

Abstract
The development of multidrug resistant (MDR) and extensively drug resistant (XDR) forms of tuberculosis (TB) has stimulated research efforts globally to expand the new drug pipeline. Nitro aromatic compounds, including 1, 3-Benzothiazin-4-ones (BTZs) and related agents, are a promising new class for the treatment of TB. Research has shown that the nitroso intermediates of BTZs that are generated in vivo cause suicide inhibition of decaprenylphosphoryl-β-D-ribose 2′ oxidase (DprE1), which is responsible for cell wall arabinogalactan biosynthesis. We have designed and synthesized novel anti-TB agents inspired from BTZs and other nitroaromatic compounds. Computational studies indicated that the unsubstituted aromatic carbons of BTZ043 and related nitroaromatic compounds are the most electron deficient and might be prone to nucleophilic attack. Our chemical studies on BTZ043 and the additional nitro aromatic compounds synthesized by us and the others confirmed the postulated reactivity. The results indicate that nucleophiles such as thiolates, cyanide and hydride induce non-enzymatic reduction of the nitro groups present in these compounds to the corresponding nitroso intermediates by addition at the unsubstituted electron deficient aromatic carbon present in these compounds. Furthermore we demonstrate here that these compounds are good candidates for the classical von Richter reaction. These chemical studies offer an alternate hypotheses for the mechanism of action of nitro aromatic anti-TB agents in that the cysteine thiol(ate) or a hydride source at the active site of DprE1 may trigger the reduction of the nitro groups in a manner similar to the von Richter reaction to the nitroso intermediates, to initiate the inhibition of DprE1.
Introduction
Tuberculosis (TB) has plagued the human race throughout history.1 In 1882, when Robert Koch published his landmark finding that TB was caused by Mycobacterium tuberculosis2 (Mtb), he also noted that 1/7 of all humans died of TB! Today, approximately 1/3 of all people are infected with Mtb, making TB persistently prevalent.3 Each year, about 8 million more are infected and 2-3 million people die of TB. Heroic efforts during the golden age of antibiotics in the 20th century led to the discovery of several anti-TB agents that temporarily stemmed the tide of lethal TB infections. While no single drug has emerged that is completely effective against Mtb, when given under carefully monitored conditions, combinations of antibiotics have saved countless lives. Still, the number of people being infected by Mtb grows every year as the increase in global population far outweighs the slow reduction in its incidence.4 The situation has been exacerbated by the emergence of multidrug resistant (MDR) and extensive drug resistant (XDR) strains.5 Thus, while the need for effective treatment of TB is dire, no new anti-TB drug has been marketed in decades and new TB drug discovery remains a formidable task.6,7
Among the very potent and most intriguing leads related to the discovery of new anti-TB agents, are new nitro aromatic compounds, including a series of nitro furans 1,8 nitroimidazoles, such as the extensively studied PA824 (2),9,10 and the benzothiazinones (BTZ) as represented by benzothiazinone 043 (BTZ043, 3, Figure 1).11,12 The mode of action and detailed structure-activity-relationships of 2 have been intensively studied in a number of laboratories around the world and it is now accepted that 2 is really a prodrug that delivers nitric oxide (NO) as the active agent from the essential nitro group.10 The chemistry is initiated through conjugate addition with a hydride from a deazaflavin cofactor F420 present in deazaflavin dependent nitroreductase (Ddn) to the electron deficient aromatic imidazole that induces a reductive process (see Figure 2). 13,14
Figure 1.
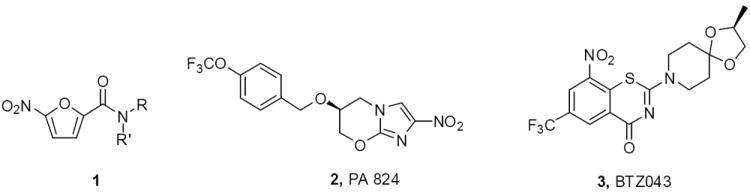
Representative structure of nitro aromatics as a class of anti-TB agents.
Figure 2.
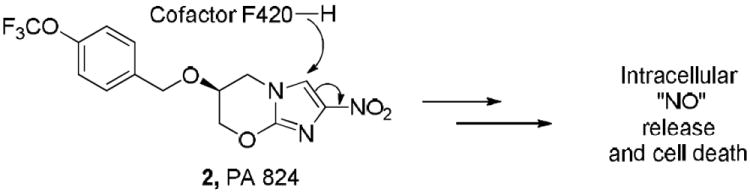
Mechanism of antitubercular action of PA824
BTZ043 has been shown to exhibit exceptional anti-TB activity in vitro and in vivo.11 It is also a prodrug, which, upon activation, results in the suicide inhibition of decaprenylphosphoryl-β-D-ribose 2′ oxidase (DprE1) of Mtb.15,16 Additional studies substantiated the importance of the nitro group by demonstrating that nitroreductase producing mycobacteria were resistant to 3.17,18 Chemical reduction of the nitro group to the hydroxylamine or completely to the corresponding amine also resulted in drastic loss of activity.11,15,16 Recent enzymatic studies with DprE1 obtained from M smegmatis (DprE1SM) with the BODIPY-X labeled analogue of 3 (compound 4, Figure 3) suggest that the presence of the DprE1SM substrate, decaprenylphosphoryl-β-D-ribosefuranose (DPR) at the active site is important for the reduction of the nitro group of 4 into the nitroso derivative. The authors hypothesize that the FADH2 generated during the oxidation of DPR to decaprenylphosphoryl-D-2′-keto-erythro-pentofuranose (DPX) is responsible for the nitro reduction of 4 into the nitroso derivative (5), which, in turn, reacts with the essential cysteine of DprE1SM to form a covalent semimercaptal adduct (6).16 DprE1 is an important enzyme required for arabinan biosynthesis, an essential process for the cell wall assembly of mycobacteria.19
Figure 3.
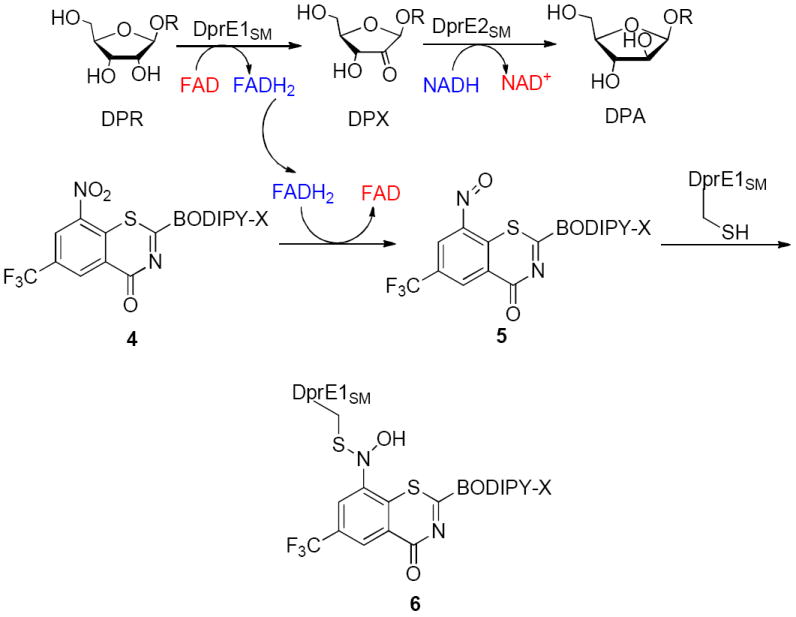
The proposed mechanism of DprE1 inhibition by BTZ-BODIPY-X.16
Crystal structures of DprE1 with BTZ043 and related analogues were recently reported.20,21 The study reported by Neres et al substantiated the formation of the “semimercaptal adduct” at the active site of DprE1 from reaction of the active site cysteine with the nitroso intermediate derived from 3 and concomitant oxidation of FADH2 to FAD.21 However, the mechanistic details related to the reduction of the nitro group of BTZ043 to the nitroso intermediate are yet to be fully elucidated. Specifically, how does FADH2, produced from FAD in DprE1 which is an oxidase (or dehydrogenase), initiate such reduction? Or if a reactive nitroso agent is generated by exogenous reductases, would it survive long enough to react with DprE1? Studies to address these questions will facilitate the rational design, syntheses and biological evaluations of new anti-TB agents. Herein, we report that the nitro aromatic compounds, design of which are based on BTZ043 and other related nitro aromatic compounds, react directly with thiolates and similarly basic and nucleophilic cyanide as well as hydrides to initiate redox chemistry that results in reduction of aromatic nitro groups to nitroso intermediates. The studies described here suggest that a cysteine thiolate or a hydride at the active site of DprE1 might induce the reduction of the nitro group of BTZ043 to the nitroso intermediate.
