

Abstract
The regulatory protein PmrA controls expression of lipopolysaccharide (LPS) modification genes in Salmonella enterica serovar Typhimurium, the etiologic agent of human gastroenteritis and murine typhoid fever. PmrA-dependent LPS modifications confer resistance to serum, Fe3+, and several antimicrobial peptides, suggesting that the pmrA gene is required for Salmonella virulence. We now report that, surprisingly, a pmrA null mutant is actually hypervirulent when inoculated i.p. into C3H/HeN mice. We establish that the PmrA protein binds to the promoter and represses transcription of ssrB, a virulence regulatory gene required for expression of the Spi/Ssa type three-secretion system inside macrophages. The pmrA mutant displayed heightened expression of SsrB-dependent genes and faster Spi/Ssa-dependent macrophage killing than wild-type Salmonella. A mutation in the ssrB promoter that abolished repression by the PmrA protein rendered Salmonella as hypervirulent as the pmrA null mutant. The antivirulence function of the PmrA protein may limit the acute phase of Salmonella infection, thereby enhancing pathogen persistence in host tissues.
Keywords: Salmonella pathogenicity island 2, cytotoxicity
Pathogens possess virulence genes that enable them to invade nonphagocytic cells, to survive within host phagocytic cells, to resist antimicrobial agents and the immune response presented by the host, and/or to kill host cells (1, 2). Paradoxically, pathogens can also have genes that actually reduce their virulence. That is, inactivation of certain genes can decrease the median lethal dose (i.e., LD50) and/or enhance pathogen growth in host tissues (3, 4). However, the function(s) of such antivirulence factors and the contribution(s) they make to a pathogen’s lifestyle remain largely unknown. Here, we establish that an ancestral Salmonella regulator is an antivirulence protein that represses expression of a horizontally acquired locus necessary for survival inside phagocytic cells.
The ancestral two-component system PmrA/PmrB is the major regulator of lipopolysaccharide (LPS) modification genes in Salmonella enterica serovar Typhimurium, the etiologic agent of human gastroenteritis and murine typhoid fever. The regulator PmrA is activated when its cognate sensor PmrB detects mildly acidic pH (5) or the presence of Fe3+ or Al3+ (6), and also when Salmonella experiences low Mg2+, which is detected by the sensor PhoQ (7). Together with the regulator PhoP, PhoQ forms a two-component system that governs virulence functions in Salmonella (8). The PhoP-activated PmrD protein (9) activates the PmrA protein at the posttranslational level (10). The PmrA/PmrB system is active when Salmonella is inside macrophages and during infection of mice (11). PmrA-dependent modifications of the LPS increase bacterial resistance to the antibiotic polymyxin B (6) and host-derived antimicrobial peptides (6, 12), to serum complement (13), and to high concentrations of Fe3+ (14); and they reduce the proinflammatory properties of the LPS (15). However, inactivation of the pmrA gene was reported to have minimal effects on Salmonella’s ability to cause a lethal infection in BALB/c mice when inoculated orally and no effect when inoculated i.p. (16).
Type-three secretion systems (T3SSs) are specialized molecular machines that certain Gram-negative bacteria use to deliver effector proteins into eukaryotic cells (17). These effectors typically manipulate host cell functions, thereby mediating bacterial entry to and/or survival within host tissues (17, 18). Salmonella encodes a T3SS—termed Spi/Ssa—on the Salmonella pathogenicity island (SPI)-2 that is unique in that it translocates effectors into and across the phagosomal membrane (19) and is necessary for bacterial survival within macrophages (20). Expression of spi/ssa genes depends on the SPI-2–encoded SsrB/SpiR two-component system (21), which consists of the sensor SpiR (also known as SsrA) and the DNA binding regulator SsrB (22). An ssrB null mutant is highly attenuated for virulence in mice inoculated by the i.p. route (23), consistent with the notion that the SsrB-dependent Spi/Ssa system is required for pathogen growth in deep tissues.
We now report that inactivation of the pmrA gene renders Salmonella hypervirulent in C3H/HeN mice. We establish that the PmrA protein down-regulates expression of spi/ssa genes by repressing transcription from the ssrB promoter. The hypervirulence of the pmrA null mutant can be recapitulated by rendering the ssrB promoter resistant to repression by PmrA. Our findings suggest that bacterial pathogens use antivirulence factors for optimal fitness in host tissues.
Results
Inactivation of the pmrA Gene Renders Salmonella Hypervirulent.
We reexamined the virulence behavior of a pmrA null mutant in C3H/HeN mice because the PmrA/PmrB system is activated in the presence of Fe3+ (6), and C3H/HeN mice harbor a functional allele of the Nramp1 (also known as Slc11a1) gene, which encodes a protein that mediates iron transport across the phagosomal membrane (24). By contrast, BALB/c mice are Nramp1−/−.
Unexpectedly, the pmrA mutant was more virulent than the isogenic wild-type strain following i.p. inoculation (Fig. 1A). The hypervirulence phenotype was manifested by an earlier time to death than animals infected with wild-type Salmonella (Fig. 1A). This result implied that the PmrA/PmrB system promotes expression of a gene(s) that antagonizes Salmonella virulence and/or represses expression of a virulence determinant(s).
Fig. 1.
