

Abstract
Cystic fibrosis (CF) patients are hypersusceptible to chronic Pseudomonas aeruginosa lung infections. Cultured human airway epithelial cells expressing the ΔF508 allele of the cystic fibrosis transmembrane conductance regulator (CFTR) were defective in uptake of P. aeruginosa compared with cells expressing the wild-type allele. Pseudomonas aeruginosa lipopolysaccharide (LPS)–core oligosaccharide was identified as the bacterial ligand for epithelial cell ingestion; exogenous oligosaccharide inhibited bacterial ingestion in a neonatal mouse model, resulting in increased amounts of bacteria in the lungs. CFTR may contribute to a host-defense mechanism that is important for clearance of P. aeruginosa from the respiratory tract.
Among the most serious manifestations of CF are chronic pulmonary infections with the bacterium P. aeruginosa. The basis for hypersusceptibility of CF patients to this bacterium is not well understood, and the role of mutant CFTR, if any, is not clear. Binding and internalization of respiratory pathogens by epithelial cells followed by desquamation could be an important mechanism for clearing bacteria from the lung. This mechanism has been shown to be important in protecting against bladder infections (1).
To investigate whether the most common and severe CFTR mutation (ΔF508) affected uptake of P. aeruginosa, we performed bacterial invasion assays (2) with four cell lines: CFT1, an airway epithelial cell line derived from a CF patient homozygous for ΔF508 CFTR and that is transformed with human papilloma virus 18 E6/E7; CFT1-ΔF508, which expresses a third copy of ΔF508 CFTR introduced by a retrovirus; CFT1-LC3, which expresses a control gene (β-galactosidase) introduced by the same retrovirus; and CFT1-LCFSN, which expresses a functional wild-type human CFTR gene (3). We tested a standard laboratory strain of P. aeruginosa, designated PAOI, and two nonmucoid, LPS-smooth clinical isolates from CF patients (4). Compared with CFT1-LCFSN cells, the three lines expressing ΔF508 CFTR internalized significantly fewer bacterial cells (Fig. 1A). The ΔF508 mutation causes inefficient processing of CFTR, a defect that is partially corrected if the cells are grown at 26°C (5). When epithelial cells were cultured for 72 hours at 26°C there were no longer significant differences in uptake of the P. aeruginosa strains by the cells expressing wild-type or mutant CFTR (Fig. 1B). Because the overall uptake of bacteria at 26°C was low, we performed additional experiments with cells grown for 72 hours at 26°C in which the invasion assay was performed at 37°C for 3 hours, conditions under which surface expression of mutant ΔF508 CFTR is maintained (5). No significant difference in bacterial cell uptake was measured (Fig. 1C), and overall amounts of internalization approached those of the CFT1-LCFSN cells at 37°C. Returning cells expressing ΔF508 CFTR to 37°C for 24 hours after growth for 72 hours at 26°C removes CFTR from the cell surface (5); under these conditions internalization of the bacterial strains was essentially identical to that shown in Fig. 1A (6). These data indicate that internalization of P. aeruginosa by airway epithelial cells correlated with membrane expression of CFTR.
Fig. 1.

Internalization of P. aeruginosa by 105 transformed airway epithelial cells (2). Cells were grown for 72 hours and bacteria were then allowed to invade the cells for 3 or 4 hours. Bars indicate the mean of the determinations and error bars the SD. Cell lines are as follows: 1, CFT1-LCFSN; 2, CFT1; 3, CFT1-LC3; and 4, CFT1-ΔF508. (A) Cells were grown at 37°C, a temperature that inhibits membrane expression of ΔF508 CFTR (5) and the assay was carried out at 37°C. Multiple comparisons for all three bacterial strains were significant (P < 0.001, ANOVA); the CFT1-LCFSN line was significantly different from any other cell line (P < 0.01, Fisher's PLSD statistic) for all three bacterial strains. (B) Cells were grown at 26°C and internalization was assessed at 26°C. (C) Cells were grown at 26°C and internalization was assessed at 37°C. When cells were grown at 26°C to promote surface expression of ΔF508 CFTR (5), there were no significant (P > 0.2, ANOVA) differences in bacterial uptake among the cell lines for any P. aeruginosa strain tested.
