Moreover, the induced formation of tau aggregates could be propagated between mouse brains. These findings suggest that once tau aggregates have formed in discrete brain areas, they become self-propagating and spread in a prion-like manner.
Soluble microtubule-associated protein tau assembles into insoluble, filamentous, and hyperphosphorylated intracellular inclusions in a variety of human neurodegenerative diseases, including Alzheimer’s disease (AD), tangle-only dementia (TD), Pick disease (PiD), argyrophilic grain disease (AGD), progressive supranuclear palsy (PSP), and corticobasal degeneration (CBD) (1). In adult human brain, six tau isoforms are expressed from a single microtubule associated protein tau (MAPT) gene through alternative mRNA splicing (2). It gives rise to three tau isoforms with three repeats each and three tau isoforms with four repeats each. The repeats are 31 or 32 amino acids in length and are located toward the carboxy terminus of tau. Adult mice express predominantly tau isoforms with four repeats (3). The repeats form the core of the tau filaments whose isoform composition can vary between diseases. Thus, in AD and TD, both three- and four-repeat tau make up the neurofibrillary lesions (4–6), whereas in PiD three-repeat tau predominates in the neuronal inclusions (7). The assembly of four-repeat tau into filaments is characteristic of PSP, CBD, and AGD (8–11).
Mutations in MAPT cause familial forms of frontotemporal dementia, establishing that tau protein dysfunction is sufficient to cause neurodegeneration and dementia (12–14). These mutations cause the development of inclusions made of hyperphosphorylated filamentous tau; depending on the mutations, the inclusions are made of all six tau isoforms, three-repeat tau or four-repeat tau. At an experimental level, these findings have led to the development of transgenic mouse models that recapitulate the essential molecular and cellular features of human tauopathies. Thus, transgenic mice expressing human mutant P301S tau show neurodegeneration and abundant filaments made of hyperphosphorylated tau protein (15, 16). By contrast, mouse lines expressing single isoforms of wild-type human tau do not produce tau filaments or develop neurodegeneration (17–19). We previously showed that the injection of brain extracts from human mutant P301S tau-expressing mice into the brains of mice transgenic for the longest human four-repeat tau isoform (ALZ17 line) induced the assembly of wild-type human tau into silver-positive inclusions and the spread of pathology from the sites of injection to neighboring brain regions (20). In conjunction with other findings (21–28), this work is consistent with the intercellular transfer of tau aggregates. Filaments from human tauopathy brains exhibit a range of morphologies (29) and distinct conformers of aggregated tau may cause distinct tauopathies, based on the involvement of specific brain regions and cell types.
Here we have injected brain extracts from humans with pathologically confirmed AD, TD, PiD, AGD, PSP, and CBD into the hippocampus and cerebral cortex of ALZ17 mice. Silver-positive inclusions formed in all cases, with the presence of pathological structures reminiscent of AGD, PSP, and CBD following the injection of the corresponding human brain extracts. Silver-positive inclusions also formed after the intracerebral injection of brain homogenates from human tauopathies into wild-type mice. Moreover, the induced formation of tau inclusions could be propagated between mouse brains. Aggregation and spreading of human tau in the ALZ17 brains were not observed following the injection of Aβ-containing extracts or the crossing of ALZ17 with human mutant amyloid precursor protein transgenic mice (APP23 line).
Results
Tau Inclusion Formation in ALZ17 Transgenic Mice Following the Intracerebral Injection of Brain Extracts from Sporadic Human Tauopathies.
To investigate the prion-like properties of aggregated human tau, we stereotaxically injected total brain homogenates prepared from affected brain regions of neuropathologically confirmed cases of AD, TD, PiD, AGD, PSP, and CBD unilaterally into the hippocampus and overlying cerebral cortex of 3-mo-old ALZ17 mice. Injected mice were killed 6, 12, and 15 mo after injection. Gallyas-Braak silver staining and immunolabeling with antitau antibody AT100 were then used to detect the presence of tau filaments. Tau hyperphosphorylation was investigated with antibody AT8. In all injected ALZ17 mice, irrespective of human tauopathy, staining with Gallyas-Braak and immunolabeling with AT100 were observed. Tau inclusions were present in the hippocampus 6 mo after the injection of AD, TD, AGD, PSP, CBD, and PiD brain homogenates (Fig. 1) and the amount of aggregated tau progressed over time (Fig. S1 and Dataset S1). The smallest number of silver-positive structures was obtained when brain homogenates from cases of PiD were injected (Dataset S1). At the injection level, Gallyas-Braak and AT100 staining was observed in the fimbria, the optic tract, the medial lemniscus, the dorsal thalamus, the cerebral peduncle, and the amygdala (Fig. S2). Anterior to the injection level, scattered tau aggregates were present in the fimbria, the thalamus, and the internal capsule. Posterior to the injection level, aggregated tau was present in the cerebral peduncle and the entorhinal cortex (Fig. S2). Structures positive with AT8, AT100, and Gallyas-Braak were also observed in the fornix, a brain region that is connected to the hippocampus, but located far distant ventrally (Fig. S2). The propagation of the induced filamentous tau pathology was similar for all injected tauopathies, with the exception of PiD, where induced tau filaments remained confined to the injection sites. No staining with Gallyas-Braak or AT100 was observed 15 mo after the injection of a brain homogenate prepared from an age-matched control (Fig. S1). Electron microscopy of sarkosyl-insoluble material from the injected brains failed to show filaments, presumably because there were too few to detect. Future experiments will analyze injected mouse brain areas as a function of time using electron microscopy.