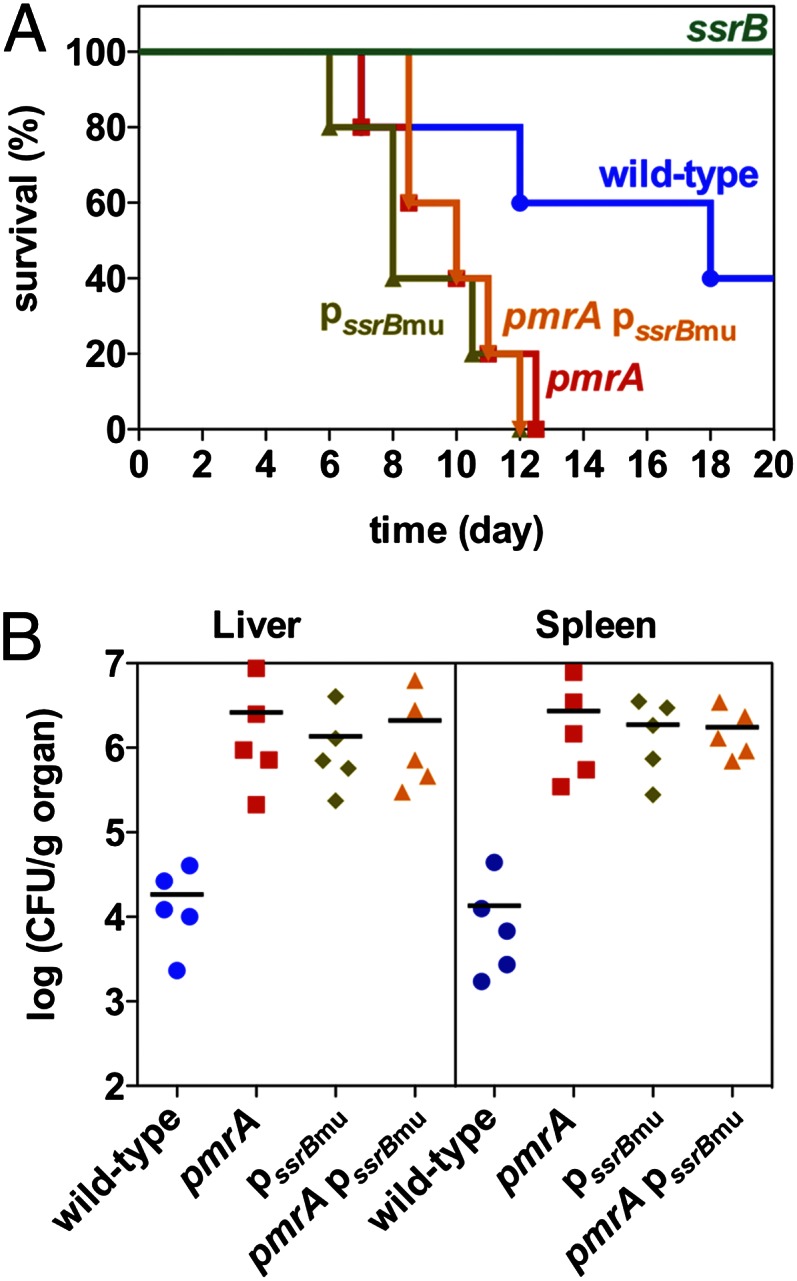
Lack of PmrA or its binding site in the ssrB promoter increases Salmonella virulence in mice. C3H/HeN mice were inoculated i.p. with ∼103 cfu of wild-type, pmrA, ssrB, ssrB promoter mutant (pssrBmu), and pmrA ssrB promoter double mutant (pmrA pssrBmu) Salmonella. Survival of mice was monitored daily (A). At 5 d after infection, the number of bacteria in the liver and spleen were determined (B).
PmrA Represses Transcription of the SPI-2 ssaG Gene.
Given that the pmrA hypervirulence phenotype was observed in Nramp1+/+ mice, we wondered whether the PmrA/PmrB system might regulate expression of SPI-2 genes. Genes located in the SPI-2 pathogenicity island are up-regulated in the spleens of Nramp1+/+ mice relative to those of congenic Nramp1−/− animals (25). Thus, we investigated expression of the SsrB-activated ssaG gene, which is the first gene of a large operon (from ssaG to ssaU) and encodes a component of the Spi/Ssa T3SS (22). We used a set of isogenic strains harboring a plasmid containing the ssaG promoter region fused to a promoterless gfp variant that specifies an unstable GFP (i.e., GFPaav) (26).
There was more fluorescence in the pmrA mutant than in the wild-type strain when bacteria were grown in minimal media at pH 4.6 with high Mg2+ (Fig. 2A), a condition reported to activate SPI-2 genes (27). This behavior was also true when bacteria were grown in minimal media at pH 4.6 with low Mg2+, although there was less of a difference between the two strains. By contrast, the expression was similar in wild-type and pmrA Salmonella in media containing neutral pH regardless of the Mg2+ concentration (Fig. 2A), which is a noninducing condition for SPI-2 genes (27, 28). Control experiments demonstrated that there was no detectable fluorescence in an ssrB mutant strain regardless of growth conditions (Fig. 2A), as anticipated by the established requirement for ssrB in ssaG transcription (21). Moreover, the elevated ssaG expression displayed by the pmrA mutant when grown in minimal media at pH 4.6 with high Mg2+ was restored to wild-type levels by a plasmid expressing the wild-type pmrA gene from a heterologous promoter but not by the plasmid vector (Fig. 2B).
Fig. 2.

PmrA decreases expression of the SPI-2 ssaG gene. (A) Fluorescence from a ssaG-gfp transcriptional fusion was determined in wild-type, pmrA, and ssrB Salmonella grown in N-minimal medium at pH 4.6 with 1 mM Mg2+ (LH) or 10 μM (LL), or pH 7.7 with 1 mM Mg2+ (HH) or 10 μM (HL) for 8 h. (B) Relative expression from a chromosomal ssaG-lacZ transcriptional fusion was determined in wild-type, pmrA, pmrA harboring the plasmid vector (pUHE21-2lacIq), and pmrA harboring a plasmid expressing pmrA from a lac promoter derivative following growth in N-minimal medium at pH 4.6 with 1 mM Mg2+ (LH). Values are normalized relative to those displayed by the wild-type strain. (C) mRNA levels of ssaG produced by wild-type, pmrA, and ssrB Salmonella grown overnight in LB media (inoculum) and 4 h after infection of J774A.1 macrophages.
Because Fe3+ is a prime activator of the PmrA/PmrB system (6), we reasoned that it should be possible to modulate expression of SPI-2 genes in wild-type Salmonella by varying the concentrations of Fe3+ in the growth media. Indeed, ssaG expression was greatly reduced as the Fe3+ concentration increased (Fig. 3A). This effect was mediated by PmrA because ssaG expression was no longer responsive to the Fe3+ concentration in a pmrA mutant background (Fig. 3B).
