

Significance
Naturally occurring regulatory T (Treg) cells suppress aberrant or excessive immune responses, thereby maintaining immune self-tolerance and homeostasis. This study shows that a combination of IL-2 repression, CTLA-4 expression, and antigenic stimulation is able to convert conventional T cells to potently immunosuppressive Treg-like cells, which are able to deprive IL-2 and CD28 signal from other T cells. Like natural Treg cells, they acquire a self-skewed T-cell receptor repertoire in the course of their thymic development, enabling them to control autoimmune responses effectively. This Treg construction by targeting IL-2 and CTLA-4 in conventional T cells is a novel way of immune suppression.
Keywords: immune tolerance, CD25, thymic selection
Abstract
Thymus-produced CD4+ regulatory T (Treg) cells, which specifically express the transcription factor forkhead box p3, are potently immunosuppressive and characteristically possess a self-reactive T-cell receptor (TCR) repertoire. To determine the molecular basis of Treg suppressive activity and their self-skewed TCR repertoire formation, we attempted to reconstruct these Treg-specific properties in conventional T (Tconv) cells by genetic manipulation. We show that Tconv cells rendered IL-2 deficient and constitutively expressing transgenic cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) were potently suppressive in vitro when they were preactivated by antigenic stimulation. They also suppressed in vivo inflammatory bowel disease and systemic autoimmunity/inflammation produced by Treg deficiency. In addition, in the thymus, transgenic CTLA-4 expression in developing Tconv cells skewed their TCR repertoire toward higher self-reactivity, whereas CTLA-4 deficiency specifically in developing thymic Treg cells cancelled their physiological TCR self-skewing. The extracellular portion of CTLA-4 was sufficient for the suppression and repertoire shifting. It interfered with CD28 signaling to responder Tconv cells via outcompeting CD28 for binding to CD80 and CD86,or modulating CD80/CD86 expression on antigen-presenting cells. Thus, a triad of IL-2 repression, CTLA-4 expression, and antigenic stimulation is a minimalistic requirement for conferring Treg-like suppressive activity on Tconv cells, in accordance with the function of forkhead box p3 to strongly repress IL-2 and maintain CTLA-4 expression in natural Treg cells. Moreover, CTLA-4 expression is a key element for the formation of a self-reactive TCR repertoire in natural Treg cells. These findings can be exploited to control immune responses by targeting IL-2 and CTLA-4 in Treg and Tconv cells.
Naturally arising CD25+CD4+ regulatory T (Treg) cells, which specifically express the transcription factor Foxp3, physiologically engage in the maintenance of immunological self-tolerance and homeostasis (1–3). The majority of them are produced by the thymus as a functionally distinct T-cell subpopulation, with the T-cell receptor (TCR) repertoire considerably skewed toward recognizing self-antigens. However, the molecular basis of Treg suppression, thymic generation, and self-skewed TCR repertoire formation is not fully understood. Foxp3 has been proposed to control the expression of putative suppressive molecules in Treg cells because ectopic expression of Foxp3 in conventional T (Tconv) cells is able to convert them to Treg-like cells with suppressive function (4, 5). Furthermore, deficiency or mutations of the Foxp3 gene hampers the development and suppressive function of natural Treg cells. A variety of molecules encoded by Foxp3-controlled genes are known to be involved in Treg cell function, although it is obscure how and to what extent each molecule contributes to Treg cell development and function (1–3, 6). One way to elucidate the key molecular mechanisms of Treg suppressive function and development would be to determine which Foxp3-controlled gene(s), when it is expressed in Tconv cells, can confer on them Treg-like in vivo and in vitro suppressive activity and/or developmental characteristics that include the acquisition of the self-reactive TCR repertoire.
IL-2 and CTLA-4, which are the molecules most stably repressed and activated, respectively, by Foxp3 in natural Treg cells, play key roles in Treg cell function and development (7, 8). In vitro, exogenous IL-2 abrogates Treg suppressive activity, indicating its involvement in Treg-mediated suppression and suggesting that Treg cells may deprive responder T cells of IL-2 via their constitutively expressed high-affinity IL-2 receptor (9–11). Treg-specific CTLA-4 deficiency produces fatal autoimmune/inflammatory disease via impairment of Treg suppressive activity (12). As possible roles of CTLA-4 in Treg-mediated suppression, several studies have shown that CTLA-4, which has much higher affinity than CD28 for their common ligands CD80 and CD86, outcompetes CD28 for binding to the ligands in the immunological synapse and also down-modulates CD80/CD86 expression on antigen-presenting cells (APCs), thereby depriving the CD28 signal from responder T cells (12–17). However, it has been shown repeatedly that Foxp3+ Treg cells from IL-2 receptor– or CTLA-4–deficient mice with systemic inflammation still exhibit substantial in vitro suppressive function (12, 18, 19). These findings, taken together, indicate that either an IL-2/IL-2 receptor– or CTLA-4–dependent suppressive mechanism alone is insufficient to produce full suppressive activity in Foxp3+ Treg cells.
Foxp3+ Treg cell development in the thymus requires both IL-2 and CD28 signals, although either IL-2 or CD28 deficiency alone resulted in only a partial reduction of the number of Treg cells (20, 21). TCR signal intensity also plays a key role in Treg cell development. It has been suggested that developing CD4+ T cells expressing TCRs highly reactive with self-peptide/MHC ligands may preferentially differentiate into Foxp3+ Treg cells, resulting in their self-skewed TCR repertoire (22–28). It remains to be determined, however, whether TCR signal intensity alone directly determines the fate of Treg cells and their self-skewed TCR repertoire in the course of thymic selection.
To address the above outstanding issues on Treg function and development, we have attempted to determine whether Treg-like suppressive activity and self-skewed TCR repertoire can be reconstructed in Tconv cells by modulating the expression of genes that are controlled by Foxp3 in natural Treg cells. We show that a combination of IL-2 nonproduction, high CTLA-4 expression, and antigenic stimulation is sufficient to convert naïve T cells to Treg-like cells with potent in vivo and in vitro suppressive activity. Furthermore, forced expression of CTLA-4 in developing T cells is able to produce self-skewed TCR repertoire in the thymus, whereas Treg-specific CTLA-4 deficiency cancels physiological acquisition of self-reactive TCR repertoire by developing Foxp3+ Treg cells. A CTLA-4 mutant form lacking the cytoplasmic signaling portion is sufficient for the suppression and repertoire skewing. These results provide key insights into the molecular mechanisms of Treg cell development and function and also delineate a minimum molecular requirement for constructing antigen-specific Treg-like suppressive T cells from Tconv cells without Foxp3.
Results
Effects of IL-2 Deficiency, CD28 Nonexpression, or Constitutive CTLA-4 Expression on T-Cell Development and Autoimmunity.
We first analyzed how T-cell development was altered by IL-2 deficiency [by IL-2 gene KO (IL2KO)], constitutive expression of full-length CTLA-4 [by CTLA-4 transgene (C4Tg) expression], or a mutant form CTLA-4 lacking the cytoplasmic portion [by tailless CTLA-4 transgene (TLC4Tg) expression], CD28 nonexpression [by CD28 gene KO (CD28KO)], or combinations of IL-2 deficiency and others. By C4Tg or TLC4Tg expression under the human CD2 promoter, all thymocytes after the CD4+CD8+ double-positive stage expressed CTLA-4 (29). Compared with WT mice, the ratio and the number of Foxp3+ cells among CD4+CD8− [CD4 single-positive (SP)] cells significantly decreased in the thymus and the periphery of C4Tg, TLC4Tg, or CD28KO mice, without significant differences in the ratio and the number of CD4SP cells (Fig. 1 A–D) (21, 29–31). When accompanied by IL-2 deficiency, the frequency of Foxp3+ cells in these mice was further reduced, especially in the periphery, to less than 0.5% of CD4+ T cells compared with ∼8% in WT mice (Fig. 1 B and D).
Fig. 1.
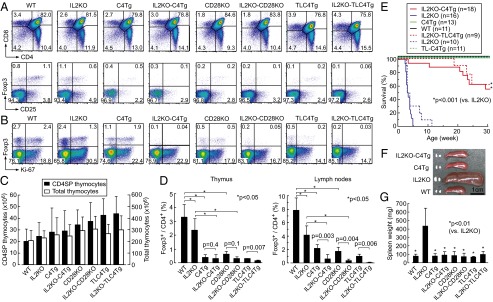
Effects of IL-2 deficiency, CD28 nonexpression, or constitutive CTLA-4 expression on T-cell development and autoimmunity. (A) Staining of CD4 and CD8 on whole thymocytes (Upper) and CD25 and intracellular Foxp3 of CD4SP thymocytes (Lower), in designated strains of mice. (B) Expression of intracellular Ki-67 and Foxp3 by lymph node CD4+ T cells in respective strains. Numbers in A and B indicate the percentages in gated areas. (C) Numbers of CD4SP (left axis, black columns) and total (right axis, white columns) thymocytes in respective strains. (D) Percentages (mean ± SD) of Foxp3+ cells among CD4SP thymocytes and CD4+ lymph node T cells in respective strains at 3–5 wk of age (n = 3–11 each). Significant difference (*P < 0.05) in post hoc comparison for ANOVA and P value in unpaired Student t tests are shown. (E) Survival of WT, IL2KO, IL2KO-C4Tg, and C4Tg mice on the BALB/c background (solid lines) and survival of TLC4Tg, IL2KO-TLC4Tg, and IL2KO mice on mixed backgrounds (dotted lines). (F) A representative picture of axillary and inguinal lymph nodes and spleens in indicated groups of mice at 4 wk of age. (G) The weight (mean ± SD) of spleens at 2–4 wk of age in respective groups (n = 4–12 each).