Fig. 1.
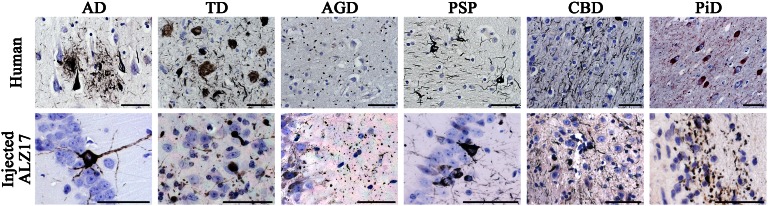
Gallyas-Braak silver-positive neuronal tau inclusions in the hippocampal region of ALZ17 mice 6 mo after the intracerebral injection of brain homogenates from sporadic human tauopathies. (Upper row) Pathological tau hallmark lesions observed in the human tissues used for brain extract preparation. From Left to Right: Gallyas-Braak silver impregnation visualized neurofibrillary tangles (NFTs) and neuropil threads (NTs), as well as dystrophic neurites in the vicinity of plaques in AD; NFTs and NTs in the absence of plaques in TD; argyrophilic grains in AGD; globose NFTs and NTs in PSP and small NFTs and abundant NTs in CBD. Pick bodies were observed in PiD using AT100 immunostaining. (Lower row) Filamentous tau lesions formed in the brain of ALZ17 mice after injection of the corresponding human brain extracts. From Left to Right: NFTs after the injection of AD and TD brain homogenates; argyrophilic grains after the injection of AGD brain homogenate; nerve cell body inclusions and NTs after the injection of PSP homogenate; small numbers of nerve cell body inclusions and numerous NTs after the injection of CBD homogenate; short, thick NTs after the injection of PiD homogenate. (Scale bars, 50 µm.) Sections were counterstained with hematoxylin.
Morphological Diversity of Induced Tau Inclusions.
Six months after the intracerebral injection of AD and TD human brain homogenates into ALZ17 mouse hippocampus and cerebral cortex, tau inclusions in the form of silver-positive staining of nerve cell bodies and neuropil threads were in evidence. AD and TD homogenates induced morphologically similar tau aggregates, which resembled the pathologies of the human tissues used for injection (Fig. 1). Aβ plaques did not form in ALZ17 mice following the injection of AD brain extract. The intracerebral injection of AGD extracts also induced the formation of silver-positive tau inclusions in ALZ17 mice. These inclusions consisted of small spherical or comma-shaped structures that closely resembled the argyrophilic grains of AGD (Fig. 1). At the injection sites, the inclusions were located predominantly in the dentate gyrus and in sectors CA1 and CA2 of the hippocampus. The argyrophilic grains were either loosely dispersed or arranged in rows, the latter apparently being located in nerve cell processes. In addition to argyrophilic grains, small numbers of neurofibrillary tangles were observed, similar to what is known to occur in AGD cases (Fig. S1). The intracerebral injection of brain homogenates from patients with PSP also induced the formation of silver-positive inclusions in ALZ17 mice. In neurons and their processes, tauopathy was present as silver-positive nerve cell body inclusions and neuropil threads (Fig. 1), similar to what was observed following the injection of AD and TD brain homogenates. The injection of CBD brain homogenates resulted in the induction of numerous neuropil threads and a smaller number of silver-positive nerve cell body inclusions (Fig. 1). The injection of PiD extract into the brain of ALZ17 mice resulted in the formation of short and thick neuropil threads that could be detected using Gallyas-Braak silver and antitau antibodies 12E8 and AT100 (Fig. 1). Typical Pick bodies (known to be negative for Gallyas-Braak silver and antibody 12E8) were not observed. In injected animals, immunohistochemistry with antiionized calcium-binding adapter molecule 1 (Iba1) antibody failed to detect neuroinflammation, similar to what was described previously following the injection of P301S mouse brain homogenates (20).
In addition to the induction of argyrophilic neuronal inclusions, all of the injected brain homogenates generated Gallyas-Braak silver-positive and AT100-immunoreactive tau inclusions in oligodendroglia in the form of coiled bodies. Similar oligodendroglial inclusions were previously observed following the intracerebral injection of brain extracts from symptomatic mice transgenic for human mutant P301S tau into ALZ17 mice (20). In addition, we observed astrocytic inclusions following the injection of PSP, CBD, and AGD brain homogenates; they closely resembled the inclusions characteristic of these human tauopathies, based on GFAP, AT8, and AT100 immunohistochemistry, and Gallyas-Braak silver impregnation (Fig. 2). Thus, ALZ17 mice injected with PSP brain homogenates showed astrocytic inclusions closely resembling tufted astrocytes, which exhibited Gallyas-Braak and/or AT100-positive lesions in proximal and distal astrocytic processes (Fig. 2). The intracerebral injection of CBD brain homogenates into ALZ17 mice led to the formation of inclusions closely resembling astrocytic plaques, with the distal tips of astrocytic processes being positive with Gallyas-Braak silver and/or antitau antibody AT100 (Fig. 2). The induced tufted astrocyte-like and astrocytic plaque-like lesions were double labeled by AT100 and anti-GFAP (Fig. 2). The intracerebral injection of AGD brain homogenates induced the formation of an AT100-positive, but Gallyas-Braak–negative, astrocytic tau pathology in ALZ17 mice (Fig. 2), closely resembling the astroglial pathology of human AGD. Noninjected ALZ17 mice were devoid of astrocytic tau inclusions; the same was true of ALZ17 mice injected with brain homogenates from AD, TD, or PiD patients.