Fig. 3.

Iron regulates expression of the SPI-2 ssaG gene in a PmrA-dependent manner. GFP activity from the ssaG-gfp transcriptional fusion was determined in wild-type Salmonella grown in N-minimal medium at pH 4.6 with 1 mM Mg2+ for 8 h with or without 100 μM Fe3+ (A); or in wild-type and pmrA Salmonella grown in N-minimal medium at pH 4.6 with 1 mM Mg2+ with various concentrations of iron (0.008, 0.04, 0.2, and 4 μM Fe3+) or without iron (B).
Next, we examined whether the enhanced ssaG expression resulting from inactivation of the pmrA gene was also observed when Salmonella was inside macrophages. This test was important because expression of SPI-2 genes greatly increases upon Salmonella phagocytosis (29, 30), and the PmrA/PmrB system is active inside macrophages (11). We found ssaG expression to be ∼30-fold higher at 4 h after internalization by J774A.1 macrophages relative to expression displayed by bacteria grown in LB (Fig. 2C), which is in agreement with previous results (22, 29). Inactivation of the pmrA gene further increased ssaG expression (Fig. 2C), as observed during growth in minimal media (Fig. 2A). We note that the increased ssaG expression does not depend on the presence of a functional Nramp1 protein because J774A.1 macrophages originated from an Nramp1−/− animal (31). Taken together, these results indicate that PmrA functions as a negative regulator of ssaG and potentially of other SPI-2 genes.
PmrA Represses ssaG Transcription Indirectly, by Hindering Expression of the ssaG Activator SsrB.
In principle, PmrA could repress ssaG transcription directly, by binding to the ssaG promoter, or, indirectly, by hindering transcription of an ssaG activator(s) or by furthering expression of an ssaG repressor(s). Sequences resembling a PmrA box (i.e., a direct repeat of the sequence CTTAAT separated by 5 nt present in PmrA-activated promoters; refs. 32–34) were not found in the ssaG promoter region. By contrast, we identified a perfect match to the consensus PmrA box downstream of the mapped transcriptional start site of the ssrB gene (Fig. 4A), which encodes an activator necessary for ssaG transcription (22). This analysis suggested that PmrA might bind to the ssrB promoter hampering ssrB transcription.
Fig. 4.

PmrA binds to the promoter of the ssrB gene. (A) DNA sequence of the promoter region of the ssrB gene. A +1 corresponds to the reported transcriptional start site, the boxed sequences indicate those matching the PmrA-binding consensus sequence, and the underlined sequence indicates the region protected by the PmrA protein in the ssrB promoter shown in B. Mutated nucleotides at the PmrA binding site in the ssrB promoter mutant strain are indicated beneath the sequence. (B) DNase I footprinting analysis of the ssrB promoter using purified PmrA-His6 protein (0, 100, 200, and 300 pmoles) was performed as described in Materials and Methods. Lane A+G corresponds to dideoxy chain-termination sequences for the ssrB promoter DNA. The black bar indicates the region protected by the PmrA-His6 protein. (C) In vivo occupancy of the ssrB, pmrC, and ssaG promoter regions by the PmrA-HA protein when Salmonella strains expressing PmrA-HA from its original chromosomal location and promoter were switched from noninducing conditions (N-minimal medium at pH 7.7 with 1 mM Mg2+) into noninducing or inducing conditions (N-minimal medium at pH 7.7 with 10 μM Mg2+ and 100 μM Fe3+). (D) β-galactosidase activity expressed by wild-type, ssrB, and pmrA Salmonella harboring a plasmid with a plac1–6-driven transcriptional fusion of the 5′ untranslated region (5′UTR) of ssrB or spiR to a promoterless lacZ gene. Bacteria were grown in N-minimal medium at pH 7.7 with 10 μM Mg2+. Values are normalized by that of the wild-type strain. (E) Fluorescence originating from a plasmid-linked ssaG-gfp transcriptional fusion was determined in wild-type, pmrA, ssrB, and ssrB pmrA Salmonella grown in N-minimal medium at pH 4.6 with 1 mM Mg2+ (LH) for 8 h.
The PmrA protein appears to repress ssrB directly because: First, the purified PmrA protein protected the −45 to −16 region from ATG of the ssrB gene in vitro (Fig. 4B), which contains the predicted PmrA binding site in the ssrB promoter (Fig. 4A). Second, chromatin immunoprecipitation experiments revealed that an HA-tagged PmrA expressed from its normal promoter and chromosomal location bound to the ssrB promoter in vivo but not to the ssaG promoter (Fig. 4C). Importantly, the enrichment in ssrB promoter DNA was comparable to that of other known PmrA-regulated genes, including pmrC (Fig. 4C). Third, substitution of four conserved nucleotides in the predicted PmrA box of the ssrB promoter (Fig. 4A) reduced PmrA binding to ssrB promoter DNA in vitro (Fig. 5A) and increased ssaG expression in vivo (Fig. 5B). Notably, the ssaG expression levels exhibited by the strain with the mutation of the PmrA-binding site in the ssrB promoter were similar to those displayed by the pmrA null mutant (Fig. 5B). Furthermore, nucleotide substitution of the PmrA-binding site in the ssrB promoter increased expression of both ssrB and ssaG within the J774A.1 macrophage-like cell line (Fig. 5C) and in bone marrow-derived macrophages prepared from C3H/HeN mice (Fig. S1). We note that the expression levels of the ssrB and ssaG genes in the ssrB promoter mutant were comparable to those in the pmrA mutant (Fig. 5C and Fig. S1) and that the ssrB promoter mutation had no effect on the expression of the PmrA-activated pmrC gene (Fig. 5C).
Fig. 5.