In contrast with IL2KO mice, which spontaneously developed lymphoproliferation and died within a month after birth, IL2KO-C4Tg or IL2KO-TLC4Tg mice failed to develop splenomegaly and lymphoadenopathy at least by 4 wk after birth, surviving much longer than IL2KO mice, despite severe deficiency of Foxp3+ Treg cells (Fig. 1 E–G) (32). In accord with these observations, actively proliferating (i.e., Ki-67+) peripheral CD4+ T cells, which were high in IL2KO mice, were substantially reduced in IL2KO-C4Tg or IL2KO-TLC4Tg mice to the levels comparable in WT, C4Tg, TLC4Tg, CD28KO, or IL2KO-CD28KO mice (Fig. 1B) (33).
Thus, high expression of the extracellular portion of CTLA-4 in T cells, together with IL-2 deficiency, inhibits thymic development of natural Foxp3+ Treg cells and peripheral activation of Foxp3− Tconv cells, as in CD28 deficiency, presumably because the CTLA-4 expression reduces CD28 signal to developing T cells via interfering with the binding of CD28 to CD80 CD86 expressed on thymic stromal cells.
Suppression of Scurfy Disease by IL-2 Nonproduction and Transgenic CTLA-4 Expression.
We next determined whether IL-2 deficiency, constitutive CTLA-4 expression, or the combination of these was able to prevent fatal systemic autoimmunity/inflammation in Foxp3-mutant Scurfy mice, which are deficient of natural Foxp3+ Treg cells. The majority of IL2KO-C4Tg BALB/c Scurfy mice lived more than 20 wk (Fig. 2A), without suffering from splenomegaly at least by 4 wk after birth (Fig. 2B). It contrasted with short life expectancy (less than 4 wk) of WT, C4Tg, or IL2KO BALB/c Scurfy mice, as previously shown with IL2KO C57BL/6 Scurfy mice (34). The fractions of CD4+ T cells activated and/or proliferating (i.e., CD25+, CD69+, or Ki-67+) or producing cytokines such as IFN-γ, IL-4, and IL-10 were significantly lower in IL2KO-C4Tg Scurfy mice compared with WT Scurfy mice; T-cell activation and cytokine production were only partially suppressed in C4Tg Scurfy mice (Fig. 2 C–F). Thus, the combination of IL-2 nonproduction and transgenic CTLA-4 expression, but not either one alone, was able to inhibit the development of systemic autoimmunity/inflammation normally consequent to Foxp3+ Treg cell deficiency.
Fig. 2.

Suppression of systemic autoimmunity/inflammation in Scurfy mice by IL-2 deficiency and transgenic CTLA-4 expression. (A) Survival of Scurfy, IL2KO Scurfy, C4Tg Scurfy, and IL2KO-C4Tg Scurfy mice. (B) The weight (mean ± SD) of spleens at 2–3 wk of age (n = 3–5 each). (C) Expression of CD25 and intracellular Ki-67 by lymph node CD4+ T cells. (D) Expression of CD69 by lymph node CD4+ and CD8+ T cells. (E) Intracellular staining of IL-4 and IFN-γ in indicated lymph node CD4+ T cells. (F) Proportions of lymph node CD4+ T cells producing indicated cytokines (n = 3–5 each). Numbers in C–E indicate the percentages in gated areas. Representative staining of three to five mice in C–E.
In Vitro Hyporesponsiveness and Suppressive Activity of IL2KO-C4Tg T Cells.
These results suggested that the combination of IL-2 deficiency and CTLA-4 overexpression rendered Tconv cells antigen hyporesponsive, suppressive, or both. By in vitro analysis, Treg-depleted CD25−CD4+ T cells [hereafter called naïve T (Tn) cells] from IL2KO, C4Tg, or TLC4Tg mice were much less proliferative than WT CD4+ Tn cells on anti-CD3 stimulation (Fig. 3A). IL2KO-C4Tg CD4+ Tn cells had even lower proliferative activity than IL2KO or C4Tg Tn cells, and the activity was equivalent to WT CD25+CD4+ Treg cells or CD28KO CD4+ Tn cells. Addition of anti-CD28 or exogenous IL-2 effectively reversed the hypoproliferation of IL2KO, C4Tg, TLC4Tg, and IL2KO-C4Tg Tn cells, and also WT Treg cells; further, addition of both anti-CD28 and IL-2 abrogated the hypoproliferation of IL2KO-C4Tg CD4+ Tn cells and WT Treg cells in an additive manner. CTLA-4 expression alone down-regulated IL-2 production substantially but not completely, as indicated by the low IL-2 concentration in the supernatant of C4Tg Tn cell culture (Fig. 3B); nevertheless, this small amount of IL-2 rendered them more proliferative than IL2KO-C4Tg Tn cells (Fig. 3A). In addition, TCR stimulation of IL2KO T cells in the presence of anti-CD28, and CD28KO T cells in the presence of IL-2, exhibited substantial proliferative responses, indicating that IL-2 and CD28 signals were complementary for T-cell proliferation to a certain extent. Collectively, these results indicate that deprivation of both IL-2 and CD28 signals is essentially required to inhibit the activation/proliferation of Tconv cells effectively. Also, similar hyporesponsiveness of C4Tg or TLC4Tg T cells, together with restoration of their responsiveness by CD28 stimulation, supports the notion that full-length or tailless CTLA-4 blocks CD28 signal to the T cells.
Fig. 3.

Proliferation of IL2KO-C4Tg CD4+ T cells. (A) Proliferation measured by 3H-thymidine incorporation of CD25+ or CD25−CD4+ lymph node T cells from designated groups of mice when stimulated for 3 d in the presence of APCs and indicated reagents. Significant difference were analyzed with post hoc comparison in ANOVA among IL2KO, IL2KO-C4Tg, and C4Tg Tn cells under anti-CD3 stimulation and among WT, IL2KO, IL2KO-C4Tg, and C4Tg Tn cells in the other conditions. cpm, count per minute. (B) IL-2 concentration assessed by ELISA in culture supernatant after 3-d incubation in the presence of APCs and anti-CD3. Representative results (mean ± SD of triplicates) from three independent experiments are shown. TLC4Tg mice used were deficient in endogenous CTLA-4 expression by mating with CTLA-4−/− mice.
We next assessed possible in vitro suppressive activity of IL2KO-C4Tg T cells when they were either freshly prepared or preactivated in vitro for 2 d by anti-CD3, anti-CD28, and IL-2. Activation resulted in up-regulation of CD25, but failed to induce Foxp3 expression (Fig. 4A). The level of intracellular CTLA-4 in activated IL2KO-C4Tg CD4+ T cells was equivalent to that in activated Foxp3+ natural Treg cells. To assess suppressive function, carboxyfluorescein diacetate succinimidyl ester (CFSE)-labeled WT CD4+ Tn cells from congenic Thy1.1+ BALB/c mice were stimulated with anti-CD3 and APCs for 3 d in the presence of WT CD25+CD4+ Treg cells or CD25−CD4+ Tn cells from WT, IL2KO, C4Tg, or IL2KO-C4Tg Thy1.2+ BALB/c mice (on the DO11.10 TCR transgenic background to avoid severe inflammatory status in TCR nontransgenic IL2KO mice). Unlike WT natural Treg cells, freshly prepared C4Tg or IL2KO-C4Tg CD4+ Tn cells only slightly suppressed the proliferation of WT Tn cells as assessed by the percentages (Fig. 4 B and C) and the number of CFSE-diluting WT Tn cells (Fig. S1). Interestingly, however, when these populations were preactivated, they suppressed WT CD4+ Tn cell proliferation to various degrees. Preactivated IL2KO-C4Tg CD4+ T cells in particular suppressed as potently as WT natural Treg cells at every cell ratio (Fig. 4D). Even WT activated T cells, especially when they ceased to produce IL-2 and still expressed CTLA-4 in the course of immune responses, suppressed the proliferation of Tn cells (Fig. 4 B and C; Fig. S2).
Fig. 4.