Fig. 2.

Induction of astrocytic tau inclusions in ALZ17 mice 12 mo after the intracerebral injection of PSP, CBD, and AGD brain homogenates. (Top row) Gallyas-Braak silver impregnation reveals a tufted astrocyte in the PSP case used for injection (Left). Following the injection of PSP brain homogenate, double labeling with anti-GFAP (dark blue) and AT100 (red) revealed a tufted-like astrocytic tau inclusion (Center) in which aggregated tau was detected by Gallyas-Braak staining in proximal and distal processes (arrows) located around the nucleus (arrowheads) of the tufted-like astrocyte (Right). (Middle row) Gallyas-Braak silver impregnation revealed an astrocytic plaque in the CBD case used for injection (Left). Following the injection of CBD brain homogenate, double labeling with anti-GFAP (dark blue) and AT100 (red) revealed an astrocytic plaque-like lesion (Center) in which silver-positive material was only found in the distal processes (arrows) of the plaque-like lesion after Gallyas-Braak staining (Right). (Bottom row) AT100 immunolabeling shows a tufted-like astrocyte in the AGD case used for injection (Left). Following the injection of AGD brain homogenate, AT100 immunolabeling revealed a tau-positive tufted-like astrocyte (Center) that was negative by Gallyas-Braak silver (Right). (Scale bars, 50 µm.) Sections were counterstained with hematoxylin.
Tau Inclusion Formation in Wild-Type Mice Following the Intracerebral Injection of Brain Extracts from Sporadic Human Tauopathies.
To investigate if overexpression of human tau is required for the induction and spreading of tau aggregates, we next injected AD, TD, AGD, and PSP human brain homogenates into the hippocampus and overlying cerebral cortex of 3-mo-old C57BL/6 mice, which were then analyzed 6 and 15 mo postinjection. Gallyas-Braak silver-positive and AT8- and AT100-immunoreactive inclusions were in evidence (Fig. 3), although they were less numerous than after the intracerebral injection of human brain homogenates into ALZ17 mice. Silver-positive nerve cell bodies, neuropil threads, and oligodendroglial coiled bodies were present at the hippocampal injection site 6 mo after the injection. The amount of silver-positive material increased over time and extended to the optic tract, the subiculum, and the dorsal thalamus (Fig. 3). Astrocytic tau inclusions, positive with antitau antibodies AT8 and AT100, but not with Gallyas-Braak silver, also formed after the injection of AGD brain extract (Fig. 3). The tau-positive material induced in C57BL/6 mice was not immunoreactive with the human tau-specific antibody T14 but was revealed by antibody MT1 that detects mouse tau (Fig. 3), confirming the aggregation of murine tau.
Fig. 3.

Induction of tau inclusions in nontransgenic C57BL/6 mice 12 mo after the intracerebral injection of brain homogenates from sporadic human tauopathies. Gallyas-Braak silver impregnation revealed the presence of neuropil threads and coiled bodies in (A) the optic tract following the injection of TD homogenate, (B) the subiculum after the injection of PSP homogenate, and (C) the dorsal thalamus following the injection of AD homogenate. AT100 immunostaining of the hippocampal region showed the presence of (D) coiled bodies (arrows) and (E) a tufted-like astrocyte following the injection of AGD homogenate. (F) MT1 immunostaining detected mouse tau aggregates after the injection of AD brain homogenates. (Scale bars, 50 µm.) Sections were counterstained with hematoxylin.
Propagation of Tau Inclusion Formation by Serial Injection into Mouse Brains.
The serial propagation of induced filamentous tau pathology was investigated following the intracerebral injection into 3-mo-old ALZ17 mice of homogenates from ALZ17 mice that had received a bilateral injection of brain extract from human P301S tau transgenic mice 18 mo earlier. One half of the brain was used for the preparation of homogenates, with the other half serving for the verification of the presence of silver-positive tau inclusions (Fig. S3A). Twelve months after the injection, Gallyas-Braak silver staining and AT100 immunostaining revealed the presence of neuronal and oligodendroglial tau inclusions at the injection sites (Fig. 4 A and B). A second set of homogenates was prepared from the brains of C57BL/6 mice that had been injected bilaterally with TD or AGD brain homogenates 18 mo earlier (Fig. S3 B and C). Twelve months after the intracerebral injection into ALZ17 mice, many neuropil threads and some nerve cell body tau aggregates were present at the injection sites (Fig. 4 C and D).
Fig. 4.

Induction of tau inclusions following the intracerebral injection of induced mouse brain homogenates, as detected by Gallyas-Braak silver impregnation. (A) Neuropil threads and coiled bodies in the alveus and (B) neurofibrillary tangle in the subiculum of an ALZ17 mouse 12 mo after the injection of brain homogenate from an ALZ17 mouse that had 18 mo previously been injected with brain homogenate from a mouse transgenic for human mutant P301S tau. (C and D) ALZ17 mouse 12 mo after the injection of brain homogenate from a C57BL/6 mouse that had 18 mo previously been injected with TD brain homogenate. (Scale bars, 50 µm.) Sections were counterstained with hematoxylin.