Results and Discussion
Intuitively, the structure of 3 suggests that it could be a substrate for classical nucleophilic aromatic substitution (Scheme 1) with the essential cysteine of DprE1. Therefore, 3 might simply be thought of as a thiol trap similar to the Sanger reagent, 2,4-dinitrofluorobenzene, or 2,4-dinitrobenzenesulfonamides that react with thiols at the carbon bearing the sulfonyl group and eventually release SO2 (see supporting information Scheme S1). The latter process has evolved into the eventual use of related sulfonamides as amine protecting groups22 and most recently as a prodrug strategy for development of new anti-TB agents that release SO2 as the active agent.23
Scheme 1.
BTZ043 possibly acting as a thiol trap at the DprE1 active site.
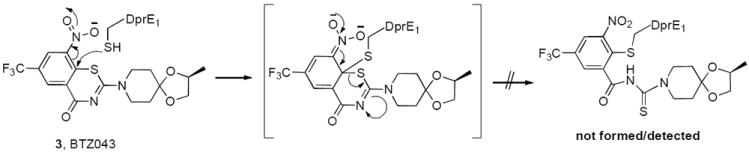
However, several separate observations were inconsistent with the standard nucleophilic aromatic substitution mechanism as the mode of action for 3 at DprE1. First, the compounds are remarkably selective and thus not general arylating agents. Secondly, no such direct nucleophilic aromatic substitution products of 3 or its analogues have been reported.11,15,16 Prior oxidation of the sulfur of BTZ043 to the sulfoxide or sulfone followed by nucleophilic substitution at the sulfur bearing carbon has been ruled out by isolation of the previously mentioned semimercaptal.21 Additionally, Mulliken charges calculated by AM1 method for 3 indicate that the unsubstituted aromatic carbons are electron deficient whereas the aromatic carbon adjacent to sulfur is electron rich. As a result, the nucleophilic attack at this position is unlikely (Figure 4, see supporting information for the Mulliken charges for all the atoms).
Figure 4.
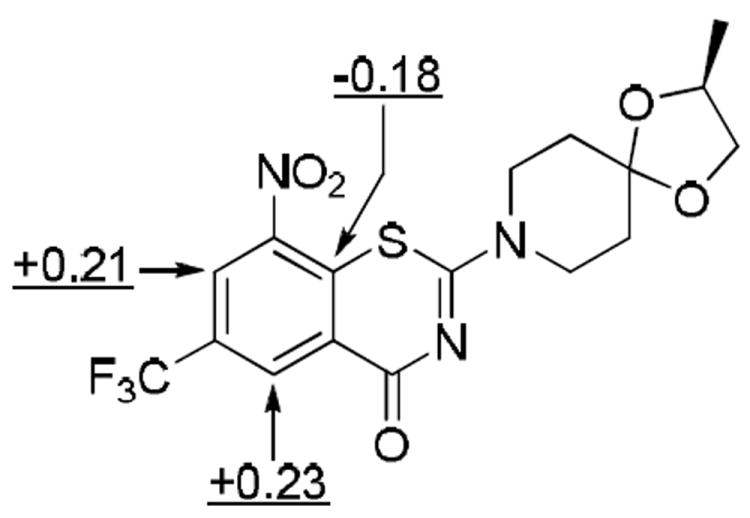
Key Mulliken charges for 3.
Other nitro aromatic compounds without the sulfur, and especially meta-trisubstituted nitro-benzamides were found to have significant anti-TB activity.24-26 This was initially thought to be surprising as they were structurally different from BTZs, yet were subsequently found to inhibit the same molecular target, DprE1.17,25 The combination of these studies and observations suggests that the molecular mode of action of 3 and simpler nitrobenzamides do not involve the simple nucleophilic aromatic substitution, such as the one shown in Scheme 1, at the active site of DprE1 with the thiol of the essential cysteine.
In order to explore reactions of these nitro aromatic anti-TB agents with thiols, we designed and synthesized simplified nitro benzamides 11-12 that can be considered to be hybrids of 3 and the nitrobenzamides (7)17,24-26 as the latter were found to have the same target as 3.17,25 Briefly, the synthesis involved the EDC-mediated coupling of nitrobenzoic acids, 8 and 9 with (S)-2-methyl-1,4-dioxa-8-azaspiro[4.5]decane (10) to obtain 11, 12, respectively (Scheme 2). For additional comparative studies we also synthesized simple nitrobenzamides 14 and 15 (Scheme 2). Compound 15 was recently discovered to have anti-TB activity using high throughput screening.15
Scheme 2.
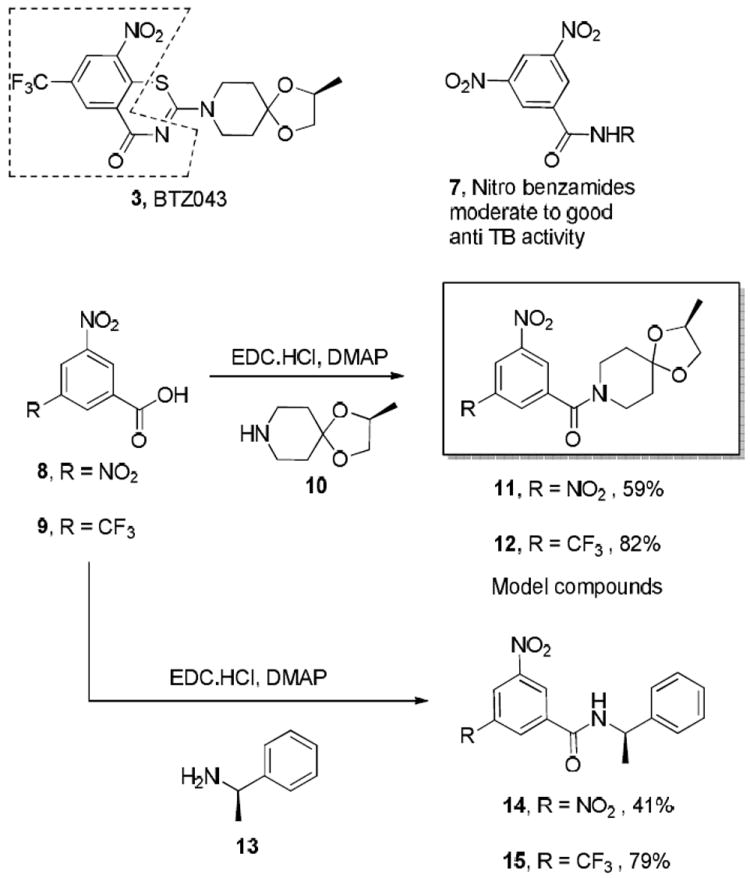
Design and synthesis of the nitroaromatic model compounds based on BTZ043 and other nitrobenzamides.
The similarity among the model and the prototype nitroaromatics, such as 3 and 7, is emphasized in Scheme 2 and we anticipated that studies of these model compounds, which do not have a built-in leaving group for classical nucleophilic aromatic substitution, would help determine if they are reactive with thiols.
Before studying the reaction with thiols, we evaluated the synthesized compounds (11, 12, 14 and 15) for their anti-mycobacterial activity in vitro (see Supporting Information Table S1). As anticipated, dinitro compounds 11 and 14 were more active than the mono nitro analogs 12 and 15, against M. vaccae and MtbH37Rv (MIC = 0.8 μM and 0.25 μM for 11 and 14, respectively vs MIC = > 50 μM and 0.95 μM for 12 and 15, respectively in 7H12 media). Although the purpose of this study was to determine inherent chemical reactivity of nitroaromatic compounds to help discern possible modes of biological activity, it was heartening to see that none of these compounds were found to be toxic as judged by VERO toxicity assay (see Supporting Information Table S1 for VERO toxicity data). Nitroaromatic compounds are often avoided in medicinal chemistry because of potential toxicity concerns; however, studies of 2 and 3 indicated that these compounds have remarkably clean profiles, yet are very potent and selective anti-TB agents. Similarly, the biological assays in H37Rv coupled with VERO toxicity data obtained for 11, 12, 14 and 15 indicated that these compounds have good safety profiles and are potential leads for development of anti-TB agents. When 3, 11 and 12 were exposed to cell cultures of M. smegmatis for 24 h, 3 and 12 were found to be completely reduced to the corresponding amines 16 and 18 respectively whereas 11 was found to be mono-reduced to 17. None of the above compounds were reduced in the non-mycobacterial control experiments in cell culture media. These experiments indicate that the nitro groups present in 3, 11 and 12 undergo similar metabolic transformations in M. smegamtis cell cultures and thus may have common target and inactivating enzymes.