Binding of PmrA to the ssrB promoter is required for PmrA-mediated repression of the SPI-2 ssaG gene. (A) Electrophoretic mobility shift assays of wild-type or mutated ssrB promoter region DNA fragments using purified PmrA-His6 protein (0, 75, 150, 300, 600, and 1,200 pmoles) were carried out as described in Materials and Methods. The shifted bands can be competed out by the corresponding cold probe (unlabeled DNA) in the presence of the maximum amount of PmrA-His6 protein used. (B) Fluorescence from a plasmid-linked ssaG-gfp transcriptional fusion was determined in wild-type, pmrA mutant, the ssrB promoter mutant (pssrBmu), and pmrA ssrB promoter double mutant (pmrA pssrBmu) Salmonella grown in N-minimal medium at pH 4.6 with 1 mM Mg2+. (C) J774A.1 macrophages were infected with wild-type, pmrA mutant, ssrB mutant, and the ssrB promoter mutant (pssrBmu) strains. mRNA levels of the ssrB, ssaG, and pmrC genes were determined at the indicated times after infection.
PmrA’s action at the ssrB promoter is not simply to antagonize activation of ssrB transcription by the SsrB protein because the levels of β-galactosidase activity originating from a plasmid-linked ssrB-lacZ transcriptional fusion were higher in the pmrA mutant than in the wild-type strain and the ssrB mutant (Fig. 4D) even though this fusion was driven by the heterologous lac promoter derivative plac1–6 (35). The only Salmonella sequences present in the plasmid-linked ssrB-lacZ fusion were those located between the ssrB transcription start site and the ssrB start codon, which include the PmrA binding site. By contrast, inactivation of pmrA had no effect on the expression of a similar transcriptional fusion that included the leader region of the spiR gene instead of that of ssrB (Fig. 4D). In addition, similarly low fluorescence levels were displayed by isogenic ssrB and ssrB pmrA strains harboring a plasmid carrying a fusion between the ssaG promoter and a promoterless gfp (aav) gene (Fig. 4E). And inactivation of pmrA in the ssrB promoter mutant background did not alter the levels of ssaG expression (Fig. 5B). These results demonstrated that SsrB is necessary to activate the ssrB promoter even in the absence of PmrA repression. Finally, PmrA’s role as a repressor of ssrB transcription is not simply to prevent the PhoP and OmpR proteins from activating ssrB transcription (21, 36) because there were similarly low levels of ssaG expression in phoP, phoP pmrA, ompR, and ompR pmrA mutants (Fig. S2). Taken together, the results presented in this section indicate that PmrA’s effects on ssaG expression result from direct repression of ssrB transcription.
PmrA Delays Salmonella-Induced Macrophage Death.
To examine the physiological consequences of PmrA repression of ssrB transcription, we examined the kinetics of SPI-2–promoted macrophage death, which is critical for Salmonella virulence (37). We infected macrophages with various Salmonella strains and evaluated cell death by measuring lactate dehydrogenase released from dead or damaged cells. Cell death took place earlier in macrophages infected with the pmrA mutant than with the wild-type strain (Fig. 6). The faster cell death kinetics was also displayed by macrophages infected with the ssrB promoter mutant (Fig. 6). Control experiments showed reduced cell death in macrophages infected with the ssrB mutant (Fig. 6), in agreement with previous reports (38). These data indicate that the PmrA protein delays macrophage death induced by wild-type Salmonella.
Fig. 6.
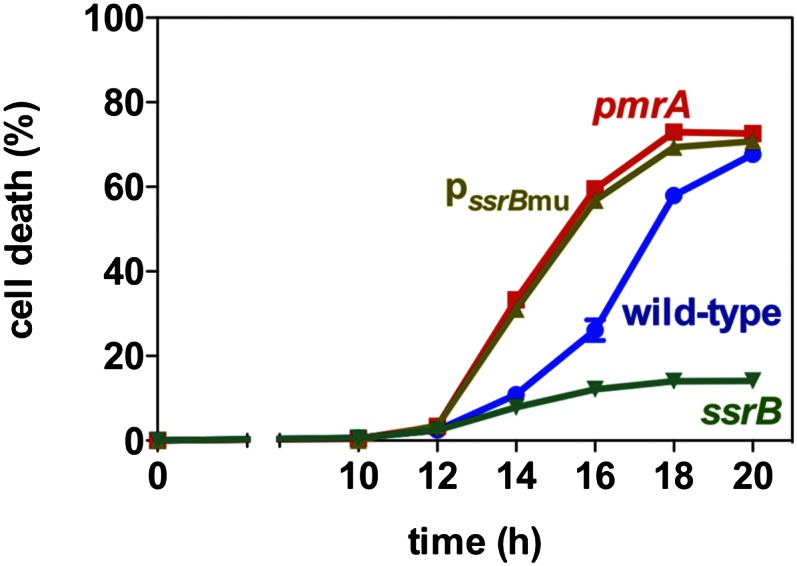
PmrA delays SPI-2–promoted macrophage death. J774A.1 macrophages were infected with wild-type, pmrA, ssrB, and the ssrB promoter mutant (pssrBmu) Salmonella. The percentage of macrophage death was determined by detecting the amount of lactate dehydrogenase released into the supernatants at the indicated times after infection.
PmrA Reduces Salmonella Virulence by Repressing ssrB Transcription.
The results presented above suggested that the hypervirulence phenotype of the pmrA null mutant might be due to PmrA repression of ssrB transcription. If this notion were the case, one would expect: First, that the ssrB promoter mutant that is refractory to repression by the PmrA protein would display the same hypervirulence phenotype as the pmrA null mutant. And second, that a pmrA ssrB promoter double mutant would behave just like pmrA or ssrB promoter single mutants.
We determined that C3H/HeN mice infected with the pmrA mutant or the ssrB promoter mutant died earlier than those infected with the wild-type strain (Fig. 1A). This behavior was also true for the pmrA ssrB promoter double mutant (Fig. 1A). As an independent assay of virulence in vivo, we determined that 5 d after infection the number of bacteria in the liver and spleen of animals infected with the pmrA mutant, the ssrB promoter mutant, and the pmrA ssrB promoter double mutant were similar and were ∼100-fold higher than those infected with the wild-type strain (Fig. 1B). These results support the notion that PmrA dampens virulence primarily by repressing transcription of the major regulator of SPI-2 genes.