Aβ Did Not Induce Tau Aggregation in ALZ17 Mice.
We investigated the presence of pathological Aβ in the human brains used for injection into ALZ17 mice. Pathological Aβ was absent in TD, PSP, CBD, PiD, and control extracts (Fig. S4). Abundant neuritic Aβ plaques were present in AD and moderate numbers of diffuse plaques were in evidence in AGD. Biochemical characterization showed the presence of both Aβ (1–40) and Aβ (1–42) in AD (Fig. S4A), with a predominance of Aβ (1–42) in AGD (Fig. S4C). To study the effect of Aβ deposition on the aggregation of wild-type human tau, we intracerebrally injected homogenates prepared from the neocortex of 24-mo-old APP23 mice into the hippocampus and neocortex of 3-mo-old ALZ17 mice. By Western blotting, both Aβ (1–40) and Aβ (1–42) were present (Fig. S4H). The intracerebral injection of the Aβ-rich APP23 brain extract failed to influence AT8-positive tau pathology in ALZ17 mice (Fig. 5 A and B). In particular, tau inclusions immunoreactive with antitau antibody AT100 or silver-positive using the Gallyas-Braak method were absent for up to 18 mo after the injection (Fig. 5C). We also failed to detect Aβ deposits following the injection of Aβ-rich extracts into ALZ17 mice (Fig. 5D). Neither tau inclusions nor Aβ deposits formed following the injection of brain homogenates from age-matched control mice.
Fig. 5.

Aβ did not induce tau aggregation in ALZ17 mice. The injection of APP23 brain homogenate into the hippocampal formation of ALZ17 mice had no effect on the pretangle tau staining with antibody AT8 at 15 mo after the injection (A), compared with a noninjected ALZ17 mouse (B). (C) Gallyas-Braak silver impregnation failed to detect tau inclusions in the hippocampal formation of an ALZ17 mouse 15 mo after the injection of APP23 brain homogenate. (D) Staining for Aβ with serum NT11 of a tissue section adjacent to that used in C failed to reveal the presence of Aβ deposits. (Scale bar, 100 µm.) Sections were counterstained with hematoxylin.
Next we crossbred APP23 with ALZ17 tau transgenic mice. This bigenic line developed a combination of the changes characteristic of each transgenic line, namely Aβ deposits with associated glial and reactive neuronal alterations and accumulation of hyperphosphorylated tau in the somatodendritic compartment (Fig. S5 A and B). Aβ and tau abnormalities coexisted, but did not appear to influence each other. In particular, AT100- and/or Gallyas-Braak–positive inclusions were not present, even in 2-y-old APP23 × ALZ17 mice (Fig. S5 C and D).
Discussion
The present findings show that brain homogenates from a number of human tauopathies induced the formation of tau inclusions, both in ALZ17 mice transgenic for wild-type human tau and in nontransgenic control mice.
In ALZ17 mice, neuronal and oligodendroglial inclusions formed following the intracerebral injection of brain homogenates from all cases of AD, TD, PiD, AGD, PSP, and CBD. The ALZ17 line expresses a single isoform of wild-type four-repeat human tau (19). With the exception of PiD, where the tau inclusions consist predominantly of three-repeat tau (7), the inclusions of the other tauopathies are made of four-repeat tau (AGD, PSP, and CBD) (8–11) or of a mixture of three-repeat and four-repeat tau (AD and TD) (4, 5). From the work on other neurodegenerative diseases, especially prionoses, it is known that the amount of misfolded protein formed is proportional to the sequence similarity between the seed and the soluble protein recruited by the seed (30). The same appears to be true of tau, because the number of inclusions induced following the intracerebral injection of PiD disease homogenates into ALZ17 mouse brains was smaller than that present following the injection of AGD, PSP, and CBD brain homogenates. However, we cannot exclude that the aggregation-inducing activity in the PiD homogenates consisted of a small amount of aggregated four-repeat tau, because PiD is only rarely a pure three-repeat tauopathy (31). This was supported by the presence of 12E8 staining following the intracerebral injection of PiD brain extract. The aggregates characteristic of AGD, PSP, and CBD are made of four-repeat tau, as is the human tau isoform the ALZ17 line is transgenic for. These findings agree with in vitro studies that have shown that seeds made of either three-repeat, three/four-repeat, or four-repeat tau can recruit soluble four-repeat tau (32).
From the study of prion diseases, it is also known that different aggregate conformations can give rise to distinct disease phenotypes and neuropathologies (30). We show here a similar phenomenon for four-repeat tauopathies. Thus, the intracerebral injection of PSP brain homogenates into ALZ17 mice resulted in the formation of silver-positive astrocytic aggregates that resembled tufted astrocytes (the hallmark lesions of PSP) (33, 34), the injection of CBD homogenates gave rise to the formation of silver-positive structures reminiscent of the astrocytic plaques found in CBD (35, 36), and the injection of AGD homogenates resulted in the formation of silver-negative astrocytic tau pathology as observed in human AGD (37, 38). All of the astrocytic inclusions were strongly immunoreactive with antitau antibodies AT8 and AT100. Following the intracerebral injection of AGD brain extracts into ALZ17 mice, numerous silver-positive grains also formed in nerve cells. It thus appears that several tau conformers made of assembled four-repeat tau exist, which can give rise to the distinct clinical phenotypes and glial and/or neuronal pathologies of several human tauopathies, including PSP, CBD, and AGD. It remains to be determined if additional tau conformers can also give rise to AD, TD, and PiD. Signs of neurodegeneration were not observed for up to 18 mo after the injection of human brain homogenates, in line with our previous findings following the injection of brain extracts from mice transgenic for human P301S tau (20). This supports the suggestion that the molecular tau species responsible for propagation and neurotoxicity may be different. Alternatively, spreading and neurodegeneration may be caused by the same molecular tau species, but the relatively short lifespan of the mouse may have precluded the development of significant neurodegeneration in the present experimental setting.