We then investigated the reaction between the hybrid compounds and a series of thiols in both organic and mixed aqueous solvents under neutral conditions. Interestingly, prolonged treatment of 11, 12, 14 or 15 with methanethiol and glutathione induced no reaction. However, upon addition of sodium methanethiolate (NaSMe) to separate solutions of 11 and 15, we observed an immediate color change to a dark red solution, reminiscent of the formation of a Meisenheimer complex during classical nucleophilic aromatic substitution reactions, even though neither 11 nor 15 contains a typical leaving group.
In order to investigate the chemistry further, we carried out LC/MS studies of the reaction between 11 and NaSMe (Scheme 4). The LC/MS analysis revealed generation of a complex mixture containing compounds with molecular weights corresponding to starting material, partially reduced compound (compound 17), and possibly the adducts with methane thiol (compounds 20-23), and the corresponding dimeric forms of the nitroso intermediate (azoxy analogue, compound 19) generated in situ from 11 (see Supporting Information for the LC/MS spectra).
Scheme 4.
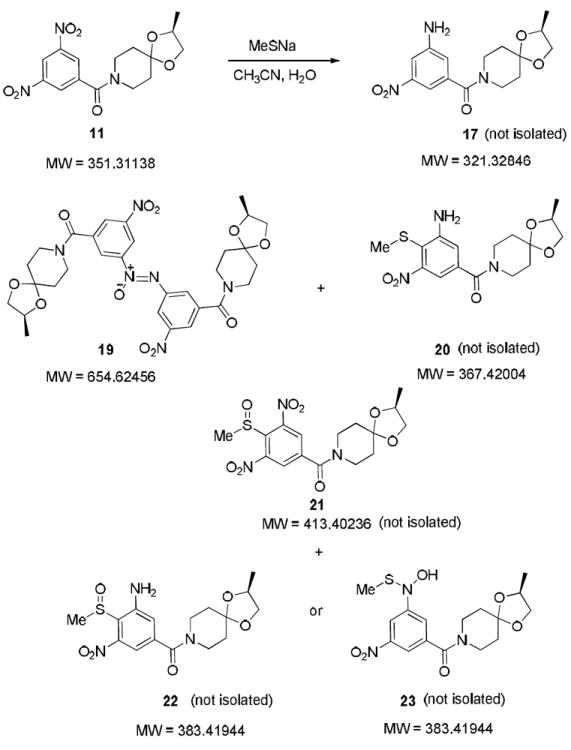
Probable structures of the molecular ions found in the LC/MS analysis of the reaction between 11 and sodium methanethiolate (see Supporting Information for LC/MS spectra).
The 1H NMR of the crude reaction mixture after neutralization and normal extractive work up indicated the proton signals corresponding to compound 19, which was later confirmed by its isolation and characterization. The 1H NMR also indicated the presence of compound(s) with substitution at the aromatic carbon between the two nitro groups indicating that some of the product(s) formed may be similar to the thiol adducts shown in Scheme 4. Subsequent chromatography of the crude material did not allow isolation of pure samples of any of the compounds with a molecular weight corresponding the thiol adducts, such as compounds 20-23.
As mentioned above, azoxy compound, 19 was isolated and fully characterized. Azoxy compounds like 19 are well known to be formed by dimerization of aryl nitroso species followed by loss of a single oxygen. In fact, many electron deficient aryl nitroso compounds are known to exist in equilibrium with their dimer,27 but subsequent loss of a single oxygen prohibits reformation of the monomeric nitroso agent. This process has plagued applications of nitroso cycloaddition and ene chemistry.28,29 It can be concluded from this study that NaSMe induced the redox chemistry of electron deficient nitro aromatic compound as verified by the formation of the compound 19. This induction of the redox chemistry by thiolates might mimic the reaction of nitro aromatic compounds such as 3 with DprE1 provided that cysteine at the active site is appropriately reactive (ionized).
The pKa of alkyl thiols are typically in the range of 10.5-11 and that of the thiol of cysteine is 10.8, but, as expected, substituents influence thiol acidity.30 Incorporation of a beta-hydroxy group on ethane thiol to give mercaptoethanol reduces the pKa value to 9.5 and in cysteine ethyl ester, the thiol pKa is 9.05. The biochemical reactivity of many functional groups in enzymes depends dramatically on their states of protonation. Ionization depends on the inherent pKa of the groups and the microenvironment created by the enzyme.30 The pKa values of functional groups found in the active sites of many enzymes often are significantly affected with changes of frequently >2 pKa units.30 Thus, it was exciting to consider that the thiol of the essential cysteine residue of DprE1 enzyme may be significantly ionized and/or nucleophilic in its microenvironment to initiate redox chemistry on 3 similar to the one observed for the reaction of 11 with methanethiolate.
There are some indications in the literature regarding the possible role of thiolate or thiols in the reduction of nitro groups to the corresponding nitroso intermediates. Leblanc et al,31 in their studies on the action of thiolate anion on nitrofluoraromatics observed that the reaction of m-fluoronitrobenzene with methanethiolate resulted only in the formation of the 3,3′-difluoroazoxybenzene, possibly via the reduction of the nitro to nitroso intermediate. Sodium dithionate,32 sodium trimethylsilanethiolate,33 chlorotrimethylsilane in combination with sodium sulfide,34 dihydrolipoamide in combination with ferrous ion,35 and thioacetate36 and ethane thiol27 have all been shown to reduce nitro aromatics to either aniline or amides (reductive amidation) presumably via the formation of a nitroso and/or hydroxylamine intermediates.33-36 Sulfides such as hydrogen sulfide, ammonium sulfide, sodium hydrosulfide and disulfide ion have been used in the Zinin reduction strategy in the past to achieve nitro aromatic reduction to anilines and benzene hydroxylamines.37-41 However, except for a study with ethane thiol in the presence of sodium hydroxide at 85°C in water27, attempts were not made to either detect and/or isolate the nitroso intermediates. The reaction conditions described in our work are milder and can essentially mimic the enzyme microenvironment. Thus, it was intuitive to hypothesize that the cysteine thiol at the active site of DprE1 may initiate the redox chemistry on 3 to convert it into the nitroso intermediate which would then be subsequently attacked by the same cysteine thiolate to form the covalent semimercaptal adduct. This hypothesis explains only the change in the oxidation state of nitro group of 3 to the corresponding nitroso intermediate. It does not, however, explain the corresponding change in oxidation state of cysteine (thiol), a process that is addressed later in this report. Nevertheless, this thiolate-induced reactivity opens up new avenues for the exploration of molecular mechanism of 3 and other anti-TB nitroaromatics.
While we encourage studies by those who have access to the enzyme, we extended the fundamental studies with additional thiolates such as Boc-cysteine, Boc-cysteamine, ethanethiol, and thiophenol (see Supporting Information for the LC/MS of the reactions of these thiols with 11). Indeed, simple changes from the reactions of the same set of previously used thiols under neutral conditions to basic conditions in mixed organic-aqueous solvents (apparent pH 9.5-11.5) induced rapid reaction with the model nitro aromatic compounds such as 11 and BTZ043 (3).
For example, reaction of 11 with Boc-cysteamine in acetonitrile/ water at an apparent pH of 11.4 resulted in the formation of the azoxy product thus indicating the transient generation of its predecessor nitroso species. It should be noted that only two products, 24, which was formed by the oxidation of the thiol, and the azoxy intermediate, 19, were isolated from the reaction mixture after work up (Scheme 5). Attempts to isolate and characterize the corresponding semimercaptal (Scheme 5, compound 25) or other thiol adducts similar to the ones shown in Scheme 4 were not fruitful because of their instability.
Scheme 5.