The effect of ΔF508 CFTR on ingestion appeared to be specific for P. aeruginosa; other bacterial pathogens tested (7) were internalized equally well by cells expressing mutant ΔF508 or wild-type CFTR. Quantitative bacterial binding assays revealed that differences in internalization by the epithelial cells were not simiply due to differences in the adherence of P. aeruginosa.
Over the course of chronic lung infection in CF patients, variants of P. aeruginosa emerge that no longer express complete LPS structures (8). To investigate the effect of LPS structure on epithelial cell internalization, we tested eight P. aeruginosa strains that differ in LPS structure (9) (Fig. 2, A and B). Maximal invasion occurred into the CFT1-LCFSN cells by bacterial strains expressing an intact LPS outer core; in addition, cells expressing wild-type CFTR ingested significantly more P. aeruginosa, regardless of LPS phenotype, than did those expressing ΔF508 CFTR. No effect on bacterial ingestion was seen when isogenic P. aeruginosa strains differing in production of pili, flagella, or mucoid exopolysaccharide were tested (6).
Fig. 2.

Role of the complete outer-core region of P. aeruginosa LPS in internalization by airway epithelial cells (2). Bars indicate the mean of the determinations and error bars the SD. (A and B) Assays with bacterial strains of defined LPS phenotype (9); 1, PAC1 R, wild-type, smooth; 2, PAC557, complete core; 3, PAC1R algC::tet, incomplete core; 4, PAC605, incomplete core; 5, PAO1, wild-type, smooth; 6, AK44, complete core; 7, PAO1 algC::tet, incomplete core; and 8, AK1012, incomplete core. All eight strains were internalized by the CFT1-LCFSN cell line significantly better than by the three cell lines expressing mutant ΔF508 CFTR (P < 0.001, ANOVA; P < 0.05 for all pair-wise comparisons, Fisher PLSD). Bacterial strains with a smooth or complete-core LPS were internalized by all of the cell lines significantly better than strains expressing an incomplete core (P < 0.01, ANOVA; P < 0.05 for all pair-wise comparisons, Fisher PLSD). (C to E) Increased internalization by airway epithelial cells of recombinant LPS-smooth P. aeruginosa strains carrying cloned rfb genes (solid bars) compared with parental clinical isolates expressing an incomplete LPS structure and carrying only the DNA cloning vector (open bars) (10). (C) Strain 2192; (D) strain 332; (E) strain 9125. Ingestion of all of the LPS-smooth strains by all of the cell lines was significantly better than ingestion of the corresponding LPS-incomplete isolate (P ≤ 0.002, unpaired t test), except for strain 9125 by the CFT1 cells. There was also significantly greater (P < 0.001, ANOVA) uptake of all of the LPS-smooth bacteria by the CFT1-LCFSN cells compared with the other three cell lines expressing ΔF508 CFTR.
We also compared the internalization of three clinical isolates of P. aeruginosa with incomplete LPS structures with that of recombinant strains converted back to expression of complete LPS structures by introduction of portions of the rfb locus of P. aeruginosa strain PA103 (10). All three recombinant LPS-smnooth bacterial strains showed the same pattern of ingestion by the cell lines (Fig. 2, C to E) as was depicted in Figs. 1 and 2, A and B, for naturally occurring LPS-smooth strains. Uptake of the recomnbinant LPS-smooth strains by all of the cell lines was significantly increased compared with the LPS-incomplete clinical isolates carrying only the vector for DNA cloning. These results suggest that the emergence of strains expressing an incomplete LPS structure during chronic infection of CF patients may further reduce epithelial cell uptake and thus contribute to bacterial survival in the airways.