In nontransgenic control mice, neuronal and oligodendroglial inclusions were present following the intracerebral injection of human brain homogenates from AD, TD, AGD, and PSP brains. Inclusions formed at the injection sites, from where they spread over time. They were positive with a mouse tau-specific antibody and negative with a human tau-specific antibody, consistent with the assembly of mouse tau. These findings establish that the overexpression of human tau is not required for inclusion formation and spreading. However, the number of tau inclusions formed following the intracerebral injection of a human brain homogenate into ALZ17 mice was approximately five times greater compared with the injection of the same homogenate into C57BL/6 mice, consistent with a quantitative effect of overexpression and/or a significant species barrier. The present findings also show that aggregates made of human tau can induce the formation of inclusions made of mouse tau, in line with recent findings (24, 25). We showed previously that the parent P301S tau transgenic line develops tau inclusions that are largely devoid of mouse tau (15) and that the injection of brain extracts from this line into wild-type mice did not lead to the significant spreading of tau aggregates (20). In conjunction with the present findings, this suggests that injected tau seeds made of human wild-type tau promote the spreading of aggregated mouse tau more efficiently than tau seeds made of human mutant P301S tau.
Serial transmission was observed in ALZ17 mice following the injection of homogenates prepared from ALZ17 brains that had been injected with brain extracts from mice transgenic for human mutant P301S tau. Silver-positive nerve cell body inclusions, neuropil threads, and coiled bodies formed at the injection sites. The same was true of brain homogenates from C57BL/6 mice that had been injected with TD or AGD brain extracts. These findings established that the abnormal protein was present in an apparently transmissible form in the brains of animals that had been injected with brain homogenates 18 mo earlier.
AD is characterized by neuritic plaques, primarily composed of extracellular deposits of Aβ, and neurofibrillary lesions, composed of intracellular inclusions made of hyperphosphorylated tau (1). The amyloid cascade hypothesis of AD pathogenesis posits that the aggregation of Aβ triggers the formation of tau inclusions (39). Consistent with this view, previous work has demonstrated an interaction between Aβ and tau pathologies in some brain regions (40–44). However, in these studies, both APP and tau were mutant, unlike what is the case in AD, where tau is wild type (1). It has previously been shown that in mice transgenic for human mutant APP, tau does not aggregate (45, 46). Here we demonstrate the same for mice double transgenic for human mutant APP and wild-type human tau. Both types of pathologies coexisted, but did not influence each other. This was also the case when brain homogenates from mouse line APP23 were intracerebrally injected into tau mouse line ALZ17. At present, there exists no in vivo model system allowing one to study the aggregation of both APP and wild-type tau. The findings reported here and in ref. 20 have demonstrated the formation of tau inclusions in ALZ17 mice following the intracerebral injection of brain homogenates from sporadic human tauopathies and mice transgenic for human mutant P301S tau.
The present work indicates that once small numbers of tau inclusions have formed in the brain, they may become self-propagating and spread in a prion-like manner, independently of other pathogenic mechanisms. What is true of aggregated human tau may also be the case of other aggregation-prone proteins that cause human neurodegenerative diseases, including α-synuclein, superoxide dismutase 1, huntingtin, trans-activator regulatory (TAR) DNA-binding protein 43 (TDP-43), and Aβ (47). The inhibition of cell-to-cell transmission of pathological aggregates, for instance by passive immunotherapy, may constitute an effective mechanism-based therapeutic strategy for most human neurodegenerative diseases.
Materials and Methods
Mice.
Line ALZ17 expressing human wild-type four-repeat tau (19), mice transgenic for human mutant P301S tau (15), line APP23 expressing human mutant amyloid precursor protein (48), and nontransgenic control mice, all females on the C57BL/6 background, were used. ALZ17 × APP23 mice were generated by crossing ALZ17 with APP23 mice for at least seven generations. All experiments were in compliance with the Basel Committee for Animal Care and Animal Use, Kantonales Veterinäramt, Basel.
Preparation of Brain Homogenates.