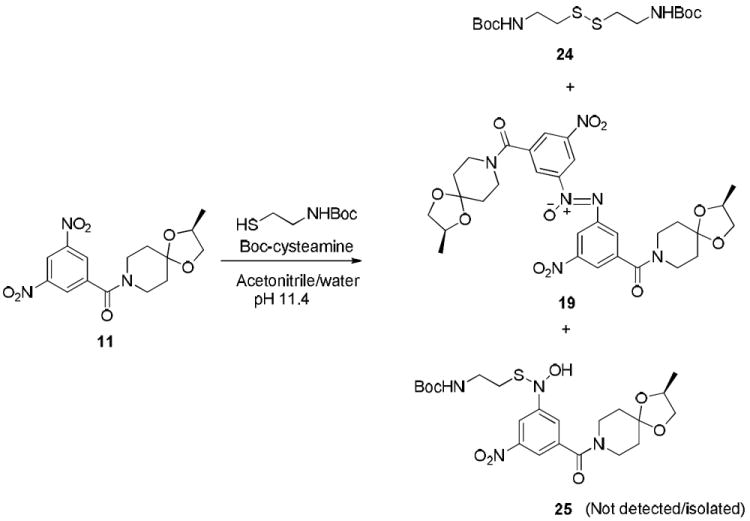
Reaction of 11 with Boc-cysteamine.
The reaction of 11 with Boc-cysteine at pH 10-11 essentially duplicated the observations above and only the Boc-cystine (oxidized cysteine) and nitroso derived azoxy intermediates were detected by LC/MS analysis (see Supporting Information). Incorporation of a smaller group at the aromatic carbon between the two nitro substituents such as a 4-methyl derivative of 11 (compound 39, see Supporting Information) did not significantly alter the outcome of this redox chemistry. Thus, reaction of 39 with NaSMe, resulted in formation of a mononitro reduced product and a corresponding azoxy dimer as judged by LC/MS analyses.
To confirm the generation of the nitroso intermediate in these reactions, we repeated the reaction of 11 with Boc-cysteamine under basic conditions (apparent pH 10-12) as a representative thiolate anion equivalent and with NaSMe in the presence α-terpinene and 1,3-cyclohexadiene, respectively, to trap any nitroso intermediate as a hetero Diels-Alder cycloadduct before it could be converted into the azoxy dimer (Scheme 6, see Supporting Information for additional reactions of 11 and Boc-cysteamine in the presence of 1,3-cyclohexadiene). The LC/MS analyses of these reactions indicated the formation of the corresponding hetero Diels-Alder cycloadducts. When BTZ043 was subjected to similar reaction conditions using thiol(ate)s, such as NaSMe or Boc-cysteamine, Boc-cysteine and ethane thiol at apparent pH 11-12 in the presence of α-terpinene, the formation of the hetero Diels-Alder cycloadduct was not detected. Only the reduced amine, 16, and the azo product, 27, were detected by LC/MS analyses and, after 24 h, 3 was completely consumed to afford 16 and 27 (Scheme 6). The formation of the reduced amine and the azo intermediate is still consistent with the in situ formation of the nitroso intermediate in the reaction42,27 and suggests that such reactions proceed by reduction of the nitro group to the nitroso intermediate prior to the formation of the amine, azoxy, or the azo intermediate. It should be noted that the outcome of the reaction of 11 with NaSMe in presence of 1,3-cyclohexadiene remained unaltered under deoxygenated conditions (see Supporting information for the experimental conditions and LC/MS spectra). Similar results were obtained when 3 was subjected to reaction with glutathione under basic conditions in the presence of 1,3-cyclohexadiene (see Supporting Information for LC/MS spectra). These experiments clearly indicate that thiolates induce redox chemistry of 3 and 11, a process that appears to mimic the redox activation of BTZ analogues at DprE1 (see Figure 2).
Scheme 6.
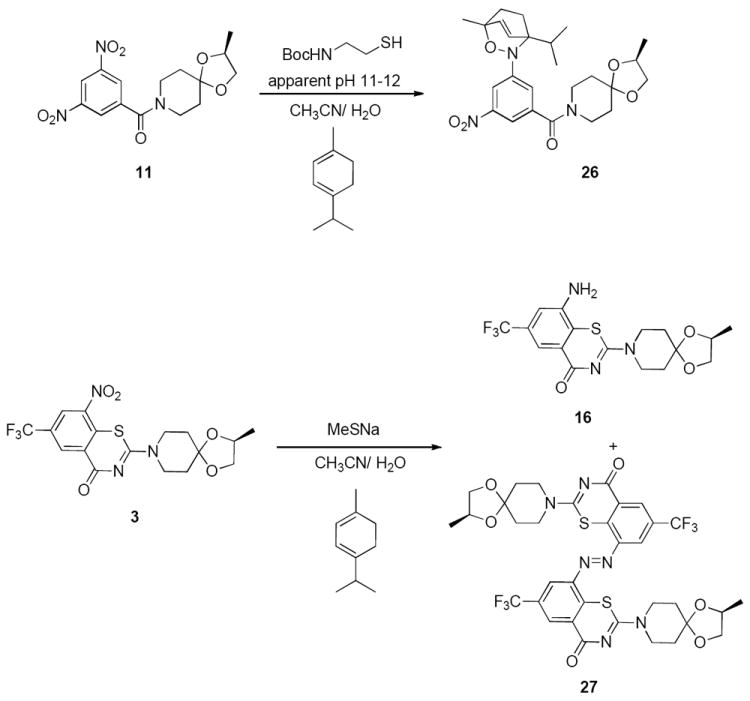
Trapping of the nitroso intermediate generated in the reaction of 11 and 3 with Boc-cysteamine.
We have extensively studied nitroso cycloaddition and ene reactions and their utility in syntheses.28 To substantiate the formation of the hetero Diels-Alder cycloadduct (26) generated in the reaction of 11 with Boc-cysteamine and verify that 3 indeed undergoes nitro reduction to the nitroso intermediate prior to the subsequent reduction to amine, we decided to subject 3 to a reaction with stannous chloride (SnCl2•2H2O) in the presence of 1,3-cyclohexadiene. The above reaction produced the expected nitroso cycloadduct 40 (see Supporting Information) which was isolated, purified, fully characterized.
While redox chemistry was shown to be induced by reaction with thiolates, attempts to isolate and individually characterize the thiol adducts such as the semimercaptal were unsuccessful, perhaps because of the well documented instability of the latter in aqueous solution and under basic conditions.43-45
Several interesting questions still remain, most notably: What is the fate of the thiol (ate) after the nitro reduction and what is the detailed mechanism of the redox chemistry? As mentioned earlier, direct LC/MS studies of the model reactions indicated the putative formation of the thiol adducts and the partially reduced form of the nitro substrates (nitroso equivalents) all of which were unstable and could not be isolated (Scheme 4). In the model experiments with Boc-cysteamine and Boc-cysteine, the corresponding disulfides were detected. While redox chemistry of thiols in solution would likely induce disulfide formation, the enzyme and model chemistries can proceed differently once the redox chemistry is initiated. The enzyme, DprE1 apparently contains a single cysteine thiol at the active site11,15 so it is unlikely that a disulfide could form. There is only one another cysteine located at ~ 15 Å from the cysteine present at the active site as observed in the X-ray crystal structure of DprE1 and only a conformational change could bring these two cysteines in close proximity to form the disulfide bond. However, oxidized forms such as a sulfenic acid or semimercaptal would cause the enzyme to be inactivated. To test for possible formation of sulfenic acid, the reaction between 11 and Boc-cysteamine was carried out with two different sulfenic acid trapping agents (see Supporting Information) namely, dimedone46 and 4-chloro-7-nitrobenzofurazan (NBD-Cl).47 However, reactions of 11 with dimedone were not conclusive and did not suggest any adduct formed with sulfenic acid under the reaction conditions whereas the reaction of 11 with NBD-Cl, as expected, resulted in nucleophilic displacement of the chlorine by Boc-cysteamine. (compound 41, see Supporting Information)
Detailed mechanistic studies are merited, and are ongoing, to explore this redox chemistry initiated by thiol(ate)s. Thiols and amino thiols are known to have a dramatic effect on redox behavior of other nitroaromatic drugs such as metronidazole 48 and even influence the enzymatic reduction of nitroimidazoles,49 but detailed mechanisms remain elusive. Thiols are known to react with aromatic nitroso agents to give unstable thionitroxide radicals [ARN(O•)SR]50 and, as mentioned earlier, form semimercaptals.44,51,52 Thiols, including glutathione, also react with one electron reduction products of nitroaromatic drugs53 but in some cases the thiol acts as a reducing agents only after alternative formation of the nitro radical anion.48
So far, the accumulated data presents a strong case for the redox chemistry between 3, 11 or an analogue of 11 with thiol(ate)s. To unambiguously synthesize a thiol adduct corresponding to addition to 11, and to demonstrate that a related appropriately substituted electron deficient aromatic compound can undergo classical nucleophilic aromatic substitution, a 4-chloro derivative of 11 (compound 42, see Supporting Information) was synthesized and subjected to reaction with Boc-cysteamine in the presence of an organic base. As expected, the reaction with Boc-cysteamine generated only the products of the nucleophilic aromatic substitution (compound 43, see Supporting Information). No such simple substitution products were detected by direct LC/MS analyses of reactions of 11 (which does not contain a classical leaving group) with either NaSMe or Boc-cysteamine.