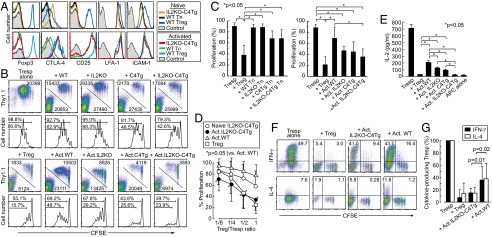
In vitro suppression by activated IL2KO-C4Tg CD4+ T cells. (A) Intracellular expression of Foxp3 and CTLA-4 and cell surface expression of CD25, LFA-1, and ICAM-1 by freshly prepared (Upper) or in vitro anti-CD3/anti-CD28/IL-2 activated T cells (Lower) prepared from indicated T-cell populations. (B) Proliferation of CFSE-labeled Thy1.1+ CD25−CD4+ responder T cells (Tresp) cocultured with Thy1.2+ CD25+CD4+ Treg or CD25−CD4+ Tn cells from depicted strains. The latter cells were freshly prepared or preactivated (Act) in vitro for 2 d by anti-CD3/anti-CD28/IL-2 stimulation. Staining for CFSE and Thy1.1 expression of live cells (Upper) and CFSE intensity of gated Thy1.1+ T cells (Lower) with the mean cell numbers and percentages in gated areas. (C) Percentages of dividing responder T cells in the coculture with indicated cells. (D) Proliferation of Thy1.1+ Tresp cells cocultured with graded numbers of designated populations of Thy1.2+ cells. Proportions of dividing Tresp cells were normalized by the proliferation of Tresp cells alone. *Significant difference from Tresp proliferation with WT activated T cells at each cell ratio. The means ± SDs of five independent experiments are shown. (E) IL-2 concentration (mean ± SD of triplicates) assessed by ELISA in the culture supernatants after 3-d culture. (F) Cytokine production by Tresp cells and designated groups of T cells cocultured under a Th1 (IL-12 and anti-IL-4) or Th2 (IL-4 and anti-IL-12) condition. Numbers indicate the percentages of cytokine-producing cells among Tresp cells and those among Thy1.1−Thy1.2+CFSE− cells. (G) Proportion of cytokine-producing Tresp cells in F is shown after normalization by the proportion without Thy1.2+ T cells. Representative results of at least three independent experiments are shown in A, E, and F.
IL-2 concentration, which was closely correlated with T-cell proliferation (Fig. 3), was equivalently reduced by coculture with WT Treg cells or activated IL2KO-C4Tg T cells and to a significant but lesser degree by coculture with activated IL2KO or C4Tg T cells (Fig. 4E). Furthermore, activated IL2KO-C4Tg T cells and natural Treg cells suppressed the differentiation of responder Tn cells into IFN-γ–producing Th1 or IL-4–producing Th2 cells under Th1 (with IL-12 and anti-IL-4) or Th2 (with IL-4 and anti–IL-12) culture conditions, respectively, although the preactivated IL2KO-C4Tg T cells, unlike natural Treg cells, produced these cytokines themselves (Fig. 4 F and G). When activated human Foxp3−CD4+ peripheral blood T cells are made IL-2 deficient and CTLA-4--expressing by retroviral gene transduction, they also exhibited similar in vitro suppressive activity comparable with that of human natural Foxp3+ Treg cells (Fig. S3).
Thus, the combination of IL-2 nonproduction and constitutive CTLA-4 expression in conjunction with TCR/CD28-stimulated preactivation is able to confer Treg-like in vitro suppressive activity on Tconv cells, irrespective of their ability to produce inflammatory cytokines.
Mechanisms of In Vitro Suppression by IL2KO-C4Tg CD4+ T Cells.
The expression levels of Treg signature genes (e.g., Cd25, Itgb8, Socs2, and Eos) and the genes related to Treg suppressive function [e.g., Il10, Tgfb1, Il35(Il12a/Ebi3), Cd39/Cd73, Lag3, Granzyme B, and Galectin 1] were similar between IL2KO-C4Tg CD4+ T cells and WT CD4+ T cells and much lower than Foxp3+ Treg cells, with or without activation by anti-CD3, anti-CD28, and IL-2 (Fig. 5A). The only difference in the gene expression profiles between activated IL2KO-C4Tg CD4+ T cells and activated WT CD4+ T cells was found in the Il-2 and Ctla-4 genes.
Fig. 5.
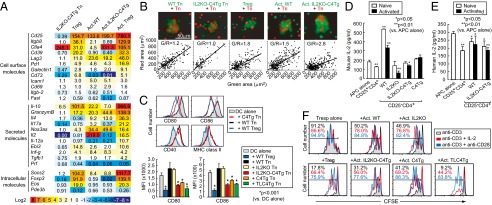
Mechanisms of suppression by preactivated IL2KO-C4Tg Foxp3−CD4+ T cells. (A) Expression of indicated genes by CD25−CD4+ T cells from WT or IL2KO-C4Tg mice and by WT CD25+CD4+ Treg cells was assessed by quantitative PCR before and after 2-d activation with anti-CD3/anti-CD28/IL-2. Numbers indicate relative expression of each gene compared with the expression by WT CD25−CD4+ Tn cells. (B) Red dye–labeled CD25−CD4+ Tn cells and green dye–labeled depicted T-cell populations from DO11.10 TCR transgenic mice were cultured for 2 d with BALB/c CD11c+ splenic DCs and OVA323–339 peptide. Green and red area in each cell aggregate is plotted with fitted curves. G/R indicates the median of green area/red area ratio. (C) Expression of CD80, CD86, CD40, and MHC class II by CD11c+ splenic DCs cocultured with depicted cell populations. The mean ± SDs of four independent experiments are also shown for expression of CD80 and CD86. TLC4Tg mice used were rendered deficient in endogenous CTLA-4 expression by mating with CTLA-4−/− mice. MFI, median fluorescence intensity. (D) IL-2 concentration in culture supernatant of depicted populations incubated in the presence of 10 U/mL (187.9 pg/mL) of exogenous IL-2, anti-CD3, and APCs for 20 h. (E) Human IL-2 concentration in culture supernatant of depicted populations stimulated in the presence of 250 pg/mL of human IL-2. (F) CFSE intensity of dividing CFSE-labeled Thy1.1+ CD25−CD4+ Tresp cells cocultured with the same number of depicted T-cell populations in the presence of indicated stimulation. Representative results of at least three independent experiments are shown in A, B, and D–F).
Activated IL2KO-C4Tg CD4+ T cells, which expressed adhesion molecules [such as lymphocyte function-associated antigen-1 (LFA-1) and intercellular adhesion molecule-1 (ICAM-1)] as highly as natural Treg cells or activated WT CD4+ T cells (Fig. 4A), aggregated more strongly than antigen-reactive CD4+ Tn cells, thus likely hampering the access of the latter to antigen-presenting dendritic cells (DCs) in in vitro coculture assay (Fig. 5B), as previously observed with natural Treg cells (14). To assess the expression levels of costimulatory ligands on APCs, WT CD25+CD4+ Treg cells or CD25−CD4+ Tn cells from WT, IL2KO, C4Tg, IL2KO-C4Tg, or TLC4Tg mice were cocultured with splenic CD11c+ DCs for 2 d in the presence of anti-CD3 and IL-2. Like natural Treg cells, C4Tg CD4+ Tn cells, whether they were IL2KO or not, down-modulated the expression of CD80 and CD86, but not CD40 or MHC class II, on live DCs (Fig. 5C). TLC4Tg Tn cells, which expressed CTLA-4 at a higher level than C4Tg T cells in part due to the lack of endocytosis of cell surface CTLA-4 (29), similarly down-modulated CD80 and CD86 on DCs.
In addition, we assessed the capacity of IL2KO-C4Tg Tn cells and natural Treg cells to absorb IL-2 (Fig. 5D). By incubating with a fixed amount of IL-2 for 20 h in the presence of anti-CD3 stimulation, preactivated IL2KO or IL2KO-C4Tg CD4+ T cells and natural Treg cells, all of which highly expressed CD25 (Fig. 4A), absorbed small but significant amounts of mouse IL-2 (Fig. 5D), whereas WT non-Treg cells secreted a large amount of the cytokine (Figs. 3B and 5D). Similar incubation of the T cells with human IL-2 to exclude possible contribution of murine IL-2 produced by the incubated cells to measured IL-2 concentration revealed that Treg cells and activated IL2KO-C4Tg or WT T cells absorbed IL-2 to a comparable extent (Fig. 5E). Importantly, WT CD25+CD4+ Treg cells and preactivated CD4+ T cells from IL2KO-C4Tg or TLC4Tg mice potently suppressed the proliferation of cocultured responder naïve CD4+ T cells; the suppression was abrogated by addition of agonistic anti-CD28 or exogenous IL-2 (Fig. 5F).
Taken together, activated IL2KO-C4Tg CD4+ T cells dominantly aggregate around APCs, deterring responder T cells from contacting the APCs, and limit the availability of CD28 ligand and IL-2 to the former in a similar manner as natural Treg cells.
In Vivo Immune Suppression by IL2KO-C4Tg CD4+ T Cells.
We next assessed in vivo function of IL2KO, C4Tg, and IL2KO-C4Tg CD4+ Tn cells using a mouse colitis model, in which transfer of Treg-depleted WT CD45RBhighCD4+ Tn cells into lymphopenic RAG2−/− mice induced inflammatory bowel disease (IBD) accompanied by loss of body weight, diarrhea, and histologically evident colitis (35). Interestingly, transfer of CD45RBhighCD4+ Tn cells from IL2KO, C4Tg, or IL2KO-C4Tg mice failed to elicit body weight loss or diarrhea, despite the presence of systemic autoimmunity/inflammation in donor IL2KO mice (Fig. 6A). Histology of colitis in these recipients was significantly less severe than the disease induced by WT CD45RBhighCD4+ Tn cell transfer (Fig. 6B). The majority of the transferred IL2KO or IL2KO-C4Tg CD4+ Tn cells were Foxp3− even after 2 mo; Ki-67 expression was equivalent between IL2KO-C4Tg T cells and WT T cells in the recipients (Fig. 6C). In contrast with less expansion of C4Tg or IL2KO-C4Tg T cells compared with WT T cells, IL2KO T cells expanded more with higher Ki-67 expression than WT T cells; however, they failed to induce colitis (Fig. 6 C and D). Taken together, these results indicate that the failure to induce colitis in RAG2−/− mice by the transfer of CD45RBhighIL2KO or IL2KO-C4Tg Tn cells is not due to their de novo generation of Foxp3+ Treg cells or their inability to expand or survive following cell transfer. The results also support a crucial contribution of IL-2, produced by transferred T cells themselves, as well as CD28 stimulus to the activation/expansion of colitogenic T cells, and suggest possible acquisition of a suppressive activity by IL2KO, C4Tg, or IL2KO-C4Tg CD4+ Tn cells activated by self-antigens or intestinal microbes.