Discussion
The genes present in a bacterial pathogen can be divided into three distinct groups based on their virulence roles. The best understood group is constituted by the virulence genes, which are genes that upon inactivation reduce the pathogenicity of a pathogen. A second group includes those genes that do not appear to affect virulence because they do not alter pathogenicity when mutated. The third group encompasses the antivirulence genes. These genes are of particular interest because, puzzlingly, their inactivation renders a pathogen hypervirulent.
We have now determined that the regulatory protein PmrA is an antivirulence factor because inactivation of the pmrA gene exacerbated Salmonella virulence in C3H/HeN mice (Fig. 1A). This result was unexpected because pmrA null mutants were known to be more susceptible to serum (13) and to polymyxin B (6), an antibiotic believed to mimic the action of mammalian cationic antimicrobial peptides (12), and also because a pmrA mutant was mildly attenuated when inoculated into BALB/c mice (16), which are exquisitely susceptible to Salmonella infection (LD50 < 10). Unlike BALB/c mice, C3H/HeN mice carry functional Nramp1, which localizes to the phagosomal membrane (39) and hinders growth of several intracellular pathogens that remain within a phagosome (40).
How PmrA Decreases Salmonella Virulence.
The antivirulence function of PmrA can be ascribed to its role as transcriptional repressor of the regulatory gene ssrB, which is essential for expression of genes required for proliferation inside macrophages and systemic infection in mice (23, 41). This antivirulence function is supported by the following: First, the hypervirulence phenotype of the pmrA null mutant could be phenocopied by mutating the PmrA binding site in the ssrB promoter (Fig. 1A). Second, a pmrA ssrB promoter double mutant exhibited the same hypervirulence phenotype of either single mutant (Fig. 1A). And third, SPI-2–dependent macrophage killing was accelerated in both the pmrA mutant and the ssrB promoter mutant (Fig. 6).
The SsrB protein is necessary for transcription of the genes specifying the T3SS encoded in the SPI-2 pathogenicity island as well as its secreted effectors (19, 20). Inactivation of the SPI-2 T3SS enhances Salmonella’s growth inside phagocytes during infection of an animal (42). These results suggest that PmrA repression of ssrB might enable Salmonella to control bacterial proliferation within a cell and to favor dissemination to neighboring cells. Alternatively or in addition, PmrA may enable proper repression of SPI-2 in the intestine, which is a PmrA-inducing environment (43, 44).
What is the identity of the SsrB-regulated gene(s) that must be tightly controlled for a normal course of Salmonella infection? We considered the possibility of the SsrB-repressed sciS being such a gene because its inactivation was reported to increase Salmonella virulence in BALB/c mice infected orally (4). However, we established that an sciS mutant is attenuated for virulence in C3H/HeN mice inoculated i.p. (Fig. S3), which is in contrast to the hypervirulence phenotype exhibited by the pmrA null mutant in the same mouse strain and route of inoculation (Fig. 1A).
Repression of ssrB, the master regulator of SPI-2 genes, constitutes a unique and unexpected function for the PmrA protein, whose best-described role was as the major activator of LPS modification genes (6, 16, 33). Whereas PmrA-dependent LPS modifications can reduce the risk of Salmonella detection by the Toll-like receptor-4 in a mammalian host (15, 45), its role in ssrB repression may reduce expression of Spi/Ssa-secreted effectors that dampen the host immune response (46, 47).
Control of the SPI-2 Virulence Locus.
Several negative regulatory factors have been implicated in transcriptional control of SPI-2 genes. These factors include H-NS (22), YdgT (48), and Hha (49), all of which are nucleoid-associated proteins that recognize and selectively silence the expression of foreign DNA with higher adenine and thymine content relative to the resident genome (50). The PmrA protein directly binds to a specific sequence in the promoter of ssrB, repressing its expression (Figs. 4 and 5). Deletion of the genes specifying the nucleoid-associated protein attenuates Salmonella’s virulence in mice (48, 49), whereas preventing PmrA-mediated negative regulation of SsrB increases virulence (Fig. 1). This behavior could perhaps reflect that the regulatory effect of the nucleoid-associated proteins is not limited to SPI-2 genes (50, 51).
That iron limitation increases expression of SPI-2 genes (25, 52) could reflect that under such conditions the PmrA protein is not active (6). Indeed, we could decrease SPI-2 expression by increasing the iron concentration in the growth media (Fig. 3A); this effect was PmrA-dependent because it was not observed in the pmrA null mutant (Fig. 3B). By contrast, the ferric uptake regulator (Fur) does not appear to be involved in this process because an increase in iron would promote Fur dimerization and repression of its target promoters. Because one of Fur’s target promoters is that corresponding to the hns gene (53), and H-NS is known to silence expression of SPI-2 genes (22), the increase in iron should increase expression of SPI-2 genes; however, we observed decreased expression (Fig. 3A).
It is remarkable that the PhoP protein, which is a major regulator of virulence functions in Salmonella (8), exerts two distinct and opposite effects on ssrB expression. It directly activates transcription of the ssrB gene (21). However, it promotes expression of PmrD, a posttranslational activator of PmrA (34), which represses ssrB transcription (Fig. 2). However, expression of PhoP-activated genes is maximal at 5–6 h after internalization, whereas repression of SPI-2 genes by PmrA was detected already 1 h after internalization (Fig. 5C). This result indicates that PmrA is probably activated in response to the signals detected by its cognate sensor PmrB (11). Moreover, it suggests that one of the roles of PhoP in promoting ssrB transcription might be to overcome PmrA-dependent repression of ssrB.
That a given regulator (e.g., PhoP) would promote expression of transcripts that favor virulence and reduce virulence is rare but not unprecedented. First, PhoP is required for transcription of the pcgL gene (7), the inactivation of which increases Salmonella virulence (3), as well as of several genes necessary for virulence (8). And second, PhoP is an activator of both the mgtCBR operon (7), which specifies the virulence protein MgtC (54), as well as of AmgR (55), an antisense RNA that down-regulates the levels of the mgtC transcript (55).