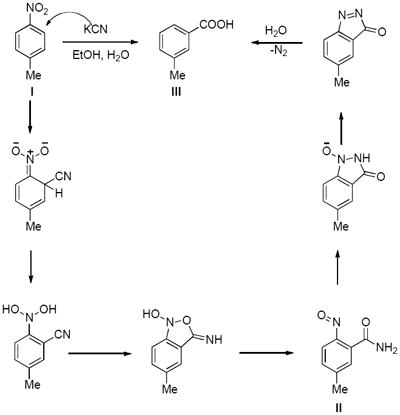
Recalling that the Mulliken charge calculations indicated that the non-substituted aromatic carbons of 3 are most electrophilic, we considered an alternative mechanism for generation of nitroso intermediates based on the well-known von Richter reaction which is initiated by cine addition to electron deficient aromatic compounds, especially nitro aromatics.54,55 Cine addition to aromatic nitro compounds that initiates reduction of the nitro group to a transient, reactive nitroso intermediate has ample precedent, (see the reaction described under reference no. 54 which illustrates the mechanism of the classic von Richter reaction for the conversion of p-nitrotoluene, I, to m-toluic acid, II via the formation of the 5-methyl-2-nitrosobenzamide, III).54,55 Also, though it has never been referenced as such, the first step of the prodrug chemistry of PA824 (2) is, in fact, a cine addition. In the von Richter reaction, the nucleophilic cyanide carbon is incorporated into the product. Mulliken charges calculated by the AM1 method for 3 and 11 (Figure 4 and see Supporting Information) indicated that the unsubstituted aromatic carbons which are ortho to the nitro substituents are electron deficient. The electronic character of the nitro aromatic portion of 3 and model hybrids reported herein appear to make them ideally suited for cine addition chemistry.
Although 2 and 3 have different targets and different modes of action, we hypothesize that the molecular mode of action of the latter also might be initiated by a cine addition of the essential cysteine sulfhydryl group either ortho or para to its essential nitro group. Mechanisms can be written to accommodate this hypothesis and also incorporate generation of a nitroso intermediate that could lead to covalent modification of the enzyme target, including generation of the corresponding semimercaptal (see Supporting Information Scheme S3).
To further demonstrate the propensity of the nitroaromatic compounds towards cine addition chemistry, we explored the possibility of 11 undergoing a von Richter reaction. Thus, it was subjected to a reaction with potassium cyanide (KCN) in acetonitrile / water at room temperature, essentially the same conditions described for the reactions with thiol(ates). The LC/MS of the reaction mixture indicated the formation of at least three identifiable reaction products. One of the reaction products corresponded to a single nucleophilic addition of a cyanide whereas the mass of the other two reaction products was related to double addition of cyanide and hence were isomeric (Scheme 7). The product corresponding to single cyanide addition was characterized by X-ray crystallography (see Supporting Information) and it turned out to be compound 31 whereas only one of the isomeric products related to double cyanide addition was characterized by X-ray and it turned out to be compound 32. The formation of 31 and 32 can only be explained via the formation of the nitroso intermediate, 29, which is formed first after the attack of the cyanide at the position between both of the nitro groups (Scheme 7). Nucleophilic substitutions of hydrogen in electron deficient nitroarenes have been reviewed.56,57 Electron deficient arenes have been shown to regenerate the aromaticity by loss of hydride after the initial attack by cyanide.58-60 (see conversion of 11 into 28). Such loss of hydride may help explain the reduction of 29 to 30 which eventually led to the formation of 31. When the reaction was repeated in the presence of α-terpinene, the LC/MS of the crude reaction mixture indicated the product corresponding to the trapped hetero-Diels-Alder cycloadduct of the nitroso intermediate with a cyanide addition (see Supporting Information). All of the above observations unequivocally substantiate the fact that cine addition of 11 results in the generation of the nitroso intermediate such as 29. This reaction still differs from the classical von Richter reaction in that one of the nitro groups becomes a part of the newly formed isooxazole ring system and the reaction was carried out at room temperature.
Scheme 7.
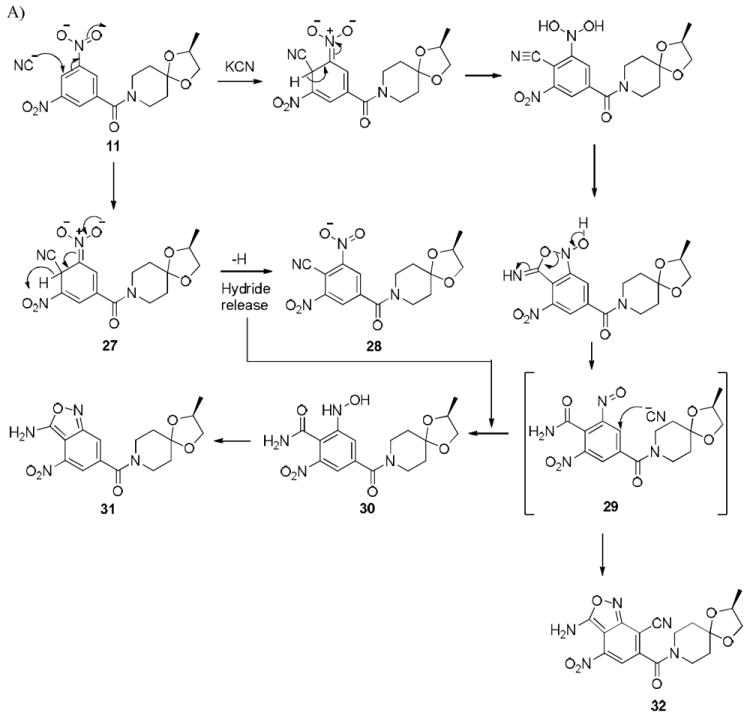
Reaction of 11 with KCN in CH3CN/H2O.
As a further indication of the susceptibility of these types of compounds to the von Richter reaction, we treated BTZ043 with K13CN under conditions similar to those described for 11 (Scheme 8). As shown in Scheme 8, two major products, 33 and 34, were isolated, corresponding to the generation of single and double cyanide additions. The isolated compounds were assigned the structures on the basis of our previous cyanide substitution experiments with 11 and 1H and 13C NMR studies (see Supporting Information).
Scheme 8.
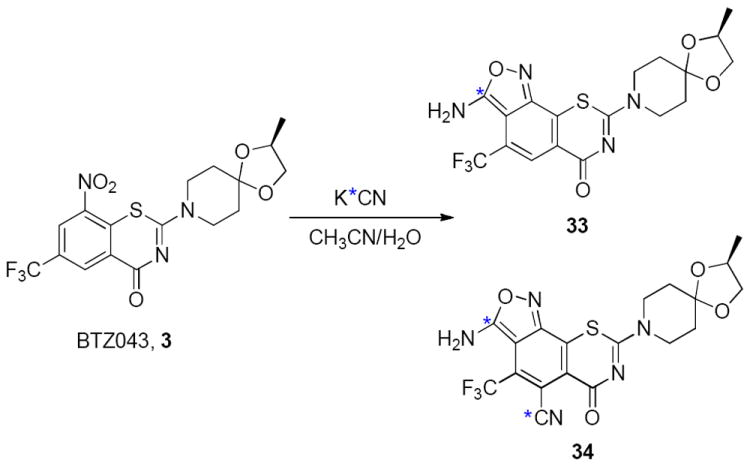
Reaction of 3 with K13CN in CH3CN/H2O. Blue asterisk indicates 99% C-13 enrichment.
All of the studies reported herein clearly indicate that the reaction with thiolates induce redox chemistry in which the nitro group is unquestionably converted to the nitroso intermediate which then undergoes further reactions, including classical dimerization. Trapping with dienes verified the generation of the nitroso moiety. Further chemistry of the nitroso and thiolate is possible. LC/MS and NMR studies of the reactions of thiolates with the nitro aromatic compounds indicate at least transient formation of adducts corresponding to the mass of the thiolate combined with the nitroso moiety, but they seem to be short lived and not yet amenable to isolation and full characterization.