Total brain homogenates (i.e., soluble and insoluble tau fractions) were prepared as described (20), resulting in a 1:5 (wt/vol) homogenate for human tissues and 1:10 (wt/vol) for murine tissues. Human brain material was derived from neuropathologically confirmed cases of nonfamilial AD [four females, 75, 79, 86, and 89 y of age, temporal cortex, postmortem delays (pmds), 2–17 h], TD (two females, 86 and 89 y of age, hippocampus, pmds 3 and 9 h), AGD (two females, 85 and 95 y of age, amygdala, pmds 9 h each), PSP (two males, 68 and 72 y of age, putamen, pmds 10 and 18 h), CBD (one male, 76 y of age, globus pallidus, pmd 9 h), and PiD (two females, one male, 67, 73, and 61 y of age, frontal cortex, pmds 8, 34, and 25 h). Brain regions were chosen because of their large burden of tau inclusions. Temporal cortex from an age-matched control (male, 76 y of age, pmd 18 h) was also used. The tissue homogenates were intracerebrally injected into 3-mo-old ALZ17 transgenic and C57BL/6 control mice. Except for PiD, M.T. reviewed all of the human cases for neuropathological changes. B.G. reviewed the cases of PiD. Informed consent was obtained from individuals who donated their brains for research. Human brain tissues were used with permission from the relevant local ethical review committees (Universities of Basel, Switzerland; Indianapolis, IN; and Toyohashi, Japan).
Histology and Immunohistochemistry.
Gallyas-Braak silver impregnation and tau immunohistochemistry were performed as described (20). Immunohistochemistry for Aβ was also performed as described (48). The following antibodies were used: T14 specific for human tau (1:1,000; Zymed); AT8 specific for tau phosphorylated at S202 and T205 (1:1,000; Pierce); AT100 specific for tau phosphorylated at T212, S214, and T217 (1:1,000; Pierce); 12E8 specific for tau phosphorylated at S262 and/or S356 (1:500; courtesy of P. Seubert, Elan Pharmaceuticals, Inc., South San Francisco, CA); MT1 specific for mouse tau (1:500, raised against amino acids 114–127 of the longest brain isoform of mouse tau); NT11, a polyclonal serum raised against purified Aβ deposits (1:1,000; courtesy of P. Paganetti, Basel, Switzerland); anti-GFAP (1;100; Abcam); and anti-Iba1 (1:300; Wako). Secondary antibodies were from Vector Laboratories (Vectastain ABC kit).
Western Blotting.
Western blotting for tau was done as described (20) using HT7, an antibody specific for human tau (1:1,000; Pierce), AT8 (1:1,000) and AT100 (1:200). Western blotting for Aβ was carried out as described (43) using the human-specific monoclonal antibody 1E8 (1:1,000; Signet).
Stereotaxic Surgery and Quantitative Analysis of Tau Pathology.
Three-month-old transgenic ALZ17 mice and nontransgenic C57BL/6 controls were operated as described (20). ALZ17 mice injected intracerebrally with human brain extract were analyzed at 6 and 12 mo postinjection (five mice per time point). Total counts of silver-positive structures at the hippocampal injection site were assessed on selected Gallyas-Braak–stained coronal sections as described (20).
Supplementary Material
Acknowledgments
We thank Sabine Ipsen, Sabina Weingärtner, Brenda Dupree, and Rose Richardson for technical support. We are grateful to Profs. Yoshio Hashizume and Akira Hori for helpful discussions and support. This work was supported by the Swiss National Science Foundation (310030_135214 and 32323B_123812), the VELUX Foundation, the UK Medical Research Council (U105184291), the US Public Health Service Grant P30 AG010133, and the Japanese Brain Bank Network for Neuroscience Research.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1301175110/-/DCSupplemental.
References
- 1.Goedert M, Clavaguera F, Tolnay M. The propagation of prion-like protein inclusions in neurodegenerative diseases. Trends Neurosci. 2010;33(7):317–325. doi: 10.1016/j.tins.2010.04.003. [DOI] [PubMed] [Google Scholar]
- 2.Goedert M, Spillantini MG, Jakes R, Rutherford D, Crowther RA. Multiple isoforms of human microtubule-associated protein tau: Sequences and localization in neurofibrillary tangles of Alzheimer’s disease. Neuron. 1989;3(4):519–526. doi: 10.1016/0896-6273(89)90210-9. [DOI] [PubMed] [Google Scholar]
- 3.Götz J, et al. Somatodendritic localization and hyperphosphorylation of tau protein in transgenic mice expressing the longest human brain tau isoform. EMBO J. 1995;14(7):1304–1313. doi: 10.1002/j.1460-2075.1995.tb07116.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Goedert M, Spillantini MG, Cairns NJ, Crowther RA. Tau proteins of Alzheimer paired helical filaments: Abnormal phosphorylation of all six brain isoforms. Neuron. 1992;8(1):159–168. doi: 10.1016/0896-6273(92)90117-v. [DOI] [PubMed] [Google Scholar]
- 5.Noda K, et al. Quantitative analysis of neurofibrillary pathology in a general population to reappraise neuropathological criteria for senile dementia of the neurofibrillary tangle type (tangle-only dementia): The Hisayama Study. Neuropathology. 2006;26(6):508–518. doi: 10.1111/j.1440-1789.2006.00722.x. [DOI] [PubMed] [Google Scholar]