Cumulatively, our data demonstrate that the iron-inducible PmrA/PmrB two-component regulatory system properly tunes the level of SPI-2 induction in phagocytes by repressing ssrB transcription. This regulation is critical for Salmonella’s pathogenicity because lack of PmrA or its binding site on the ssrB promoter results in hypervirulence. Thus, bacterial pathogenicity is determined not only by virulence proteins, but also by antivirulence factors, which may contribute to the overall fitness of a pathogen.
Materials and Methods
Bacterial Strains, Plasmids, Primers, and Growth Conditions.
S. enterica serovar Typhimurium strains were derived from wild-type strain 14028s. Unless otherwise stated, bacteria were grown at 37 °C in LB broth or in N-minimal medium (pH 7.4 or 4.6) (56) supplemented with 0.1% casamino acids, 38 mM glycerol, 1 mM or 10 μM of MgCl2, and the indicated concentration of FeSO4. When necessary, antibiotics were added at the following final concentrations: ampicillin, 50 μg/mL; chloramphenicol, 20 μg/mL; kanamycin, 50 μg/mL, and tetracycline, 10 μg/mL P22 transduction of Salmonella strains was performed as described (57). Escherichia coli DH5α was used as a host for the preparation of plasmid DNA. Bacterial strains and plasmids used in this study are listed in Table S1. Primers used in this study are listed in Table S2.
GFP Assay Salmonella.
Cells expressing a gfp variant from the ssaG promoter were grown in minimal media. The expression of the GFP was measured by using multidetector (VICTOR3; PerkinElmer) with OD600 values. The measured values for GFP expression were divided by 1,000 times the OD600 values.
Identification of PmrA’s Binding to the ssrB Promoter.
To identify the binding of PmrA to the promoter region of the ssrB gene, chromatin immunoprecipitation (55), electrophoretic mobility shift assay (55), and DNase I footprinting assay (34) were carried out as described.
Detailed methods are described in SI Materials and Methods.
Supplementary Material
Acknowledgments
We thank Tammy Latifi for the DNase I footprinting experiment; Ephraim Fass for constructing plasmid pFPV25AAV-pssaG; and Eunjin Lee, John May, and Nathan Schwalm for comments on the manuscript. This research was supported, in part, by National Institutes of Health Grant AI042236 (to E.A.G.). E.A.G. is an Investigator of the Howard Hughes Medical Institute.
Footnotes
The authors declare no conflict of interest.
This article is a PNAS Direct Submission.
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1303420110/-/DCSupplemental.
References
- 1.Groisman EA, Ochman H. How Salmonella became a pathogen. Trends Microbiol. 1997;5(9):343–349. doi: 10.1016/S0966-842X(97)01099-8. [DOI] [PubMed] [Google Scholar]
- 2.Dobrindt U, Hochhut B, Hentschel U, Hacker J. Genomic islands in pathogenic and environmental microorganisms. Nat Rev Microbiol. 2004;2(5):414–424. doi: 10.1038/nrmicro884. [DOI] [PubMed] [Google Scholar]
- 3.Mouslim C, Hilbert F, Huang H, Groisman EA. Conflicting needs for a Salmonella hypervirulence gene in host and non-host environments. Mol Microbiol. 2002;45(4):1019–1027. doi: 10.1046/j.1365-2958.2002.03070.x. [DOI] [PubMed] [Google Scholar]
- 4.Parsons DA, Heffron F. sciS, an icmF homolog in Salmonella enterica serovar Typhimurium, limits intracellular replication and decreases virulence. Infect Immun. 2005;73(7):4338–4345. doi: 10.1128/IAI.73.7.4338-4345.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Perez JC, Groisman EA. Acid pH activation of the PmrA/PmrB two-component regulatory system of Salmonella enterica. Mol Microbiol. 2007;63(1):283–293. doi: 10.1111/j.1365-2958.2006.05512.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wösten MM, Kox LF, Chamnongpol S, Soncini FC, Groisman EA. A signal transduction system that responds to extracellular iron. Cell. 2000;103(1):113–125. doi: 10.1016/s0092-8674(00)00092-1. [DOI] [PubMed] [Google Scholar]
- 7.Soncini FC, García Véscovi E, Solomon F, Groisman EA. Molecular basis of the magnesium deprivation response in Salmonella typhimurium: Identification of PhoP-regulated genes. J Bacteriol. 1996;178(17):5092–5099. doi: 10.1128/jb.178.17.5092-5099.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Miller SI, Kukral AM, Mekalanos JJ. A two-component regulatory system (phoP phoQ) controls Salmonella typhimurium virulence. Proc Natl Acad Sci USA. 1989;86(13):5054–5058. doi: 10.1073/pnas.86.13.5054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kox LF, Wösten MM, Groisman EA. A small protein that mediates the activation of a two-component system by another two-component system. EMBO J. 2000;19(8):1861–1872. doi: 10.1093/emboj/19.8.1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kato A, Groisman EA. Connecting two-component regulatory systems by a protein that protects a response regulator from dephosphorylation by its cognate sensor. Genes Dev. 2004;18(18):2302–2313. doi: 10.1101/gad.1230804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Merighi M, Ellermeier CD, Slauch JM, Gunn JS. Resolvase-in vivo expression technology analysis of the Salmonella enterica serovar Typhimurium PhoP and PmrA regulons in BALB/c mice. J Bacteriol. 2005;187(21):7407–7416. doi: 10.1128/JB.187.21.7407-7416.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vaara M. Agents that increase the permeability of the outer membrane. Microbiol Rev. 1992;56(3):395–411. doi: 10.1128/mr.56.3.395-411.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Delgado MA, Mouslim C, Groisman EA. The PmrA/PmrB and RcsC/YojN/RcsB systems control expression of the Salmonella O-antigen chain length determinant. Mol Microbiol. 2006;60(1):39–50. doi: 10.1111/j.1365-2958.2006.05069.x. [DOI] [PubMed] [Google Scholar]