Fig. 6.
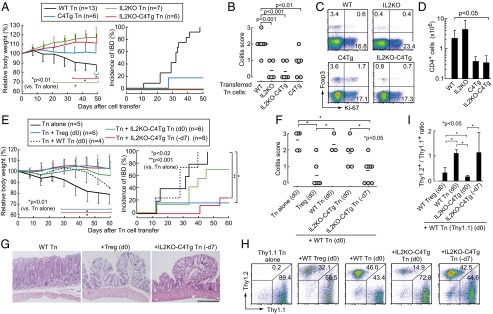
Suppression of colitis in RAG2−/− mice by IL2KO-C4Tg CD4+ T cells. (A) Body weight change (mean ± SDs) and incidence of IBD in RAG2−/− mice transferred with 1 × 105 CD45RBhighCD25−CD4+ Tn cells from indicated groups of mice. (B) Severity of colitis based on histological evaluation in each group of recipient mice 50 d after cell transfer. Circles represent individual mice. Horizontal bars indicate the means. (C) Intracellular Ki-67 and Foxp3 expression of transferred CD4+ T cells in mesenteric lymph nodes of recipient mice 50 d after cell transfer. (D) Numbers (mean ± SDs) of CD4+ T cells in mesenteric lymph nodes in the groups of mice shown in B. (E–I) Indicated numbers of RAG2−/− mice received 1 × 105 Thy1.1+ CD45RBhighCD4+ Tn cells on day 0 along with the same numbers of WT CD25+CD4+ Treg cells or CD45RBhighCD4+ Tn cells from designated groups of mice. Pretransfer of indicated Tn cell populations was performed on day −7 (−d7) in some mice as depicted. (E) Body weight change (mean ± SD) and incidence of IBD, (F) histological score of colitis, and (G) representative histology (H&E staining) of the colon from each group of recipient mice. (Scale bar, 20 μm.) (H and I) Thy1.1/Thy1.2 staining (H) and the ratio in CD4+ T cells (I) in mesenteric lymph nodes of recipient mice (shown in F) 60 d after cell transfer.
To determine whether IL2KO-C4Tg CD45RBhighCD4+ Tn cells were suppressive in vivo, we transferred them to RAG2−/− mice together with the same number of WT CD45RBhighCD4+ Tn cells as a cell mixture or with the latter inoculated 7 d later. By the simultaneous cotransfer, IL2KO-C4Tg CD45RBhighCD4+ Tn cells failed to effectively suppress IBD induction (Fig. 6 E–G). They exhibited less expansion than cotransferred WT T cells (Fig. 6 H and I), contrasting with cotransfer of Foxp3+ Treg cells, which proliferated more extensively than Foxp3− T cells during the first week after transfer (Fig. S4). In contrast, the development of IBD was markedly suppressed both clinically and histologically when IL2KO-C4Tg CD45RBhigh CD4+ Tn cells were inoculated 7 d before the transfer of WT CD45RBhigh CD4+ Tn cells (Fig. 6 E–G); 2 mo after the transfer, both the transferred populations had expanded to an equivalent level in the recipients (Fig. 6 H and I). Similarly, 7-d prior transfer of IL2KO or C4Tg CD4+ T cells effectively suppressed colitis development (Fig. S5 A and B). In addition, in vitro–activated ovalbumin (OVA)-specific TCR transgenic IL2KO CD4+ Tn cells suppressed OVA-specific Tn cells to mediate delayed type hypersensitivity reaction against OVA, as did OVA-specific natural Treg cells (Fig. S5C). Thus, IL2KO-C4Tg Tn cells are able to suppress the development of IBD dominantly when they become activated and expand to a certain extent before the activation of effector Tn cells.
CTLA-4–Dependent Formation of Self-Skewed TCR Repertoire in Developing T Cells and Treg Cells.
Whereas transgenic overexpression of CTLA-4 and IL-2 deficiency impaired the development of Foxp3+ Treg cells, it did not significantly alter the number of CD25−Foxp3−CD4SP thymocytes (Fig. 1 C and D). However, transgenic CTLA-4 expression significantly increased the proportion of Foxp3−CD4SP thymocytes expressing deletional TCR Vβ subfamilies (e.g., Vβ3, 5, 11, or 12) reactive with endogenous superantigens, with no increase in nondeletional Vβ subfamilies (e.g., Vβ2, 6, 8, or 14) on the BALB/c background (Fig. 7A). IL-2 deficiency also significantly increased the proportion of Foxp3−CD4SP thymocytes with deletional TCRVβ5, 11, or 12. Consequently, Foxp3−CD4SP thymocytes in IL2KO-C4Tg mice contained a higher proportion and a larger number of T cells with self-reactive TCR Vβ than those in WT mice (Fig. 7 A and B). Analysis of TCR transgenic mice for the repertoire of T cells expressing endogenous TCR α-chains together with the transgenic β-chain (i.e., frequency of Jα segment used by T cells expressing a particular endogenous Vα) revealed that the TCR repertoire of WT CD25+CD4SP Treg thymocytes was closer to IL2KO-C4Tg CD25−CD4SP thymocytes than to WT CD25−CD4SP thymocytes (Fig. S6). This self-skewing of the TCR repertoire in Foxp3−CD4SP thymocytes also occurred in TLC4Tg mice, indicating that the CD80/CD86-binding extracellular portion of CTLA-4, without the intracellular signaling domain, was sufficient for the skewing (Fig. 7C). Functionally, IL2KO-C4Tg CD25−CD4SP thymocytes and WT CD25+CD4SP Treg thymocytes showed higher proliferative activity to autologous splenic APCs than WT CD25−CD4SP thymocytes in an in vitro autologous mixed lymphocyte reaction (AMLR) in the presence of IL-2 and agonistic anti-CD28 (Fig. 7D). It contrasted with equivalent proliferative responses of these three populations to polyclonal stimulation with anti-CD3, anti-CD28, and IL-2 as shown in Fig. 3. Blockade of MHC class II inhibited the proliferation of IL2KO-C4Tg CD25−CD4SP thymocytes and WT CD25+CD4SP Treg thymocytes in AMLR, indicating that they proliferated through high reactivity to self-peptide/class II MHC expressed on autologous APCs.
Fig. 7.
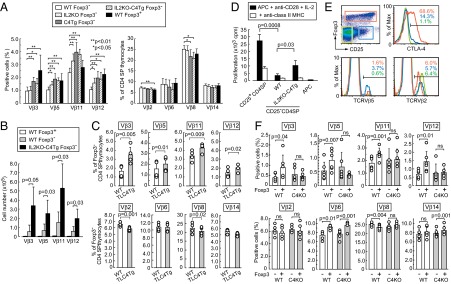
CTLA-4–dependent self-skewing of TCR repertoire in developing T cells and Foxp3+ Treg cells. (A and B) Proportions (A) and absolute numbers (B) (mean ± SD) of Foxp3+ or Foxp3− CD4SP thymocytes expressing each TCR Vβ subfamily in indicated groups of mice at 2–4 wk of age (n = 6–12 each). (C) Proportions of Foxp3− CD4SP thymocytes expressing indicated TCR Vβ subfamilies in 2- to 3-wk-old F1 mice of TLC4Tg C57BL/6 and normal BALB/c mice compared with WT F1 littermates. (D) Proliferation of indicated CD25+ or CD25− CD4SP BALB/c thymocytes cultured for 5 d with BALB/c splenic APCs, anti-CD28, and IL-2. Representative result (mean ± SD of triplicates) of three independent experiments is shown. (E) Subpopulations of BALB/c CD4SP thymocytes defined by the expression of CD25 and Foxp3 (Upper Left) and their expression of CTLA-4 and TCR Vβ5 or Vβ2. Representative of five independent stainings. (F) Proportions of Foxp3+ or Foxp3− CD4SP thymocytes expressing indicated TCR Vβ subfamilies in Treg-specific CTLA-4 conditional KO (C4KO) or WT BALB/c mice at 6 wk of age (n = 4–6 each). Circles represent individual mice; each column indicates the mean ± SD. ns, not significant.
In normal mice, some of the Foxp3−CD25+CD4SP thymocytes, which include Treg precursors with an immature (CD69high, HSAhigh) and Treg-like (CD25+, GITRhigh) phenotype (36), expressed CTLA-4 at a low level, whereas the majority of Foxp3+CD25+CD4SP thymocytes were CTLA-4high (Fig. 7E). The frequency of BALB/c T cells expressing the deletional TCR Vβ subfamilies were higher in Foxp3+ CD4SP thymocytes and Foxp3−CD25+CD4SP thymocytes, especially in the latter, than Foxp3−CD25−CD4SP thymocytes, with no significant increase in the proportions of nondeletional TCR Vβ subfamilies (Fig. 7 A, E, and F). Notably, this physiological self-skewing in the TCR Vβ repertoire of Foxp3+CD4SP thymocytes was absent in Treg-specific CTLA-4–deficient BALB/c mice (Fig. 7F).