A consistent mechanism (Scheme 9) for the generation of the nitroso intermediate invokes initial attack of thiolate in a “cine” addition to give 35. In the model chemical reaction, an additional thiolate can directly attack the thioether sulfur of 35 to induce rearomatization with simultaneous formation of the disulfide. The aromatized product 36 is at the nitroso oxidation state and can thus generate nitroso intermediate, 37 by dehydration. In the absence of other reagents, nitroso moieties are well known to rearrange to azoxy (38) or azo compounds (27). The cofactor FADH2 present near the active site of the enzyme may help regenerate the thiol functionality by acting as a hydride donor (see Scheme 9 for the regeneration of thiol from disulfide).
Scheme 9.
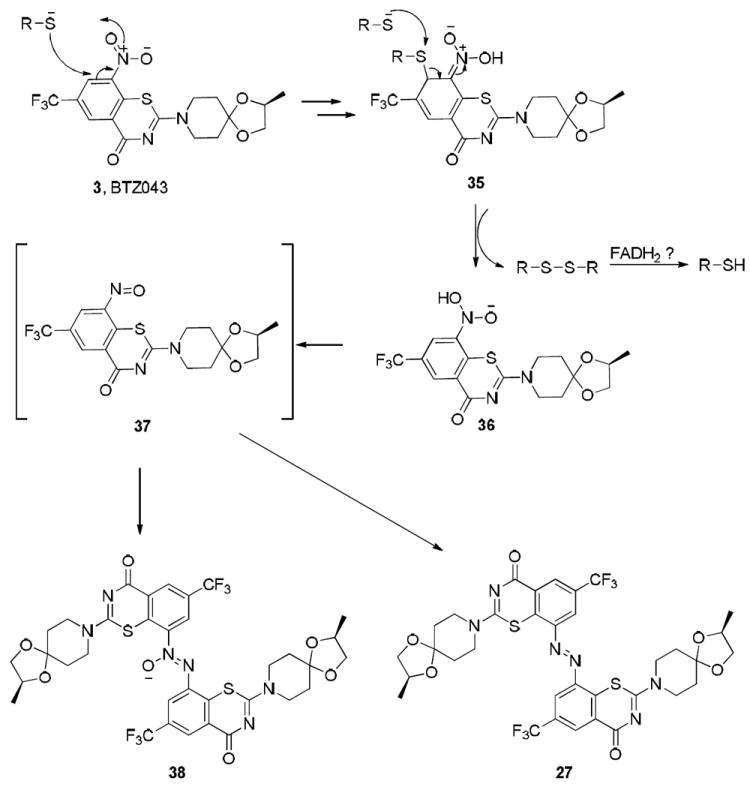
Potential mechanism for the reaction of thiolates with BTZ043 to initiate redox chemistry that mimics the essential cysteine-containing enzymatic process.
This chemical scheme accounts for the redox chemistry but not the formation of isolable adducts such as the reported semimercaptal. The model experiments reported by Makarov11,15 and others illustrated that separately generated nitroso agents do react with thiols to produce adducts as reported by many others in the literature. Recent additional crystallographic studies with DprE1SM with 3 are consistent with the formation of the semimercaptal adduct.20,21 Our model von Richter reactions on 3 and 11 with KCN substantiated our proposed mechanism for the conversion of nitro groups present in these compounds to the putative nitroso moiety as demonstrated by the observed products (Schemes 7 and 8). Specifically two products from the reaction of 11 with KCN were also analyzed by X-ray crystallography to unequivocally support the proposed structures and thereby the hypothesis behind the mechanism of their formation. Again, while the chemical mechanism in Scheme 9 is consistent with the model reactions, it remains to be seen if the only other cysteine thiolate in the enzyme is available for formation of the disulfide in a manner similar to that suspected for the chemical model reactions. However, related redox chemistry, especially any oxidation of the enzyme’s essential cysteine, would inactivate the enzyme. Mechanisms can be written that again start with the cine addition of the active site cysteine to 3 followed by oxidative elimination to generate a sulfenic acid with concomitant formation of the nitroso intermediate of 3 (see Supporting Information Scheme S3). While attempts to trap related sulfenic acids with dimedone (which is well known to react with sulfenic acids to form characterizable adducts) failed in the chemical model studies, their formation might have been circumvented by the rapid formation of disulfide. Recently, use of labeled dimedone derivatives to detect generation of trace amounts of sulfenic acids from cysteine residues in enzymatic reactions has been reported.61-63 This would be an appropriate study for those with access to the enzyme system.
Alternatively, the attack of the hydride from FADH2 in a manner similar to the attack of the thiolates and cyanide described herein to generate the nitroso intermediate can not be ruled out. To explore the possibility of such hydride reduction from FADH2 of the nitro group to the nitroso intermediate by cine addition mechanism, we subjected 11 to reaction with sodium triaceotxy borohydride (NaBH(OAc)3) in the presence of 1,3-cyclohexadiene in THF. As shown in Scheme 6, LC/MS analysis of the crude reaction mixture indicated the formation of hetero Diels-Alder cycloadduct, 26 (see Supporting Information for the LC/MS). We anticipated that the reduction of the nitro group to the nitroso intermediate prior to the formation of the hetero Diels-Alder cycloadduct occurred by the cine addition of the hydride and not by the direct reduction of nitro group by NaBH(OAc)3 as the later reagent is not known to cause a direct nitro reduction.64 Such reduction of the nitroaromatic compounds by nucleophilic attack of the hydride has been studied by deuterium labeling experiment in the past and known to involve the hydride attack on a carbon atom β to the nitro group (cine addition).65
Conclusion
The current anti-TB treatment regimen is 40 years old and suffers from poor patient compliance due to the long term drug administration, side effects and treatment costs that led to the emergence of MDR and XDR strains. There is a growing demand for new agents effective against TB.64 BTZ043 (3) and PA 824 (2) have been shown to kill M tuberculosis in vitro, ex vivo and in mouse models of TB. The simple compounds designed, synthesized and described here have promising anti-TB activity, are non-toxic and our work shows that these may share a similar mode of action with that of BTZ043. Our chemical studies described herein indicate that the unsubstituted aromatic carbon between the electron withdrawing groups of 11 and BTZ043 is susceptible to addition of the nucleophiles such as thiolates, cyanide and even hydrides. Thiolates as such can be responsible for the reduction of the nitro group present in hitherto mentioned compounds to the corresponding nitroso intermediates. The formation of the nitroso intermediates under such reaction conditions were verified either by isolation of the hetero Diels-Alder cycloadducts or by the isolation of the azoxy or azo intermediates. Additionally, reactions of KCN with 11 and BTZ043 gave products whose formation could only be explained by the in situ generation of the nitroso intermediates, thus verifying that structures such as 11 and BTZ043 are potential substrates for cine addition chemistry. Therefore, our work offers an alternate hypotheses for the activation of these compounds in that the cysteine thiol(ate) at the active site of DprE1 may induce the nitro reduction to the nitroso intermediates which subsequently cause inhibition of DprE1. Nevertheless, FADH2 may also be responsible for this particular reduction as demonstrated by our preliminary studies with hydride that are consistent with the cine addition chemistry described herein.
Supplementary Material
Scheme 3.
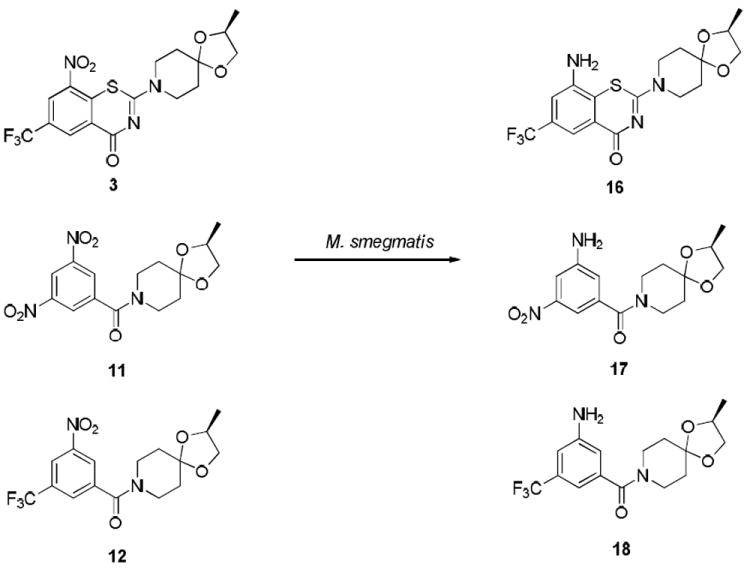
Cell culture experiments of compounds 3, 11 and 12 with M. smegmatis.