To determine if LPS could inhibit the internalization of P. aeruginosa, we isolalted LPS and the oligosaccharide fraggment lacking lipid A from strains PAC557 and PACIR algC∷tet and tested their inhibitory activity in the invasion assay. Strain PAC557 produces an LPS with a comnplete core but no O side chains, and the isogenic strain PACIR algC∷tet expresses an incomplete LPS core (11). LPS from the former, but not the latter, strain inhibited ingestion by the CFT1-LCFSN cells (Fig. 3). LPS with a complete core also inhibited bacterial adherence to the CFT1-LCFSN cell line (12). These results suggest that the complete LPS outer core is an important bacterial ligand involved in the ingestion by airway epithelial cells expressing wild-type CFTR. LPS has a similar role in the invasion of epithelial cells by Neisseria gonorrhocae (13).
Fig. 3.

Effect of intact LPS (A) and an oligosaccharide with a lipid A–free core (B) on internalization of P. aeruginosa into CFT1-LCFSN cells. Each point is the mean of three to seven replicates; error bars are SD. Pseudomonas aeruginosa strains: (□), PAO1; (Δ), 149; and (Ο), 324. Solid symbols in (B) represent the mean CFU of bacteria internalized in the presence of intact LPS (100 μg/ml) from strain PAC1R algC::tet, which has an incomplete-core oligosaccharide. Amounts of internalization < 104 CFU differed significantly (P < 0.01, ANOVA and Fisher PLSD for pair-wise comparisons) from amounts of internalization in the presence of incomplete-core LPS (100 μg/ml).
To assess the physiological significance of these observations, we used a neonatal mouse model for infection by P. aeruginosa wherein epithelial cell internalization of bacteria has been observed (14). Inocula containing P. aeruginosa PAO1 plus complete-core oligosaccharide produced fewer intracellular P. aeruginosa 1 and 24 hours after infection compared with controls inoculated with PAOI alone or PAOI plus incomiplete-core oligosaccharide (Fig. 4). The total number of bacteria in the lung at 1 hour was the same for all groups, but at 24 hours there were ≥28 million more P. aeruginosa per gram of lung tissue when the complete-core oligosaccharide was added to the inoculum. Forty-eight hours after infection, the mice given the complete-core inhibitor still had substantially more bacteria in the lungs compared with controls. Mice given the complete-core inhibitor had more internalized bacteria at 48 hours, although this was not significantly different (P > 0.05) from mice given incomplete-core inhibitor; presumably this higher level of internalized bacteria reflected the fact that there were more total bacteria in these mouse lungs and there would be no effect from the inhibitor at this time point. In a separate experiment, addition of intact LPS to the bacterial inoculum produced similar results (6). Thus, additioin of an exogenous ligand to block bacterial ingestion by epithelial cells resulted in less bacterial clearance from the lungs of neonatal mice.
Fig. 4.