Thus, conferment of a self-reactive TCR repertoire on Tconv cells by CTLA-4 overexpression, together with cancellation of physiological TCR self-skewing in Treg cells by Treg-specific CTLA-4 deficiency, indicates that CTLA-4 expression in developing Treg cells is essential for the formation of their self-skewed TCR repertoire with functional self-reactivity.
Discussion
In this study, we showed that Treg-like hyporesponsiveness and suppressive activity can be produced in Tconv cells simply by maintaining high expression of CTLA-4 and repressing IL-2 production. Once preactivated by antigenic stimulation, such IL-2 nonproducing and CTLA-4–expressing T cells exert potent in vivo and in vitro suppression in a similar manner to Foxp3+ natural Treg cells. Furthermore, a Treg-like self-skewed TCR repertoire can be bestowed on developing Tconv cells by CTLA-4 overexpression, whereas Treg-specific CTLA-4 deficiency cancels physiological acquisition of a self-reactive TCR repertoire by developing Foxp3+ Treg cells in the thymus. Together with the requirement of IL-2 for thymic Treg development, these findings illustrate essential roles of IL-2 and CTLA-4 for Treg function and development in the thymus and the periphery.
Depriving responder T cells of CD28 and/or IL-2 signal, especially both, is a potent mechanism of suppression. CTLA-4 expression is able to deprive CD28 signal from responder T cells as demonstrated by the acquisition of suppressive activity by CTLA-4–expressing Tconv cells and impaired Treg suppressive function in CTLA-4–deficient Treg cells (12, 37). CTLA-4, which is highly and constitutively expressed on C4Tg T cells and Foxp3+ Treg cells, inhibits the up-regulation and down-modulates the expression of CD80/CD86 expressed on APCs (12–14). Potent in vitro suppressive activity exhibited by TLC4Tg T cells and C4Tg T cells following preactivation indicates that interaction of the extracellular portion of CTLA-4 with CD80/CD86 on APCs, without intracellular signaling via CTLA-4, is sufficient for suppression. This CTLA-4–dependent hindrance of CD28 signal to responder T cells may also operate synergistically with other cell-intrinsic and -extrinsic functions of CTLA-4 in controlling immune responses. For example, CTLA-4 may increase T-cell motility and override the TCR-induced stop signal required for stable conjugate formation between T cells and APCs, although this CTLA-4 function did not affect motility of Treg cells (38, 39). CTLA-4 ligation may induce the tryptophan catabolizing enzyme indoleamine 2,3-dioxygenase, which locally depletes tryptophan and produces immunosuppressive tryptophan catabolites, although inhibition of the enzyme failed to abrogate suppression by activated IL2KO-C4Tg CD4+ T cells in these experiments (40, 41). Soluble CTLA-4 derived from activated C4Tg, TLC4Tg, or Foxp3+ Treg cells may block CD80/CD86 (42). The precise contribution of each mechanism of CTLA-4–dependent APC modulation is not fully understood and is currently under investigation.
In addition to these CTLA-4–dependent suppressive mechanisms, activated IL2KO-C4Tg T cells and Foxp3+ Treg cells are likely to exhibit suppressive activity via reducing IL-2 concentration in the surroundings by a combination of IL-2 absorption, their nonproduction of IL-2, and suppressed production by responder T cells. Deprivation of IL-2, a cytokine crucial for T-cell activation, differentiation, and survival, appears to induce apoptosis in partially activated responder T cells (11, 43). Although the exact extent of the contribution of IL-2 absorption to in vivo suppression remains to be determined, it is worth emphasizing that IL-2 nonproduction in itself by IL2KO-C4Tg T cells and natural Treg cells is a critical prerequisite for their in vivo suppressive function; if activated Treg cells could secrete IL-2, this would attenuate suppression.
Stimulation of IL2KO-C4Tg T cells via TCR is required for their effective exertion of suppression, as is the case with Foxp3+ natural Treg cells (9, 10). This TCR prestimulation of IL2KO-C4Tg T cells and Foxp3+ natural Treg cells up-regulates their expression of adhesion molecules including LFA-1 and ICAM-1 (with a conformational change of LFA-1), which are critically required for outcompeting naïve T cells for interaction with APCs and also for augmenting the interaction of CTLA-4 with CD80/CD86 on APCs (14). It also up-regulates the expression of the IL-2 receptor and may enhance their IL-2 absorption. Moreover, TCR stimulation by self-antigens or commensal microbial antigens induces in vivo expansion of antigen-specific IL2KO-C4Tg T cells and Foxp3+ Treg cells, leading to their local numerical dominance over antigen-specific effector T cells (44). Thus, TCR stimulation is able to expand IL-2–hypoproducing and CTLA-4–expressing T cells and evoke their suppressive activity in vivo and in vitro.
These roles of IL-2, CTLA-4, and TCR stimulation for T cell–mediated suppression support the notion that the IL-2– and CTLA-4–dependent suppressive mechanism, which is evoked by TCR stimulation, can be a core and ubiquitous mechanism of suppression mediated by natural Foxp3+ Treg cells. Some of the other functions of natural Foxp3+ Treg cells may augment the core suppressive mechanism (6). For example, IL-10 (and TGF-β), which is secreted by chronically stimulated Treg cells and other T cells, is able to down-modulate CD80/CD86 expression by DCs and reduce IL-2 production by responder T cells, thus acting in synergy with the IL-2– and CTLA-4–dependent suppressive mechanism (45, 46). In addition, expression of specific transcriptional factors of Th1, Th2, Tfh, and Th17 cells may be required for Treg cells to effectively suppress each type of inflammation (e.g., via controlling their migration to inflammation sites) (3, 47–51). It is under investigation whether installment of additional Treg cell properties, including the expression of Th type–specific transcriptional factors, would enable IL2KO-C4Tg T cells to control specific inflammatory responses more efficiently.
CTLA-4 is also a key molecule for forming a self-skewed TCR repertoire in developing T cells, as indicated by the following findings. Forced expression of full length or tailless CTLA-4 and resulting reduction of CD28 signaling skewed the TCR repertoire of developing Foxp3− Tconv cells toward higher self-reactivity without significantly affecting their cell number (29, 52, 53). In line with this finding, the TCR repertoire of CD28-deficient Tconv cells was reported to be slightly more self-reactive than WT Tconv cells (29, 52, 53). On the other hand, Treg-specific CTLA-4 deficiency cancelled physiological self-skewing of their TCR repertoire. In addition, CTLA-4–deficient TCR transgenic mice bore an altered TCR repertoire in both Treg and Tconv cells (54). These findings when taken together indicate that a common CTLA-4–dependent mechanism of TCR self-skewing operates in developing Foxp3− Tconv cells overexpressing CTLA-4, as well as Foxp3+ natural Treg cells, which physiologically express the molecule in the course of their thymic development from the Foxp3−CD25+ Treg precursor stage to the Foxp3+ stage. That is, given that CD28 signaling physiologically amplifies TCR signal (55), attenuation of TCR signal due to the CTLA-4–dependent CD28 signal reduction allows some of those normally deleted highly self-reactive T cells to escape negative selection while inhibiting positive selection of some non- or low self-reactive T cells; these events together shift the whole TCR repertoire toward higher self-reactivity (Fig. S7). It is also of note that, without CD28-dependent amplification of the TCR signal in CD28-deficient mice and C4Tg or TLC4Tg mice, developing T cells fail to generate a TCR signal sufficiently potent to induce Foxp3 expression, resulting in specific reduction of Foxp3+ Treg cells in these mice (Fig. 1). In contrast, in Treg-specific CTLA-4–deficient mice, a TCR signal amplified by a CD28 signal (without CTLA-4–dependent signal reduction) is sufficiently potent to induce Foxp3 expression in some T cells, leading to the development of Foxp3+ cells without self-skewing in their TCR repertoire. This model of CTLA-4–dependent Treg development and self-skewing of their TCR repertoire explains how Treg and Tconv cells acquire respective TCR repertoires that are apparently distinct but overlapping to a certain extent (22). It remains to be determined how Foxp3-dependent and -independent mechanisms control CTLA-4 expression and IL-2 nonproduction, which also appears to contribute somehow to Treg TCR repertoire skewing (56).
We have thus shown that self-reactive Treg-like suppressive T cells can be constructed from naïve T cells simply by targeting IL-2 and CTLA-4, without Foxp3. This minimalistic construct of Treg-like cells implies that their suppressive properties could be shared by other reported Foxp3− suppressive T cells, which generally appear to be IL-2 hypoproducing and readily express CTLA-4 on antigenic stimulation, and are suppressive in short-term in vitro suppression assays (46, 57–64) and in an experimental IBD model (65). Furthermore, any antigen-stimulated non-Treg cells are potentially able to temporarily exhibit suppressive activity on naïve T cells in a negative feedback manner when they cease to produce IL-2 and still retain CTLA-4 expression (or when CTLA-4 expression inhibits IL-2 production) in the course of immune responses (Fig. S2). However, a crucial difference between such Foxp3− suppressive T cells and Foxp3+ natural Treg cells is that, besides the latter’s self-skewed TCR repertoire and repression of inflammatory cytokine production, natural Treg cells have more stable IL-2 repression and CTLA-4 expression because of their continuous Foxp3 expression and Treg-specific epigenome status (7, 8, 56, 66, 67). Another implication of our results is that antigen-specific effector T cells can be converted to antigen-specific suppressive T cells by manipulating only two molecular species: IL-2 and CTLA-4. This finding can be exploited to develop novel ways of immune suppression in a variety of clinical settings by targeting these molecules not only in Foxp3+ Treg cells but also in Tconv cells.