Acknowledgments
This research was supported in part by grant 2R01AI054193 from the National Institutes of Health (NIH). The same grant supported the anti-tuberculosis assays which were performed at the Institute for Tuberculosis Research, College of Pharmacy, University of Illinois at Chicago, under the direction of Prof. Scott G. Franzblau. We thank Dr. Albert Hinnen of Alere Technologies GmbH and Alere Inc. for providing BTZ043 and partial financial support, Dr. Ute Möllmann at the Leibniz Institute for Natural Product Research and Infection Biology – Hans Knöll Institute, Beutenbergstrasse 11a, D-07745 Jena, Germany for helpful discussions, and Dr. Anthony Serianni, University of Notre Dame, for providing K13CN. MCM thanks CBBI for financial support. We would like to thank the Office of the Vice President of Research at the University of Notre Dame for financial support for the purchase of the copper micro-focus source used in this research. We thank the University of Notre Dame, especially the Mass Spectrometry and Proteomics Facility (Bill Boggess, Michelle Joyce, and Nonka Sevova), which is supported by Grant CHE-0741793 from the National Science Foundation (NSF).
ABBREVIATIONS
- DMF
N,N-dimethyl formamide
- DprE1
decaprenylphosphoryl-β-D-ribose 2′ oxidase
- DprE1SM
decaprenylphosphoryl-β-D-ribose 2′ oxidase obtained from M. smegmatis
- LC/MS
liquid chromatography coupled to mass spectrometry
- KCN
potassium cyanide
- MDR
multi-drug resistant
- M tuberculosis
Mycobacterium tuberculosis
- M. smegmatis
Mycobacterium smegmatis
- XDR
extensively drug resistant
- TB
tuberculosis
- TLC
thin layer chromatography
Footnotes
Supporting Information
Complete experimental section along with the details of all biological assays and cell culture experiments, Mulliken charges for 3 and 11, LC/MS spectra and experimental procedures related to the nitroso trapping experiments, details related to the sulfenic acid trapping experiments, characterization of the compounds and crystallographic information files (CIF) for compounds 31 and 32. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Zink AR, Sola C, Reischl U, Grabner W, Rastogi N, Wolf H, Nerlich AG. J Clin Microbiol. 2003;41:359–367. doi: 10.1128/JCM.41.1.359-367.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Koch R. Berliner Klinischen Wochenschrift. 1882;19:221–230. [Google Scholar]
- 3.Dye C, Williams BG. Science. 2010;328:856–861. doi: 10.1126/science.1185449. [DOI] [PubMed] [Google Scholar]
- 4.World Health Organization (WHO) report. 2009 [Google Scholar]
- 5.Gandhi NR, Nunn P, Dheda K, Schaaf HS, Zignol M, van Soolingen D, Jensen P, Bayona J. The Lancet. 2010;375:1830–1843. doi: 10.1016/S0140-6736(10)60410-2. [DOI] [PubMed] [Google Scholar]
- 6.Koul A, Arnoult E, Lounis N, Guillemont J, Andries K. Nature. 2011;469:483–490. doi: 10.1038/nature09657. [DOI] [PubMed] [Google Scholar]
- 7.Villemagne B, Crauste C, Flipo M, Baulard AR, Deprez B, Willand N. Eur J Med Chem. 2012;51:1–16. doi: 10.1016/j.ejmech.2012.02.033. [DOI] [PubMed] [Google Scholar]
- 8.Hevener KE, Ball DM, Buolamwini JK, Lee RE. Bioorg Med Chem. 2008;16:8042–8053. doi: 10.1016/j.bmc.2008.07.070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marsini MA, Reider PJ, Sorensen EJ. J Org Chem. 2010;75:7479–7482. doi: 10.1021/jo1015807. [DOI] [PubMed] [Google Scholar]
- 10.Singh R, Manjunatha U, Boshoff HIM, Ha YH, Niyomrattanakit P, Ledwidge R, Dowd CS, Lee IY, Kim P, Zhang L, Kang S, Keller TH, Jiricek J, Barry CE., 3rd Science. 2008;322:1392–1395. doi: 10.1126/science.1164571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Makarov V, Manina G, Mikusova K, Möllmann U, Ryabova O, Saint-Joanis B, Dhar N, Pasca MR, Buroni S, Lucarelli AP, Milano A, De Rossi E, Belanova M, Bobovska A, Dianiskova P, Kordulakova J, Sala C, Fullam E, Schneider P, McKinney JD, Brodin P, Christophe T, Waddell S, Butcher P, Albrethsen J, Rosenkrands I, Brosch R, Nandi V, Bharath S, Gaonkar S, Shandil RK, Balasubramanian V, Balganesh T, Tyagi S, Grosset J, Riccardi G, Cole ST. Science. 2009;324:801–804. doi: 10.1126/science.1171583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Karoli T, Becker B, Zuegg J, Möllmann U, Ramu S, Huang JX, Cooper MA. J Med Chem. 2012;55:7940–7944. doi: 10.1021/jm3008882. [DOI] [PubMed] [Google Scholar]
- 13.Cellitti Susan E, Shaffer J, Jones David H, Mukherjee T, Gurumurthy M, Bursulaya B, Boshoff Helena I, Choi I, Nayyar A, Lee Yong S, Cherian J, Niyomrattanakit P, Dick T, Manjunatha Ujjini H, Barry Clifton E, Spraggon G, Geierstanger Bernhard H. Structure (London, England : 1993) 2012;20:101–112. doi: 10.1016/j.str.2011.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Manjunatha UH, Boshoff H, Dowd CS, Zhang L, Albert TJ, Norton JE, Daniels L, Dick T, Pang SS, Barry CE. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:431–436. doi: 10.1073/pnas.0508392103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Trefzer C, Rengifo-Gonzalez M, Hinner MJ, Schneider P, Makarov V, Cole ST, Johnsson K. J Am Chem Soc. 2010;132:13663–13665. doi: 10.1021/ja106357w. [DOI] [PubMed] [Google Scholar]
- 16.Trefzer C, Skovierova H, Buroni S, Bobovska A, Nenci S, Molteni E, Pojer F, Pasca MR, Makarov V, Cole ST, Riccardi G, Mikusova K, Johnsson K. J Am Chem Soc. 2012;134:912–915. doi: 10.1021/ja211042r. [DOI] [PubMed] [Google Scholar]
- 17.Ribeiro AL, Degiacomi G, Ewann F, Buroni S, Incandela ML, Chiarelli LR, Mori G, Kim J, Contreras-Dominguez M, Park YS, Han SJ, Brodin P, Valentini G, Rizzi M, Riccardi G, Pasca MR. PLoS One. 2011;6:e26675. doi: 10.1371/journal.pone.0026675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Manina G, Bellinzoni M, Pasca MR, Neres J, Milano A, De Jesus Lopes Ribeiro AL, Buroni S, Škovierová H, Dianišková P, Mikušová K, Marák J, Makarov V, Giganti D, Haouz A, Lucarelli AP, Degiacomi G, Piazza A, Chiarelli LR, De Rossi E, Salina E, Cole ST, Alzari PM, Riccardi G. Molecular Microbiology. 2010;77:1172–1185. doi: 10.1111/j.1365-2958.2010.07277.x. [DOI] [PubMed] [Google Scholar]
- 19.Wolucka BA. FEBS J. 2008;275:2691–2711. doi: 10.1111/j.1742-4658.2008.06395.x. [DOI] [PubMed] [Google Scholar]