- 14.Chamnongpol S, Dodson W, Cromie MJ, Harris ZL, Groisman EA. Fe(III)-mediated cellular toxicity. Mol Microbiol. 2002;45(3):711–719. doi: 10.1046/j.1365-2958.2002.03041.x. [DOI] [PubMed] [Google Scholar]
- 15.Cullen TW, et al. Helicobacter pylori versus the host: Remodeling of the bacterial outer membrane is required for survival in the gastric mucosa. PLoS Pathog. 2011;7(12):e1002454. doi: 10.1371/journal.ppat.1002454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gunn JS, Ryan SS, Van Velkinburgh JC, Ernst RK, Miller SI. Genetic and functional analysis of a PmrA-PmrB-regulated locus necessary for lipopolysaccharide modification, antimicrobial peptide resistance, and oral virulence of Salmonella enterica serovar typhimurium. Infect Immun. 2000;68(11):6139–6146. doi: 10.1128/iai.68.11.6139-6146.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Galán JE, Wolf-Watz H. Protein delivery into eukaryotic cells by type III secretion machines. Nature. 2006;444(7119):567–573. doi: 10.1038/nature05272. [DOI] [PubMed] [Google Scholar]
- 18.Cornelis GR. The type III secretion injectisome. Nat Rev Microbiol. 2006;4(11):811–825. doi: 10.1038/nrmicro1526. [DOI] [PubMed] [Google Scholar]
- 19.Kuhle V, Hensel M. SseF and SseG are translocated effectors of the type III secretion system of Salmonella pathogenicity island 2 that modulate aggregation of endosomal compartments. Cell Microbiol. 2002;4(12):813–824. doi: 10.1046/j.1462-5822.2002.00234.x. [DOI] [PubMed] [Google Scholar]
- 20.Hensel M. Salmonella pathogenicity island 2. Mol Microbiol. 2000;36(5):1015–1023. doi: 10.1046/j.1365-2958.2000.01935.x. [DOI] [PubMed] [Google Scholar]
- 21.Deiwick J, Nikolaus T, Erdogan S, Hensel M. Environmental regulation of Salmonella pathogenicity island 2 gene expression. Mol Microbiol. 1999;31(6):1759–1773. doi: 10.1046/j.1365-2958.1999.01312.x. [DOI] [PubMed] [Google Scholar]
- 22.Walthers D, et al. The response regulator SsrB activates expression of diverse Salmonella pathogenicity island 2 promoters and counters silencing by the nucleoid-associated protein H-NS. Mol Microbiol. 2007;65(2):477–493. doi: 10.1111/j.1365-2958.2007.05800.x. [DOI] [PubMed] [Google Scholar]
- 23.Yoon H, McDermott JE, Porwollik S, McClelland M, Heffron F. Coordinated regulation of virulence during systemic infection of Salmonella enterica serovar Typhimurium. PLoS Pathog. 2009;5(2):e1000306. doi: 10.1371/journal.ppat.1000306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Goswami T, et al. Natural-resistance-associated macrophage protein 1 is an H+/bivalent cation antiporter. Biochem J. 2001;354(Pt 3):511–519. doi: 10.1042/0264-6021:3540511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zaharik ML, Vallance BA, Puente JL, Gros P, Finlay BB. Host-pathogen interactions: Host resistance factor Nramp1 up-regulates the expression of Salmonella pathogenicity island-2 virulence genes. Proc Natl Acad Sci USA. 2002;99(24):15705–15710. doi: 10.1073/pnas.252415599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Andersen JB, et al. New unstable variants of green fluorescent protein for studies of transient gene expression in bacteria. Appl Environ Microbiol. 1998;64(6):2240–2246. doi: 10.1128/aem.64.6.2240-2246.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Beuzón CR, Banks G, Deiwick J, Hensel M, Holden DW. pH-dependent secretion of SseB, a product of the SPI-2 type III secretion system of Salmonella typhimurium. Mol Microbiol. 1999;33(4):806–816. doi: 10.1046/j.1365-2958.1999.01527.x. [DOI] [PubMed] [Google Scholar]
- 28.Xu X, Hensel M. Systematic analysis of the SsrAB virulon of Salmonella enterica. Infect Immun. 2010;78(1):49–58. doi: 10.1128/IAI.00931-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cirillo DM, Valdivia RH, Monack DM, Falkow S. Macrophage-dependent induction of the Salmonella pathogenicity island 2 type III secretion system and its role in intracellular survival. Mol Microbiol. 1998;30(1):175–188. doi: 10.1046/j.1365-2958.1998.01048.x. [DOI] [PubMed] [Google Scholar]
- 30.Pfeifer CG, Marcus SL, Steele-Mortimer O, Knodler LA, Finlay BB. Salmonella typhimurium virulence genes are induced upon bacterial invasion into phagocytic and nonphagocytic cells. Infect Immun. 1999;67(11):5690–5698. doi: 10.1128/iai.67.11.5690-5698.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ralph P, Prichard J, Cohn M. Reticulum cell sarcoma: An effector cell in antibody-dependent cell-mediated immunity. J Immunol. 1975;114(2 pt 2):898–905. [PubMed] [Google Scholar]
- 32.Wösten MM, Groisman EA. Molecular characterization of the PmrA regulon. J Biol Chem. 1999;274(38):27185–27190. doi: 10.1074/jbc.274.38.27185. [DOI] [PubMed] [Google Scholar]
- 33.Aguirre A, Lejona S, Véscovi EG, Soncini FC. Phosphorylated PmrA interacts with the promoter region of ugd in Salmonella enterica serovar typhimurium. J Bacteriol. 2000;182(13):3874–3876. doi: 10.1128/jb.182.13.3874-3876.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kato A, Latifi T, Groisman EA. Closing the loop: The PmrA/PmrB two-component system negatively controls expression of its posttranscriptional activator PmrD. Proc Natl Acad Sci USA. 2003;100(8):4706–4711. doi: 10.1073/pnas.0836837100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cromie MJ, Shi Y, Latifi T, Groisman EA. An RNA sensor for intracellular Mg(2+) Cell. 2006;125(1):71–84. doi: 10.1016/j.cell.2006.01.043. [DOI] [PubMed] [Google Scholar]