Materials and Methods
Mice.
CTLA-4 transgenic (C4Tg) and tailless CTLA-4 transgenic (TLC4Tg) mice express full-length and intracellular domain–deleted CTLA-4, respectively, under the control of the human CD2 promoter (29). The following mouse strains were maintained in our animal facility: Foxp3-Ires-Cre (FIC) mice that express Cre under the Foxp3 promoter, CTLA-4 conditional KO (C4KO) mice that are FIC mice with floxed CTLA-4 genes (12), IL-2–deficient (IL2KO) mice, CD28-deficient (CD28KO) mice, DO11.10 TCR transgenic mice, RAG2−/− mice, Thy1.1+ mice, and Scurfy mice all on the BALB/c background, and C4Tg mice backcrossed to BALB/c mice for at least six times. TLC4Tg mice were on the C57BL/6 background and made deficient in endogenous CTLA-4 by mating with C57BL/6 CTLA-4−/− mice in experiments in Figs. 3 and 5. In Fig. 1, IL2KO-TLC4Tg and TLC4Tg mice were on a mixed background generated from C57BL/6 TLC4Tg mice and BALB/c IL2KO mice. All animal experiments were conducted according to the institutional guidelines for animal welfare under approved protocols from the Animal Experiment Committee of Osaka University or Kyoto University.
Flow Cytometric Analysis and Reagents.
Before staining with specific antibodies, Fc receptors were blocked with anti-CD16/CD32 (2.4G2). For analysis of cell surface antigens, 0.1 μg/mL of propidium iodide (PI) solution (Dojindo) was added to exclude dead cells. For intracellular Foxp3, Ki-67, or CTLA-4 staining, cells were fixed and permeabilized with the Foxp3-staining kit from eBioscience. For intracellular cytokine staining, cells were restimulated with 20 ng/mL phorbol 12-myristate 13-acetate (PMA) and 1 μM ionomycin in the presence of GolgiStop (BD Biosciences) for 4 h. Stained cells were analyzed with FACS Calibur or Canto (BD Biosciences). Flowjo software (Tree Star) was used for data analysis. See SI Materials and Methods for additional details of reagents.
Cell Preparation.
Cell suspensions from lymph nodes and thymus were stained with specific antibodies and sorted with MoFlo (Beckman Coulter) with a typical purity of more than 97%. Cells were cultured in RPMI 1640 medium with 10% (vol/vol) FCS, 100 U/mL penicillin, 100 μg/mL streptomycin, and 50 μM 2-mercaptoethanol. For activated T cells, CD25−CD4+ T cells were stimulated with plate-bound anti-CD3 and anti-CD28 (1 μg/mL each) in the presence of 100 U/mL recombinant IL-2 for 2 d. After preactivation, live cells were separated by density gradient centrifugation with lympholyte-M (Cedarlane). Dendritic cells were isolated by magnetic sorting with magnetic bead–conjugated anti-CD11c (Milteny Biotec) from spleens treated with Liberase Blendzyme II (Roche Diagnostics). T cells were depleted from splenocytes by magnetic sorting with bead-conjugated anti-Thy1.
Proliferation and Suppression Assays.
For the proliferation assay, CD4+ T cells (3.5 × 104) were cultured in a round-bottom 96-well plate with twice the number of 18.5 Gy–irradiated splenocytes (APCs) and 0.5 μg/mL anti-CD3 for 3 d. In some cases, 0.5 μg/mL anti-CD28 and/or 200 U/mL IL-2 were added. For the suppression assay, Thy1.1+CD25−CD4+ responder T cells (5 × 104) were labeled with 1 μM CFSE (Dojindo) and cultured with 0.5 μg/mL anti-CD3 and 1 × 105 X-ray–irradiated splenocytes for 3 d in the presence of the same or graded numbers of Thy1.2+ T cells. Cells were then stained with anti-Thy1.1, anti-Thy1.2, and PI, and fluorescence intensity and cell numbers were determined with FACS Calibur. For suppression assays with anti-CD3 stimulation (Figs. 4 A–E and 5 A and B), CD25−CD4+ Thy1.2+ Tn cells were obtained from WT, IL2KO, C4Tg, or IL2KO-C4Tg mice on DO11.10 TCR transgenic background to avoid cell activation due to systemic autoimmunity in WT IL2KO mice. In autologous mixed lymphocyte reactions, 5 × 104 thymocytes were cultured with 1 × 105 Thy1+ cell-depleted splenocytes, 0.5 μg/mL anti-CD28, and 200 U/mL IL-2 for 5 d with or without 1 μg/mL anti-MHC class II (M5/114.15.2). 3H-thymidine incorporation during the last 6 h of culture was measured. For differentiation to Th1 and Th2 cells, CFSE-labeled Thy1.1+ CD25−CD4+ Tn cells were cocultured with Thy1.2+ CD25+CD4+ WT Treg cells or preactivated CD25−CD4+ T cells from WT or IL2KO-C4Tg mice, for 4 d under Th1 (2 ng/mL IL-12 and 10 μg/mL anti–IL-4) or Th2 (2 ng/mL IL-4 and 10 μg/mL anti–IL-12) conditions in the presence of anti-CD3 (0.5 μg/mL) and X-ray–irradiated splenocytes. For the suppression assay with CTLA-4–transduced IL-2 knocked down human T cells, CD25−CD45RA+CD4+ naive T cells from peripheral blood were activated with CD3/CD28-coated beads and infected with lentiviral vector pLenti6/V5 (Invitrogen) containing human CTLA-4 gene or microRNA sequences against human IL-2. See also SI Materials and Methods.
ELISA.
T cells (1 × 105) were cultured with 1 × 105 T cell–depleted X-ray--irradiated splenocytes, anti-CD3, and 10 U/mL of recombinant mouse IL-2 in 96-well round-bottom plates for 20 h. To analyze absorption of human IL-2, 250 pg/mL of recombinant human IL-2 was added instead of mouse IL-2. Mouse IL-2 and IFN-γ concentration in the culture supernatant was determined using the mouse IL-2 and IFN-γ detection kit (eBioscience), respectively. Human IL-2 concentration was determined using the human IL-2 detection kit (BD Biosciences).
Quantitative PCR Analysis.
RNA was extracted from cell lysates with RNAeasy (QIAGEN), and cDNAs were prepared with random primers and reverse transcriptase III (Invitrogen). The amount of each cDNA was determined with LightCycler 480 (Roche) with the specific primers listed in Table S1. The quantity of RNA was normalized with the amounts of mRNA of Cd4.
Fluorescence Microscopy.
Cells from mice on a DO11.10 TCR transgenic background were stained with 1.67 μM Cell Trace Far Red DDAO-SE (Invitrogen) or 3 μM CFSE. Far Red DDAO-SE–stained T cells (2 × 104) and CFSE-stained T cells (2 × 104) were incubated with 4 × 103 splenic DCs and 1 μM OVA peptide in 96-well round-bottom plates. Two days later, cell nuclei were stained with 5 μg/mL Hoechest 33342 (Invitrogen), gently transferred to a 96-well thin Nunc plate, and analyzed with Cellomics ArrayScan (Thermo Fisher Scientific). Colonies were identified with clusters of nuclei, and the area stained with green (CFSE) or red (Far Red DDAO-SE) dye in the colonies was determined using Cellomics software.
Flow Cytometric Analysis of DCs.
CD11c+ splenic DCs from WT BALB/c mice were cocultured for 2 d with CD25+CD4+ Treg cells or CD25−CD4+ Tn cells from WT, C4Tg, IL2KO, IL2KO-C4Tg, or TLC4Tg mice in the presence of anti-CD3 and 100 U/mL IL-2 in 96-well round-bottom plates. On day 2, collected cells were stained with specific antibodies for CD11c, CD80, CD86, CD40 and I-Ad (MHC class II), and PI.
Colitis Model.
CD45RBhighCD25−CD4+ Tn cells from WT, IL2KO, C4Tg, IL2KO-C4Tg, and Thy1.1 mice and CD45RBlow CD25+CD4+ WT Treg cells were sorted with MoFlo. In each group, 1 × 105 cells were transferred into RAG2−/− mice. Body weight and signs of IBD were monitored twice a week for 2 mo. Signs of IBD include diarrhea, reduction in body weight to less than 90% of initial weight, or death. After 2 mo, mice were killed, and colons were fixed in 10% formalin. Lymphocytes in mesenteric lymph nodes were counted, stained with antibodies for Thy1.1, Thy1.2, CD4, Foxp3, and Ki-67, and analyzed with FACS Calibur. Colon sections were stained with H&E, and histological severity of colitis was scored (0 = intact colon indistinguishable from control BALB/c mice, 1 = mild colitis with limited cellular infiltration and slight epithelial cell hyperplasia, 2 = moderate colitis with cellular infiltration and mild distraction of mucosa, 3 = severe colitis with extensive cellular infiltration and marked distraction of mucosa).
TCR Repertoire Analysis.