- 6.Schmidt ML, Zhukareva V, Newell KL, Lee VMY, Trojanowski JQ. Tau isoform profile and phosphorylation state in dementia pugilistica recapitulate Alzheimer’s disease. Acta Neuropathol. 2001;101(5):518–524. doi: 10.1007/s004010000330. [DOI] [PubMed] [Google Scholar]
- 7.Delacourte A, et al. Specific pathological Tau protein variants characterize Pick’s disease. J Neuropathol Exp Neurol. 1996;55(2):159–168. doi: 10.1097/00005072-199602000-00004. [DOI] [PubMed] [Google Scholar]
- 8.Flament S, Delacourte A, Verny M, Hauw JJ, Javoy-Agid F. Abnormal Tau proteins in progressive supranuclear palsy. Similarities and differences with the neurofibrillary degeneration of the Alzheimer type. Acta Neuropathol. 1991;81(6):591–596. doi: 10.1007/BF00296367. [DOI] [PubMed] [Google Scholar]
- 9.Ksiezak-Reding H, et al. Ultrastructure and biochemical composition of paired helical filaments in corticobasal degeneration. Am J Pathol. 1994;145(6):1496–1508. [PMC free article] [PubMed] [Google Scholar]
- 10.Togo T, et al. Argyrophilic grain disease is a sporadic 4-repeat tauopathy. J Neuropathol Exp Neurol. 2002;61(6):547–556. doi: 10.1093/jnen/61.6.547. [DOI] [PubMed] [Google Scholar]
- 11.Tolnay M, et al. Argyrophilic grain disease and Alzheimer’s disease are distinguished by their different distribution of tau protein isoforms. Acta Neuropathol. 2002;104(4):425–434. doi: 10.1007/s00401-002-0591-z. [DOI] [PubMed] [Google Scholar]
- 12.Poorkaj P, et al. Tau is a candidate gene for chromosome 17 frontotemporal dementia. Ann Neurol. 1998;43(6):815–825. doi: 10.1002/ana.410430617. [DOI] [PubMed] [Google Scholar]
- 13.Hutton M, et al. Association of missense and 5′-splice-site mutations in tau with the inherited dementia FTDP-17. Nature. 1998;393(6686):702–705. doi: 10.1038/31508. [DOI] [PubMed] [Google Scholar]
- 14.Spillantini MG, et al. Mutation in the tau gene in familial multiple system tauopathy with presenile dementia. Proc Natl Acad Sci USA. 1998;95(13):7737–7741. doi: 10.1073/pnas.95.13.7737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Allen B, et al. Abundant tau filaments and nonapoptotic neurodegeneration in transgenic mice expressing human P301S tau protein. J Neurosci. 2002;22(21):9340–9351. doi: 10.1523/JNEUROSCI.22-21-09340.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yoshiyama Y, et al. Synapse loss and microglial activation precede tangles in a P301S tauopathy mouse model. Neuron. 2007;53(3):337–351. doi: 10.1016/j.neuron.2007.01.010. [DOI] [PubMed] [Google Scholar]
- 17.Ishihara T, et al. Age-dependent emergence and progression of a tauopathy in transgenic mice overexpressing the shortest human tau isoform. Neuron. 1999;24(3):751–762. doi: 10.1016/s0896-6273(00)81127-7. [DOI] [PubMed] [Google Scholar]
- 18.Spittaels K, et al. Prominent axonopathy in the brain and spinal cord of transgenic mice overexpressing four-repeat human tau protein. Am J Pathol. 1999;155(6):2153–2165. doi: 10.1016/S0002-9440(10)65533-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Probst A, et al. Axonopathy and amyotrophy in mice transgenic for human four-repeat tau protein. Acta Neuropathol. 2000;99(5):469–481. doi: 10.1007/s004010051148. [DOI] [PubMed] [Google Scholar]
- 20.Clavaguera F, et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Biol. 2009;11(7):909–913. doi: 10.1038/ncb1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Frost B, Jacks RL, Diamond MI. Propagation of tau misfolding from the outside to the inside of a cell. J Biol Chem. 2009;284(19):12845–12852. doi: 10.1074/jbc.M808759200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nonaka T, Watanabe ST, Iwatsubo T, Hasegawa M. Seeded aggregation and toxicity of α-synuclein and tau: Cellular models of neurodegenerative diseases. J Biol Chem. 2010;285(45):34885–34898. doi: 10.1074/jbc.M110.148460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Guo JL, Lee VMY. Seeding of normal Tau by pathological Tau conformers drives pathogenesis of Alzheimer-like tangles. J Biol Chem. 2011;286(17):15317–15331. doi: 10.1074/jbc.M110.209296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liu L, et al. Trans-synaptic spread of tau pathology in vivo. PLoS ONE. 2012;7(2):e31302. doi: 10.1371/journal.pone.0031302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.de Calignon A, et al. Propagation of tau pathology in a model of early Alzheimer’s disease. Neuron. 2012;73(4):685–697. doi: 10.1016/j.neuron.2011.11.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kfoury N, Holmes BB, Jiang H, Holtzman DM, Diamond MI. Trans-cellular propagation of Tau aggregation by fibrillar species. J Biol Chem. 2012;287(23):19440–19451. doi: 10.1074/jbc.M112.346072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Santa-Maria I, et al. Paired helical filaments from Alzheimer disease brain induce intracellular accumulation of Tau protein in aggresomes. J Biol Chem. 2012;287(24):20522–20533. doi: 10.1074/jbc.M111.323279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lasagna-Reeves CA, et al. Alzheimer brain-derived tau oligomers propagate pathology from endogenous tau. Sci Rep. 2012;2:700. doi: 10.1038/srep00700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Crowther RA, Goedert M. Abnormal tau-containing filaments in neurodegenerative diseases. J Struct Biol. 2000;130(2-3):271–279. doi: 10.1006/jsbi.2000.4270. [DOI] [PubMed] [Google Scholar]