- 36.Feng X, Oropeza R, Kenney LJ. Dual regulation by phospho-OmpR of ssrA/B gene expression in Salmonella pathogenicity island 2. Mol Microbiol. 2003;48(4):1131–1143. doi: 10.1046/j.1365-2958.2003.03502.x. [DOI] [PubMed] [Google Scholar]
- 37.Lindgren SW, Stojiljkovic I, Heffron F. Macrophage killing is an essential virulence mechanism of Salmonella typhimurium. Proc Natl Acad Sci USA. 1996;93(9):4197–4201. doi: 10.1073/pnas.93.9.4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.van der Velden AW, Lindgren SW, Worley MJ, Heffron F. Salmonella pathogenicity island 1-independent induction of apoptosis in infected macrophages by Salmonella enterica serotype typhimurium. Infect Immun. 2000;68(10):5702–5709. doi: 10.1128/iai.68.10.5702-5709.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gruenheid S, Pinner E, Desjardins M, Gros P. Natural resistance to infection with intracellular pathogens: The Nramp1 protein is recruited to the membrane of the phagosome. J Exp Med. 1997;185(4):717–730. doi: 10.1084/jem.185.4.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Govoni G, Gros P. Macrophage NRAMP1 and its role in resistance to microbial infections. Inflamm Res. 1998;47(7):277–284. doi: 10.1007/s000110050330. [DOI] [PubMed] [Google Scholar]
- 41.Ochman H, Soncini FC, Solomon F, Groisman EA. Identification of a pathogenicity island required for Salmonella survival in host cells. Proc Natl Acad Sci USA. 1996;93(15):7800–7804. doi: 10.1073/pnas.93.15.7800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Grant AJ, et al. Attenuated Salmonella Typhimurium lacking the pathogenicity island-2 type 3 secretion system grow to high bacterial numbers inside phagocytes in mice. PLoS Pathog. 2012;8(12):e1003070. doi: 10.1371/journal.ppat.1003070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Glover J, Jacobs A. Observations on iron in the jejunal lumen after a standard meal. Gut. 1971;12(5):369–371. doi: 10.1136/gut.12.5.369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.MacKenzie EL, Iwasaki K, Tsuji Y. Intracellular iron transport and storage: From molecular mechanisms to health implications. Antioxid Redox Signal. 2008;10(6):997–1030. doi: 10.1089/ars.2007.1893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.van Mourik A, et al. Altered linkage of hydroxyacyl chains in lipid A of Campylobacter jejuni reduces TLR4 activation and antimicrobial resistance. J Biol Chem. 2010;285(21):15828–15836. doi: 10.1074/jbc.M110.102061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Le Negrate G, et al. Salmonella secreted factor L deubiquitinase of Salmonella typhimurium inhibits NF-kappaB, suppresses IkappaBalpha ubiquitination and modulates innate immune responses. J Immunol. 2008;180(7):5045–5056. doi: 10.4049/jimmunol.180.7.5045. [DOI] [PubMed] [Google Scholar]
- 47.Guiney DG, Fierer J. The role of the spv genes in Salmonella pathogenesis. Front Microbiol. 2011;2:129. doi: 10.3389/fmicb.2011.00129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Coombes BK, Wickham ME, Lowden MJ, Brown NF, Finlay BB. Negative regulation of Salmonella pathogenicity island 2 is required for contextual control of virulence during typhoid. Proc Natl Acad Sci USA. 2005;102(48):17460–17465. doi: 10.1073/pnas.0505401102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Silphaduang U, Mascarenhas M, Karmali M, Coombes BK. Repression of intracellular virulence factors in Salmonella by the Hha and YdgT nucleoid-associated proteins. J Bacteriol. 2007;189(9):3669–3673. doi: 10.1128/JB.00002-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Navarre WW, McClelland M, Libby SJ, Fang FC. Silencing of xenogeneic DNA by H-NS-facilitation of lateral gene transfer in bacteria by a defense system that recognizes foreign DNA. Genes Dev. 2007;21(12):1456–1471. doi: 10.1101/gad.1543107. [DOI] [PubMed] [Google Scholar]
- 51.Vivero A, et al. Modulation of horizontally acquired genes by the Hha-YdgT proteins in Salmonella enterica serovar Typhimurium. J Bacteriol. 2008;190(3):1152–1156. doi: 10.1128/JB.01206-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kim CC, Falkow S. Delineation of upstream signaling events in the salmonella pathogenicity island 2 transcriptional activation pathway. J Bacteriol. 2004;186(14):4694–4704. doi: 10.1128/JB.186.14.4694-4704.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Troxell B, et al. Fur negatively regulates hns and is required for the expression of HilA and virulence in Salmonella enterica serovar Typhimurium. J Bacteriol. 2011;193(2):497–505. doi: 10.1128/JB.00942-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Blanc-Potard AB, Groisman EA. The Salmonella selC locus contains a pathogenicity island mediating intramacrophage survival. EMBO J. 1997;16(17):5376–5385. doi: 10.1093/emboj/16.17.5376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lee EJ, Groisman EA. An antisense RNA that governs the expression kinetics of a multifunctional virulence gene. Mol Microbiol. 2010;76(4):1020–1033. doi: 10.1111/j.1365-2958.2010.07161.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Snavely MD, Miller CG, Maguire ME. The mgtB Mg2+ transport locus of Salmonella typhimurium encodes a P-type ATPase. J Biol Chem. 1991;266(2):815–823. [PubMed] [Google Scholar]
- 57.Watanabe T, Ogata Y, Chan RK, Botstein D. Specialized transduction of tetracycline resistance by phage P22 in Salmonella typhimurium. I. Transduction of R factor 222 by phage P22. Virology. 1972;50(3):874–882. doi: 10.1016/0042-6822(72)90441-2. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.