Thymocytes were stained with anti-CD4, anti-CD8, anti-Foxp3, and antibodies specific for TCRVβ2, Vβ3, Vβ5, Vβ6, Vβ8.1/8.2, Vβ8.3, Vβ11, Vβ12, and Vβ14, and proportions of CD4SP thymocytes expressing each TCRVβ subfamily among Foxp3− and Foxp3+ CD4SP thymocytes were determined with FACS Canto II (BD Biosciences). For sequencing of TCRJα, CDR3 regions with TCRVα10 were amplified from CD4SP thymocytes expressing DO11.10 transgenic TCR Vβ. See also SI Materials and Methods.
Statistical Method.
One-way ANOVA followed by Tukey-Kramer’s test was used to analyze the difference among more than two groups in Figs. 1D, 3A, 4 C and E, and 6 F and I. A paired t test was applied in Fig. 7 C and F. Log-rank statistics were applied to analyze survival and incidence of IBD. An unpaired Student t test was used for other statistical analyses.
Supplementary Material
Acknowledgments
We thank J. B. Wing and D. Adeegbe for critically reading the manuscript; M. Matsushita for histological preparation; A. Shibata for maintaining mice; R. Ishii for expertise in cell sorting; T. Ueno for analysis with fluorescence microscopy; and A. Tanaka, T. Murase, H. Uryu, H. Noda, N. Ohkura, and N. Sakaguchi for technical help and valuable discussion. This study was supported by Grants-in-Aid for Specially Promoted Research 20002007 from the Ministry of Education, Sports, and Culture and the Ministry of Human Welfare of Japan and CREST from the Japan Science and Technology Agency.
Footnotes
The authors declare no conflict of interest.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1307185110/-/DCSupplemental.
References
- 1.Sakaguchi S, Yamaguchi T, Nomura T, Ono M. Regulatory T cells and immune tolerance. Cell. 2008;133(5):775–787. doi: 10.1016/j.cell.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 2.Vignali DA, Collison LW, Workman CJ. How regulatory T cells work. Nat Rev Immunol. 2008;8(7):523–532. doi: 10.1038/nri2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Josefowicz SZ, Lu LF, Rudensky AY. Regulatory T cells: Mechanisms of differentiation and function. Annu Rev Immunol. 2012;30:531–564. doi: 10.1146/annurev.immunol.25.022106.141623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hori S, Nomura T, Sakaguchi S. Control of regulatory T cell development by the transcription factor Foxp3. Science. 2003;299(5609):1057–1061. doi: 10.1126/science.1079490. [DOI] [PubMed] [Google Scholar]
- 5.Fontenot JD, Gavin MA, Rudensky AY. Foxp3 programs the development and function of CD4+CD25+ regulatory T cells. Nat Immunol. 2003;4(4):330–336. doi: 10.1038/ni904. [DOI] [PubMed] [Google Scholar]
- 6.Shevach EM. Mechanisms of foxp3+ T regulatory cell-mediated suppression. Immunity. 2009;30(5):636–645. doi: 10.1016/j.immuni.2009.04.010. [DOI] [PubMed] [Google Scholar]
- 7.Ono M, et al. Foxp3 controls regulatory T-cell function by interacting with AML1/Runx1. Nature. 2007;446(7136):685–689. doi: 10.1038/nature05673. [DOI] [PubMed] [Google Scholar]
- 8.Wu Y, et al. FOXP3 controls regulatory T cell function through cooperation with NFAT. Cell. 2006;126(2):375–387. doi: 10.1016/j.cell.2006.05.042. [DOI] [PubMed] [Google Scholar]
- 9.Takahashi T, et al. Immunologic self-tolerance maintained by CD25+CD4+ naturally anergic and suppressive T cells: Induction of autoimmune disease by breaking their anergic/suppressive state. Int Immunol. 1998;10(12):1969–1980. doi: 10.1093/intimm/10.12.1969. [DOI] [PubMed] [Google Scholar]
- 10.Thornton AM, Shevach EM. CD4+CD25+ immunoregulatory T cells suppress polyclonal T cell activation in vitro by inhibiting interleukin 2 production. J Exp Med. 1998;188(2):287–296. doi: 10.1084/jem.188.2.287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Pandiyan P, Zheng L, Ishihara S, Reed J, Lenardo MJ. CD4+CD25+Foxp3+ regulatory T cells induce cytokine deprivation-mediated apoptosis of effector CD4+ T cells. Nat Immunol. 2007;8(12):1353–1362. doi: 10.1038/ni1536. [DOI] [PubMed] [Google Scholar]
- 12.Wing K, et al. CTLA-4 control over Foxp3+ regulatory T cell function. Science. 2008;322(5899):271–275. doi: 10.1126/science.1160062. [DOI] [PubMed] [Google Scholar]
- 13.Oderup C, Cederbom L, Makowska A, Cilio CM, Ivars F. Cytotoxic T lymphocyte antigen-4-dependent down-modulation of costimulatory molecules on dendritic cells in CD4+ CD25+ regulatory T-cell-mediated suppression. Immunology. 2006;118(2):240–249. doi: 10.1111/j.1365-2567.2006.02362.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Onishi Y, Fehervari Z, Yamaguchi T, Sakaguchi S. Foxp3+ natural regulatory T cells preferentially form aggregates on dendritic cells in vitro and actively inhibit their maturation. Proc Natl Acad Sci USA. 2008;105(29):10113–10118. doi: 10.1073/pnas.0711106105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Qureshi OS, et al. Trans-endocytosis of CD80 and CD86: A molecular basis for the cell-extrinsic function of CTLA-4. Science. 2011;332(6029):600–603. doi: 10.1126/science.1202947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Teft WA, Kirchhof MG, Madrenas J. A molecular perspective of CTLA-4 function. Annu Rev Immunol. 2006;24:65–97. doi: 10.1146/annurev.immunol.24.021605.090535. [DOI] [PubMed] [Google Scholar]
- 17.Yokosuka T, et al. Spatiotemporal basis of CTLA-4 costimulatory molecule-mediated negative regulation of T cell activation. Immunity. 2010;33(3):326–339. doi: 10.1016/j.immuni.2010.09.006. [DOI] [PubMed] [Google Scholar]
- 18.Fontenot JD, Rasmussen JP, Gavin MA, Rudensky AY. A function for interleukin 2 in Foxp3-expressing regulatory T cells. Nat Immunol. 2005;6(11):1142–1151. doi: 10.1038/ni1263. [DOI] [PubMed] [Google Scholar]
- 19.Tang Q, et al. Distinct roles of CTLA-4 and TGF-β in CD4+CD25+ regulatory T cell function. Eur J Immunol. 2004;34(11):2996–3005. doi: 10.1002/eji.200425143. [DOI] [PubMed] [Google Scholar]
- 20.Almeida AR, Legrand N, Papiernik M, Freitas AA. Homeostasis of peripheral CD4+ T cells: IL-2R α and IL-2 shape a population of regulatory cells that controls CD4+ T cell numbers. J Immunol. 2002;169(9):4850–4860. doi: 10.4049/jimmunol.169.9.4850. [DOI] [PubMed] [Google Scholar]
- 21.Tai X, Cowan M, Feigenbaum L, Singer A. CD28 costimulation of developing thymocytes induces Foxp3 expression and regulatory T cell differentiation independently of interleukin 2. Nat Immunol. 2005;6(2):152–162. doi: 10.1038/ni1160. [DOI] [PubMed] [Google Scholar]
- 22.Hsieh CS, Lee HM, Lio CW. Selection of regulatory T cells in the thymus. Nat Rev Immunol. 2012;12(3):157–167. doi: 10.1038/nri3155. [DOI] [PubMed] [Google Scholar]
- 23.Bautista JL, et al. Intraclonal competition limits the fate determination of regulatory T cells in the thymus. Nat Immunol. 2009;10(6):610–617. doi: 10.1038/ni.1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hsieh CS, Zheng Y, Liang Y, Fontenot JD, Rudensky AY. An intersection between the self-reactive regulatory and nonregulatory T cell receptor repertoires. Nat Immunol. 2006;7(4):401–410. doi: 10.1038/ni1318. [DOI] [PubMed] [Google Scholar]
- 25.Itoh M, et al. Thymus and autoimmunity: Production of CD25+CD4+ naturally anergic and suppressive T cells as a key function of the thymus in maintaining immunologic self-tolerance. J Immunol. 1999;162(9):5317–5326. [PubMed] [Google Scholar]
- 26.Jordan MS, et al. Thymic selection of CD4+CD25+ regulatory T cells induced by an agonist self-peptide. Nat Immunol. 2001;2(4):301–306. doi: 10.1038/86302. [DOI] [PubMed] [Google Scholar]
- 27.Pacholczyk R, Ignatowicz H, Kraj P, Ignatowicz L. Origin and T cell receptor diversity of Foxp3+CD4+CD25+ T cells. Immunity. 2006;25(2):249–259. doi: 10.1016/j.immuni.2006.05.016. [DOI] [PubMed] [Google Scholar]
- 28.Wong J, et al. Adaptation of TCR repertoires to self-peptides in regulatory and nonregulatory CD4+ T cells. J Immunol. 2007;178(11):7032–7041. doi: 10.4049/jimmunol.178.11.7032. [DOI] [PubMed] [Google Scholar]
- 29.Takahashi S, et al. In vivo overexpression of CTLA-4 suppresses lymphoproliferative diseases and thymic negative selection. Eur J Immunol. 2005;35(2):399–407. doi: 10.1002/eji.200324746. [DOI] [PubMed] [Google Scholar]
- 30.Engelhardt JJ, Sullivan TJ, Allison JP. CTLA-4 overexpression inhibits T cell responses through a CD28-B7-dependent mechanism. J Immunol. 2006;177(2):1052–1061. doi: 10.4049/jimmunol.177.2.1052. [DOI] [PubMed] [Google Scholar]