- 30.Colby DW, Prusiner SB. Prions. Cold Spring Harb Perspect Biol. 2011;3:a006883. doi: 10.1101/cshperspect.a006833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Arai T, et al. Distinct isoforms of tau aggregated in neurons and glial cells in brains of patients with Pick’s disease, corticobasal degeneration and progressive supranuclear palsy. Acta Neuropathol. 2001;101(2):167–173. doi: 10.1007/s004010000283. [DOI] [PubMed] [Google Scholar]
- 32.Dinkel PD, Siddiqua A, Huynh H, Shah M, Margittai M. Variations in filament conformation dictate seeding barrier between three- and four-repeat tau. Biochemistry. 2011;50(20):4330–4336. doi: 10.1021/bi2004685. [DOI] [PubMed] [Google Scholar]
- 33.Yamada T, McGeer PL, McGeer EG. Appearance of paired nucleated, Tau-positive glia in patients with progressive supranuclear palsy brain tissue. Neurosci Lett. 1992;135(1):99–102. doi: 10.1016/0304-3940(92)90145-w. [DOI] [PubMed] [Google Scholar]
- 34.Nishimura M, Namba Y, Ikeda K, Oda M. Glial fibrillary tangles with straight tubules in the brains of patients with progressive supranuclear palsy. Neurosci Lett. 1992;143(1-2):35–38. doi: 10.1016/0304-3940(92)90227-x. [DOI] [PubMed] [Google Scholar]
- 35.Feany MB, Dickson DW. Widespread cytoskeletal pathology characterizes corticobasal degeneration. Am J Pathol. 1995;146(6):1388–1396. [PMC free article] [PubMed] [Google Scholar]
- 36.Komori T, et al. Astrocytic plaques and tufts of abnormal fibers do not coexist in corticobasal degeneration and progressive supranuclear palsy. Acta Neuropathol. 1998;96(4):401–408. doi: 10.1007/s004010050911. [DOI] [PubMed] [Google Scholar]
- 37.Botez G, Probst A, Ipsen S, Tolnay M. Astrocytes expressing hyperphosphorylated tau protein without glial fibrillary tangles in argyrophilic grain disease. Acta Neuropathol. 1999;98(3):251–256. doi: 10.1007/s004010051077. [DOI] [PubMed] [Google Scholar]
- 38.Tolnay M, Clavaguera F. Argyrophilic grain disease: A late-onset dementia with distinctive features among tauopathies. Neuropathology. 2004;24(4):269–283. doi: 10.1111/j.1440-1789.2004.00591.x. [DOI] [PubMed] [Google Scholar]
- 39.Hardy J, Selkoe DJ. The amyloid hypothesis of Alzheimer’s disease: Progress and problems on the road to therapeutics. Science. 2002;297(5580):353–356. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 40.Lewis J, et al. Enhanced neurofibrillary degeneration in transgenic mice expressing mutant tau and APP. Science. 2001;293(5534):1487–1491. doi: 10.1126/science.1058189. [DOI] [PubMed] [Google Scholar]
- 41.Götz J, Chen F, van Dorpe J, Nitsch RM. Formation of neurofibrillary tangles in P301l tau transgenic mice induced by Abeta 42 fibrils. Science. 2001;293(5534):1491–1495. doi: 10.1126/science.1062097. [DOI] [PubMed] [Google Scholar]
- 42.Oddo S, Billings L, Kesslak JP, Cribbs DH, LaFerla FM. Abeta immunotherapy leads to clearance of early, but not late, hyperphosphorylated tau aggregates via the proteasome. Neuron. 2004;43(3):321–332. doi: 10.1016/j.neuron.2004.07.003. [DOI] [PubMed] [Google Scholar]
- 43.Bolmont T, et al. Induction of tau pathology by intracerebral infusion of amyloid-β -containing brain extract and by amyloid-β deposition in APP x Tau transgenic mice. Am J Pathol. 2007;171(6):2012–2020. doi: 10.2353/ajpath.2007.070403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Coomaraswamy J, et al. Modeling familial Danish dementia in mice supports the concept of the amyloid hypothesis of Alzheimer’s disease. Proc Natl Acad Sci USA. 2010;107(17):7969–7974. doi: 10.1073/pnas.1001056107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Xu G, Gonzales V, Borchelt DR. Abeta deposition does not cause the aggregation of endogenous tau in transgenic mice. Alzheimer Dis Assoc Disord. 2002;16(3):196–201. doi: 10.1097/00002093-200207000-00011. [DOI] [PubMed] [Google Scholar]
- 46.Boutajangout A, et al. Characterisation of cytoskeletal abnormalities in mice transgenic for wild-type human tau and familial Alzheimer’s disease mutants of APP and presenilin-1. Neurobiol Dis. 2004;15(1):47–60. doi: 10.1016/j.nbd.2003.09.007. [DOI] [PubMed] [Google Scholar]
- 47.Holmes BB, Diamond MI. Cellular mechanisms of protein aggregate propagation. Curr Opin Neurol. 2012;25(6):721–726. doi: 10.1097/WCO.0b013e32835a3ee0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sturchler-Pierrat C, et al. Two amyloid precursor protein transgenic mouse models with Alzheimer disease-like pathology. Proc Natl Acad Sci USA. 1997;94(24):13287–13292. doi: 10.1073/pnas.94.24.13287. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.