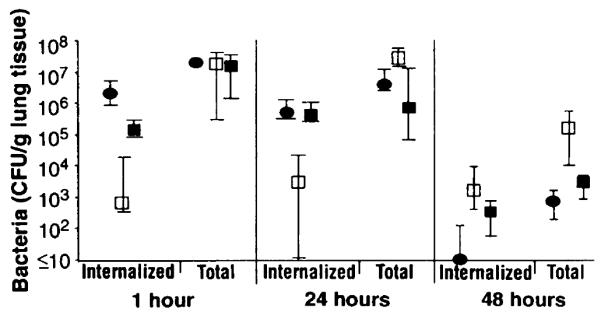
Effect of complete-core oligosaccharide on P. aeruginosa infection in neonatal mouse lungs. Each point indicates the median bacterial CFU for 8 to 10 lungs obtained from each group of mice and the bars indicate the upper and lower quartiles. Inhibitor delivered with inoculum: (●), none; (□), complete-core oligosaccharide; and (■), incomplete-core oligosaccharide. Groups of 7-day-old neonatal BALB/c mice were infected intranasally with ~ 108 CFU of strain PAO1 (14) alone or mixed with either complete-core oligosaccharide (50 μg) or incomplete-core oligosaccharide (50 μg) (11). After 1, 24, or 48 hours, four to five mice were killed and their lungs were removed, weighed, and dispersed into single-cell suspensions. The total CFU of bacteria present in each lung was determined from a sample treated with Triton X-100 (0.5%) to release intracellular bacteria. The remaining cells were treated with gentamicin (300 μg/ml) for 60 min to kill extracellular P. aeruginosa, then the cells were centrifuged, washed twice in tissue-culture medium, and resuspended in 200 μl of 0.5% Triton X-100 to release intracellular bacteria. These suspensions were diluted and plated. Differences among groups were analyzed by nonparametric statistics: P < 0.0001, Kruskall-Wallis nonparametric ANOVA; P < 0.001, Dunn procedure for individual pair-wise differences between the groups at 1 and 24 hours. Also, at 1 hour the group receiving the incomplete-core oligosaccharide had a reduced amount (P = 0.05, Dunn procedure) of intracellular bacteria compared with the group receiving nothing with the inoculum. At 48 hours, the group treated initially with complete-core inhibitor had significantly more bacteria in their lungs (P = 0.003, Kruskall-Wallis; P < 0.05, Dunn procedure for all pair-wise comparisons) than did the other groups.
Histological analyses of affected lung tissue removed from CF patients at autopsy or for transplantation routinely show that P. aeruginosa cannot be seen internalized with-in airway cells and are rarely attached to epithelial cells. Instead, bacteria are observed as microcolonies encased in the extracellular mucus layer lying in the airway lumen (15). Thus, our findings are consistent with the observed histopathology. Some studies have implicated increased receptors for P. aeruginosa on epithelial cells from CF patients as the basis for the hypersusceptibility of CF patients to chronic infection (16), whereas others have reported no differences in P. aeruginosa adherence to nasal polyp epithelial cells cultured from individuals with and without CF (17). Further studies are needed to determine the relation between binding of P. aeruginosa and internalization by epithelial cells and the pathogenesis of chronic lung infection in CF patients.
Our results suggest that P. aeruginosa can initiate infection in CF patients because airway epithelial cells expressing a mutant CFTR are defective in internalization of the bacterium, a process that may be an important host defense. The emergence during chronic infection of P. aeruginosa variants with an incomplete LPS structure, which would further impair their internalization, may contribute to the maintenance of the infectious state. Conceivably, genetic or other therapies that promote expression of CFTR on epithelial cell surfaces could enhance elimination of P. aeruginosa by cellular uptake and alter the otherwise relentless clinical course of CF.
Acknowledgments
We thank D. J. Evans for constructing LPS-smooth recombinant P. aeruginosa strains and S. M. J. Fleiszig for advice. Supported by NIH grants to G.B.P. (AI22806), to J.C.O. (HL42384), and to J.B.G. (AI35674).
REFERENCES AND NOTES
- 1.Aronson M, Medalia O, Amichay D, Nativ O. Infect. Immun. 1988;56:1615. doi: 10.1128/iai.56.6.1615-1617.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]; Dalal E, Medalia O, Harari O, Aronson M. ibid. 1994;62:5505. doi: 10.1128/iai.62.12.5505-5510.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]; Orikasa S, Hinman F., Jr. Invest. Urol. 1977;15:185. [PubMed] [Google Scholar]
- 2.Cells were released from monolayers in tissue culture flasks by 5-min incubation with a trypsin-versene mixture (BioWhitaker, Walkersville, MD) and washed, counted, and seeded into 96-well tissue culture plates at a concentration of 105 cells per well in supplemented F-12 medium (3). Plates were incubated at either 26° or 37°C in 5% CO2. Fresh cultures of P. aeruginosa grown overnight at 37°C on a tryptic soy agar plate were suspended in supplemented F-12 medium to prepare the bacterial inoculum; then ~106 colony-forming units (CFU; range from 6 × 105 to 2.3 × 106 CFU) were added per well of 105 epithelial cells. Bacteria were allowed to be ingested by the epithelial cells for 3 or 4 hours at either 26° or 37°C, after which nonadherent bacteria were removed by washing. The remainder of the assay and controls were as described [S. M. J. Fleiszig, T. S. Zaidi, G. B. Pier, Infect. Immun. 63, 4072 (1995)]. Three to nine replicates were obtained per point and were analyzed by analysis of variance (ANOVA) and the Fisher PLSD statistic to determine pair-wise differences.