- 31.Tang Q, et al. Cutting edge: CD28 controls peripheral homeostasis of CD4+CD25+ regulatory T cells. J Immunol. 2003;171(7):3348–3352. doi: 10.4049/jimmunol.171.7.3348. [DOI] [PubMed] [Google Scholar]
- 32.Hwang KW, et al. Transgenic expression of CTLA-4 controls lymphoproliferation in IL-2-deficient mice. J Immunol. 2004;173(9):5415–5424. doi: 10.4049/jimmunol.173.9.5415. [DOI] [PubMed] [Google Scholar]
- 33.Hoyer KK, Wolslegel K, Dooms H, Abbas AK. Targeting T cell-specific costimulators and growth factors in a model of autoimmune hemolytic anemia. J Immunol. 2007;179(5):2844–2850. doi: 10.4049/jimmunol.179.5.2844. [DOI] [PubMed] [Google Scholar]
- 34.Zheng L, Sharma R, Gaskin F, Fu SM, Ju ST. A novel role of IL-2 in organ-specific autoimmune inflammation beyond regulatory T cell checkpoint: Both IL-2 knockout and Fas mutation prolong lifespan of Scurfy mice but by different mechanisms. J Immunol. 2007;179(12):8035–8041. doi: 10.4049/jimmunol.179.12.8035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Powrie F, Leach MW, Mauze S, Caddle LB, Coffman RL. Phenotypically distinct subsets of CD4+ T cells induce or protect from chronic intestinal inflammation in C. B-17 scid mice. Int Immunol. 1993;5(11):1461–1471. doi: 10.1093/intimm/5.11.1461. [DOI] [PubMed] [Google Scholar]
- 36.Lio CW, Hsieh CS. A two-step process for thymic regulatory T cell development. Immunity. 2008;28(1):100–111. doi: 10.1016/j.immuni.2007.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zheng Y, et al. Acquisition of suppressive function by activated human CD4+ CD25- T cells is associated with the expression of CTLA-4 not FoxP3. J Immunol. 2008;181(3):1683–1691. doi: 10.4049/jimmunol.181.3.1683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Schneider H, et al. Reversal of the TCR stop signal by CTLA-4. Science. 2006;313(5795):1972–1975. doi: 10.1126/science.1131078. [DOI] [PubMed] [Google Scholar]
- 39.Lu Y, Schneider H, Rudd CE. Murine regulatory T cells differ from conventional T cells in resisting the CTLA-4 reversal of TCR stop-signal. Blood. 2012;120(23):4560–4570. doi: 10.1182/blood-2012-04-421420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fallarino F, et al. Modulation of tryptophan catabolism by regulatory T cells. Nat Immunol. 2003;4(12):1206–1212. doi: 10.1038/ni1003. [DOI] [PubMed] [Google Scholar]
- 41.Grohmann U, et al. CTLA-4-Ig regulates tryptophan catabolism in vivo. Nat Immunol. 2002;3(11):1097–1101. doi: 10.1038/ni846. [DOI] [PubMed] [Google Scholar]
- 42.Gerold KD, et al. The soluble CTLA-4 splice variant protects from type 1 diabetes and potentiates regulatory T-cell function. Diabetes. 2011;60(7):1955–1963. doi: 10.2337/db11-0130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Boyman O, Sprent J. The role of interleukin-2 during homeostasis and activation of the immune system. Nat Rev Immunol. 2012;12(3):180–190. doi: 10.1038/nri3156. [DOI] [PubMed] [Google Scholar]
- 44.Fisson S, et al. Continuous activation of autoreactive CD4+ CD25+ regulatory T cells in the steady state. J Exp Med. 2003;198(5):737–746. doi: 10.1084/jem.20030686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Steinbrink K, Wölfl M, Jonuleit H, Knop J, Enk AH. Induction of tolerance by IL-10-treated dendritic cells. J Immunol. 1997;159(10):4772–4780. [PubMed] [Google Scholar]
- 46.Vieira PL, et al. IL-10-secreting regulatory T cells do not express Foxp3 but have comparable regulatory function to naturally occurring CD4+CD25+ regulatory T cells. J Immunol. 2004;172(10):5986–5993. doi: 10.4049/jimmunol.172.10.5986. [DOI] [PubMed] [Google Scholar]
- 47.Koch MA, et al. The transcription factor T-bet controls regulatory T cell homeostasis and function during type 1 inflammation. Nat Immunol. 2009;10(6):595–602. doi: 10.1038/ni.1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zheng Y, et al. Regulatory T-cell suppressor program co-opts transcription factor IRF4 to control T(H)2 responses. Nature. 2009;458(7236):351–356. doi: 10.1038/nature07674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Linterman MA, et al. Foxp3+ follicular regulatory T cells control the germinal center response. Nat Med. 2011;17(8):975–982. doi: 10.1038/nm.2425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chung Y, et al. Follicular regulatory T cells expressing Foxp3 and Bcl-6 suppress germinal center reactions. Nat Med. 2011;17(8):983–988. doi: 10.1038/nm.2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chaudhry A, et al. CD4+ regulatory T cells control TH17 responses in a Stat3-dependent manner. Science. 2009;326(5955):986–991. doi: 10.1126/science.1172702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Buhlmann JE, Elkin SK, Sharpe AH. A role for the B7-1/B7-2:CD28/CTLA-4 pathway during negative selection. J Immunol. 2003;170(11):5421–5428. doi: 10.4049/jimmunol.170.11.5421. [DOI] [PubMed] [Google Scholar]
- 53.Pobezinsky LA, et al. Clonal deletion and the fate of autoreactive thymocytes that survive negative selection. Nat Immunol. 2012;13(6):569–578. doi: 10.1038/ni.2292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Verhagen J, et al. CTLA-4 controls the thymic development of both conventional and regulatory T cells through modulation of the TCR repertoire. Proc Natl Acad Sci USA. 2013;110(3):E221–E230. doi: 10.1073/pnas.1208573110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Acuto O, Mise-Omata S, Mangino G, Michel F. Molecular modifiers of T cell antigen receptor triggering threshold: The mechanism of CD28 costimulatory receptor. Immunol Rev. 2003;192:21–31. doi: 10.1034/j.1600-065x.2003.00034.x. [DOI] [PubMed] [Google Scholar]
- 56.Ohkura N, et al. T cell receptor stimulation-induced epigenetic changes and Foxp3 expression are independent and complementary events required for Treg cell development. Immunity. 2012;37(5):785–799. doi: 10.1016/j.immuni.2012.09.010. [DOI] [PubMed] [Google Scholar]
- 57.Esplugues E, et al. Control of TH17 cells occurs in the small intestine. Nature. 2011;475(7357):514–518. doi: 10.1038/nature10228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jago CB, Yates J, Câmara NO, Lechler RI, Lombardi G. Differential expression of CTLA-4 among T cell subsets. Clin Exp Immunol. 2004;136(3):463–471. doi: 10.1111/j.1365-2249.2004.02478.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lombardi G, Sidhu S, Batchelor R, Lechler R. Anergic T cells as suppressor cells in vitro. Science. 1994;264(5165):1587–1589. doi: 10.1126/science.8202711. [DOI] [PubMed] [Google Scholar]
- 60.Shimizu J, Moriizumi E. CD4+CD25- T cells in aged mice are hyporesponsive and exhibit suppressive activity. J Immunol. 2003;170(4):1675–1682. doi: 10.4049/jimmunol.170.4.1675. [DOI] [PubMed] [Google Scholar]
- 61.Kim HJ, Verbinnen B, Tang X, Lu L, Cantor H. Inhibition of follicular T-helper cells by CD8(+) regulatory T cells is essential for self tolerance. Nature. 2010;467(7313):328–332. doi: 10.1038/nature09370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Okamura T, et al. CD4+CD25-LAG3+ regulatory T cells controlled by the transcription factor Egr-2. Proc Natl Acad Sci USA. 2009;106(33):13974–13979. doi: 10.1073/pnas.0906872106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zhang ZX, Yang L, Young KJ, DuTemple B, Zhang L. Identification of a previously unknown antigen-specific regulatory T cell and its mechanism of suppression. Nat Med. 2000;6(7):782–789. doi: 10.1038/77513. [DOI] [PubMed] [Google Scholar]
- 64.Rifa’i M, Kawamoto Y, Nakashima I, Suzuki H. Essential roles of CD8+CD122+ regulatory T cells in the maintenance of T cell homeostasis. J Exp Med. 2004;200(9):1123–1134. doi: 10.1084/jem.20040395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Barthlott T, Kassiotis G, Stockinger B. T cell regulation as a side effect of homeostasis and competition. J Exp Med. 2003;197(4):451–460. doi: 10.1084/jem.20021387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Marson A, et al. Foxp3 occupancy and regulation of key target genes during T-cell stimulation. Nature. 2007;445(7130):931–935. doi: 10.1038/nature05478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zheng Y, et al. Genome-wide analysis of Foxp3 target genes in developing and mature regulatory T cells. Nature. 2007;445(7130):936–940. doi: 10.1038/nature05563. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.