- 20.Batt SM, Jabeen T, Bhowruth V, Quill L, Lund PA, Eggeling L, Alderwick LJ, Futterer K, Besra GS. Proc Natl Acad Sci U S A. 2012;109:11354–11359. doi: 10.1073/pnas.1205735109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Neres J, Pojer F, Molteni E, Chiarelli LR, Dhar N, Boy-Röttger S, Buroni S, Fullam E, Degiacomi G, Lucarelli AP, Read RJ, Zanoni G, Edmondson DE, De Rossi E, Pasca MR, McKinney JD, Dyson PJ, Riccardi G, Mattevi A, Cole ST, Binda C. Science Translational Medicine. 2012;4:150ra121–150ra121. doi: 10.1126/scitranslmed.3004395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fukuyama T, Cheung M, Jow C-K, Hidai Y, Kan T. Tetrahedron Lett. 1997;38:5831–5834. [Google Scholar]
- 23.Malwal SR, Sriram D, Yogeeswari P, Konkimalla VB, Chakrapani H. J Med Chem. 2012;55:553–557. doi: 10.1021/jm201023g. [DOI] [PubMed] [Google Scholar]
- 24.Kappes B, Neumann T, Unger E, Dahse H-M, Möllmann U, Schlegel B. Application: DE 1015805. Hans-Knoell-Institut fuer Naturstoff-Forschung E.V., Germany; Institut fuer Molekulare Biotechnologie (IMB); Institut Fuer Molekulare Biotechnologie E.V.; 2003. p. 16. [Google Scholar]
- 25.Christophe T, Jackson M, Jeon HK, Fenistein D, Contreras-Dominguez M, Kim J, Genovesio A, Carralot J-P, Ewann F, Kim EH, Lee SY, Kang S, Seo MJ, Park EJ, Śckovierová H, Pham H, Riccardi G, Nam JY, Marsollier L, Kempf M, Joly-Guillou M-L, Oh T, Shin WK, No Z, Nehrbass U, Brosch R, Cole ST, Brodin P. PLoS Pathog. 2009;5:e1000645. doi: 10.1371/journal.ppat.1000645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Brodin P, Christophe T, No Z, Kim J, Genovesio A, Fenistein DPC, Jeon H, Ewann FA, Kang S, Lee S, Seo MJ, Park E, Contreras Dominguez M, Nam JY, Kim EH. Application: WO2010003533. Institut Pasteur Korea, S. Korea; Institut National de la Sante et de la Recherche Medicale; 2010. p. 328. [Google Scholar]
- 27.Errede LA, Davis HR. J Org Chem. 1963;28:1430–1431. [Google Scholar]
- 28.Bodnar BS, Miller MJ. Angew Chem Int Ed Engl. 2011;50:5630–5647. doi: 10.1002/anie.201005764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Adam W, Krebs O, Orfanopoulos M, Stratakis M, Vougioukalakis GC. J Org Chem. 2003;68:2420–2425. doi: 10.1021/jo0266240. [DOI] [PubMed] [Google Scholar]
- 30.Harris TK, Turner GJ. IUBMB Life. 2002;53:85–98. doi: 10.1080/15216540211468. [DOI] [PubMed] [Google Scholar]
- 31.Leblanc ME, Peach ME, Winter HM. J Fluorine Chem. 1981;17:233–248. [Google Scholar]
- 32.Scheuerman RA, Tumelty D. Tetrahedron Lett. 2000;41:6531–6535. [Google Scholar]
- 33.Hwu JR, Wong FF, Shiao MJ. J Org Chem. 1992;57:5254–5255. [Google Scholar]
- 34.Shiao MJ, Lai LL, Ku WS, Lin PY, Hwu JR. J Org Chem. 1993;58:4742–4744. [Google Scholar]
- 35.Kijima M, Nambu Y, Endo T, Okawara M. J Org Chem. 1983;48:2407–2409. [Google Scholar]
- 36.Bhattacharya A, Purohit VC, Suarez V, Tichkule R, Parmer G, Rinaldi F. Tetrahedron Lett. 2006;47:1861–1864. [Google Scholar]
- 37.Haworth RD, Lapworth A. J Chem Soc Trans. 1921;119:768–777. [Google Scholar]
- 38.Lapworth A, Pearson LK. J Chem Soc Trans. 1921;119:765–768. [Google Scholar]
- 39.Porter HK. Org React. 1973;20:455–481. [Google Scholar]
- 40.Scribner JD, Miller JA. J Chem Soc. 1965:5377–5380. [Google Scholar]
- 41.Zinin N. J Prakt Chem. 1842;27:140–153. [Google Scholar]
- 42.Zuman P, Shah B. Chem Rev. 1994;94:1621–1641. [Google Scholar]
- 43.Eyer P. Chem Biol Interact. 1979;24:227–239. doi: 10.1016/0009-2797(79)90011-5. [DOI] [PubMed] [Google Scholar]
- 44.Kazanis S, McClelland RA. J Am Chem Soc. 1992;114:3052–3059. [Google Scholar]
- 45.Klehr H, Eyer P, Schafer W. Biol Chem Hoppe Seyler. 1985;366:755–760. doi: 10.1515/bchm3.1985.366.2.755. [DOI] [PubMed] [Google Scholar]
- 46.Benitez LV, Allison WS. J Biol Chem. 1974;249:6234–6243. [PubMed] [Google Scholar]
- 47.Ellis HR, Poole LB. Biochemistry. 1997;36:15013–15018. doi: 10.1021/bi972191x. [DOI] [PubMed] [Google Scholar]
- 48.Tocher JH, Edwards DI. Biochem Pharmacol. 1995;50:1367–1371. doi: 10.1016/0006-2952(95)02010-1. [DOI] [PubMed] [Google Scholar]
- 49.Ka-Ho W, Agrawal KC. Biochem Pharmacol. 1988;37:473–479. doi: 10.1016/0006-2952(88)90217-1. [DOI] [PubMed] [Google Scholar]
- 50.Waters WA. J Chem Soc, Perkin Trans. 1979;2:1078–1083. [Google Scholar]
- 51.Ellis MK, Hill S, Foster PM. Chem Biol Interact. 1992;82:151–163. doi: 10.1016/0009-2797(92)90107-v. [DOI] [PubMed] [Google Scholar]
- 52.Eyer P, Schneller M. Biochem Pharmacol. 1983;32:1029–1036. doi: 10.1016/0006-2952(83)90621-4. [DOI] [PubMed] [Google Scholar]
- 53.Núñez-Vergara LJ, Díaz-Araya G, Olea-Azar C, Atria AM, Bollo-Dragnic S, Squella JA. Pharmaceut Res. 1998;15:1690–1695. doi: 10.1023/a:1011948410183. [DOI] [PubMed] [Google Scholar]
-
54.Suwinski J, Swierczek K. Tetrahedron. 2001;57:1639–1662.
- 55.von Richter V. Deut Chem Ges Ber. 1871;iv:21. [Google Scholar]
- 56.Makosza M, Wojciechowski K. Chem Rev. 2004;104:2631–2666. doi: 10.1021/cr020086+. [DOI] [PubMed] [Google Scholar]
- 57.Makosza M. Chem Soc Rev. 2010;39:2855–2868. doi: 10.1039/b822559c. [DOI] [PubMed] [Google Scholar]
- 58.Okamoto T, Takahashi H. Chem Pharm Bull. 1968;16:1700–1704. [Google Scholar]
- 59.Okamoto T, Takahashi H. Chem Pharm Bull. 1971;19:1809–1814. [Google Scholar]
- 60.Okamoto T, Takahashi H, Takayama H, Kitagawa T, Ikeda M. Chem Pharm Bull. 1969;17:140–144. [Google Scholar]
- 61.Leonard SE, Carroll KS. Current Opinion Chem Biol. 2010;15:88–102. doi: 10.1016/j.cbpa.2010.11.012. [DOI] [PubMed] [Google Scholar]
- 62.Seo YH, Carroll KS. Angew Chem Int Ed. 2011;50:1342–1345. doi: 10.1002/anie.201007175. [DOI] [PubMed] [Google Scholar]
- 63.Truong TH, Garcia FJ, Seo YH, Carroll KS. Bioorg Med Chem Lett. 2011;21:5015–5020. doi: 10.1016/j.bmcl.2011.04.115. [DOI] [PubMed] [Google Scholar]
- 64.Abdel-Magid AF, Carson KG, Harris BD, Maryanoff CA, Shah RD. J Org Chem. 1996;61:3849–3862. doi: 10.1021/jo960057x. [DOI] [PubMed] [Google Scholar]
- 65.Suwiński J, Wagner P, Holt EM. Tetrahedron. 1996;52:9541–9552. [Google Scholar]
- 66.Rivers EC, Mancera RL. Curr Med Chem. 2008;15:1956–1967. doi: 10.2174/092986708785132906. [DOI] [PubMed] [Google Scholar]
- 67.Huang S-H, Bai Z-W, Feng J-W. Mag Reson Chem. 2009;47:423–427. doi: 10.1002/mrc.2406. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.