- 3.Olsen JC, et al. Hum. Gene Ther. 1992;3:253. doi: 10.1089/hum.1992.3.3-253. [DOI] [PubMed] [Google Scholar]; Sarkadi B, et al. J. Biol. Chem. 1992;267:2087. [PubMed] [Google Scholar]
- 4.Pier GB, DesJardins D, Aguilar T, Barnard M, Speert DP. J. Clin. Microbiol. 1986;24:189. doi: 10.1128/jcm.24.2.189-196.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Denning GM, et al. Nature. 1992;358:761. doi: 10.1038/358761a0. [DOI] [PubMed] [Google Scholar]; Denning GM, Ostedgaard LS, Welsh MJ. J. Cell Biol. 1992;118:551. doi: 10.1083/jcb.118.3.551. [DOI] [PMC free article] [PubMed] [Google Scholar]; Denning GM, Ostedgaard LS, Cheng SH, Smith AE, Welsh MJ. J. Clin. Invest. 1992;89:339. doi: 10.1172/JCI115582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grout M, Pier GB. unpublished data.
- 7.Human clinical isolates of bacterial pathogens that failed to be ingested by epithelial cells expressing wild-type CFTR differently from cells expressing ΔF508 mutant CFTR included the following: Escherichia coli (two strains), Burkholderia cepacia (three strains), Neisseria meningitidis (one strain), Staphylococcusaureus (two strains), Staphylococcus epidermidis (two strains), Streptococcus pneumoniae type XIV (one strain), and Streptococcus agalactiae (one strain).
- 8.Ohman DE, Goldberg JB, Flynn JL. 1990. In: Silver S, Chakrabarty A, Iglewski B, Kaplan S, editors. Pseudo-monas: Biotransformations, Pathogenesis, and Evolving Biotechnology. American Society for Microbiology; Washington, DC: pp. 28–35. [Google Scholar]; Penketh A, Pitt T, Roberts D, Hodson ME, Batten JC. Am. Rev. Respir. Dis. 1983;127:605. doi: 10.1164/arrd.1983.127.5.605. [DOI] [PubMed] [Google Scholar]; Hancock REW, et al. Infect. Immun. 1983;42:170. doi: 10.1128/iai.42.1.170-177.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]; Mahenthiralingam E, Campbell ME, Speert DP. ibid. 1994;62:596. doi: 10.1128/iai.62.2.596-605.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kropinski AM, Chan LC, Milazzo FH. Can. J. Microbiol. 1979;25:390. doi: 10.1139/m79-060. [DOI] [PubMed] [Google Scholar]; Koval SF, Meadow PM. J. Gen. Microbiol. 1977;98:387. doi: 10.1099/00221287-98-2-387. [DOI] [PubMed] [Google Scholar]; Rowe PS, Meadow PM. Eur. J. Biochem. 1983;132:329. doi: 10.1111/j.1432-1033.1983.tb07366.x. [DOI] [PubMed] [Google Scholar]; Jarrell K, Kropinski AM. Microbios. 1977;19:103. [PubMed] [Google Scholar]; Coyne MJ, Russell KS, Coyle CL, Goldberg JB. J. Bacteriol. 1994;176:3500. doi: 10.1128/jb.176.12.3500-3507.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Evans DJ, Pier GB, Coyne MJ, Goldberg JB. Mol. Microbiol. 1994;13:427. doi: 10.1111/j.1365-2958.1994.tb00437.x. [DOI] [PubMed] [Google Scholar]; Goldberg JB, Hatano K, Meluleni GS, Pier GB. Proc. Natl. Acad. Sci. U.S.A. 1992;89:10716. doi: 10.1073/pnas.89.22.10716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.LPS was released from these strains by lysing bacterial cells grown on 25 petri plates containing tryptic soy agar with 1 % sarcosyl in deionized water. Cellular debris was removed by centrifugation at 20,000g for 15 min, and the LPS in the supernate purified as described [G. B. Pier, H. F. Sidberry, S. Zolyomi, J. C. Sadoff, Infect. Immun. 22, 908 (1978)]. Purity and migration characteristics were confirmed in silver-stained SDS-polyacrylamide gels. Lipid A was removed from LPS by suspending LPS in 1 % acetic acid and heating the suspension for 3 hours at 95°C. Inhibition of ingestion in the presence of LPS was evaluated by adding LPS directly into wells during the bacterial ingestion assay. The highest doses of LPS were added to uninfected wells of epithelial cells to monitor for toxicity due to LPS. Staining with trypan blue revealed no overt toxicity of the LPS preparations.
- 12.Although epithelial cells expressing mutant ΔF508 CFTR ingested the P. aeruginosa expressing a complete LPS core better than they ingested bacteria expressing an incomplete LPS core (Fig. 2), there was no statistically significant reduction in bacterial ingestion by mutant CFTR epithelial cells with complete-core LPS as an inhibitor. This could be attributed either to an inability to measure further reductions in ingestion under circumstances in which the initial uptake is already quite low or to alternative ligand-receptor interactions between LPS-rough P. aeruginosa and ΔF508 CFTR–expressing cells. Purified O polysaccharides from LPS-smooth wild-type strains did not inhibit bacterial ingestion by the airway epithelial cell lines.
- 13.Schwan ET, Robertson BD, Brade H, Vanputten JPM. Mol. Microbiol. 1995;15:267. doi: 10.1111/j.1365-2958.1995.tb02241.x. [DOI] [PubMed] [Google Scholar]
- 14.Tang H, Kays M, Prince A. Infect. Immun. 1995;63:1278. doi: 10.1128/iai.63.4.1278-1285.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baltimore RS, Christie CDC, Smith GJW. Am. Rev. Respir. Dis. 1989;140:1650. doi: 10.1164/ajrccm/140.6.1650. [DOI] [PubMed] [Google Scholar]; Nelson JW, et al. Infect. Immun. 1990;58:1489. doi: 10.1128/iai.58.6.1489-1495.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]; Lam J, Chan R, Lam K, Costerton JW. ibid. 1980;28:546. doi: 10.1128/iai.28.2.546-556.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]; Jeffery PK, Brain PR. Scanning Microsc. 1988;2:553. [PubMed] [Google Scholar]
- 16.Saiman L, Prince A. J. Clin. Invest. 1993;92:1875. doi: 10.1172/JCI116779. [DOI] [PMC free article] [PubMed] [Google Scholar]; Zar H, Saiman L, Quittell L, Prince A. J. Pediatr. 1995;126:230. doi: 10.1016/s0022-3476(95)70549-x. [DOI] [PubMed] [Google Scholar]; Imundo L, Barasch J, Prince A, Al-Awqati Q. Proc. Natl. Acad. Sci. U.S.A. 1995;92:3019. doi: 10.1073/pnas.92.7.3019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Plotkowski MC, Chevillard M, Pierrot D, Altemayer D, Puchelle E. J. Med. Microbiol. 1992;36:104. doi: 10.1099/00222615-36-2-104. [DOI] [PubMed] [Google Scholar]; Cervin MA, Simpson DA, Smith AL, Lory S. Microb. Pathog. 1994;17:291. doi: 10.1006/mpat.1994.1075. [DOI] [PubMed] [Google Scholar]