

Abstract
The HIV-1 protein Tat is a critical regulator of viral transcription and has also been implicated as a mediator of HIV-1 induced neurotoxicity. Here using a high throughput screening assay, we identified the GSK-3 inhibitor 6BIO, as a Tat-dependent HIV-1 transcriptional inhibitor. Its ability to inhibit HIV-1 transcription was confirmed in TZM-bl cells, with an IC50 of 40 nM. Through screening 6BIO derivatives, we identified 6BIOder, which has a lower IC50 of 4 nM in primary macrophages and 0.5 nM in astrocytes infected with HIV-1. 6BIOder displayed an IC50 value of 0.03 nM through in vitro GSK-3β kinase inhibition assays. Finally, we demonstrated 6BIO and 6BIOder have neuroprotective effects on Tat induced cell death in rat mixed hippocampal cultures. Therefore 6BIO and its derivatives are unique compounds which, due to their complex mechanisms of action, are able to inhibit HIV-1 transcription as well as to protect against Tat induced neurotoxicity.
Keywords: HIV-1, transcriptional inhibition, GSK-3, Tat, HAND, neuroprotection, indirubin, therapeutic target
INTRODUCTION
Although current antiretroviral therapy has had a major impact on prolonging the life span of HIV-1 infected individuals it also has important limitations. Most of these therapies prevent viral entry into the cells or formation of proviral DNA. However, once the proviral DNA is integrated into the chromosome, the protease inhibitors are the only class of drugs available that prevents the formation of infectious viral particles. These compounds do not prevent the formation of the early viral proteins such as Tat. The HIV-1 Tat protein is a critical regulator of HIV replication and can be released extracellularly where it may have a multitude of effects. This is particularly important within the brain, where the virus enters early in the course of infection and establishes reservoirs in glial cells. Extracellular Tat is a potent neurotoxin and may be an important mediator of neuronal dysfunction associated with HIV-1 –associated neurocognitive disease (HAND) (Magnuson et al., 1995; Maragos et al., 2003; Sabatier et al., 1991; Weeks et al., 1995). Recent studies suggest that despite excellent control of HIV replication in the periphery, nearly 50-70% of individuals develop neurocognitive impairment. Hence approaches to prevent the effects of HIV replication in the brain are critically necessary.
Elongation of HIV-1 transcripts is dependent on the association of HIV-1 Tat with the nascent RNA stemloop structure of the transactivation response element (TAR). This process is accompanied by the binding of several cellular proteins, in particular the P-TEFb complex comprising of cyclin T1 or CDK9 to the RNAP II complex associated with the HIV-1 LTR (Fujinaga et al., 1998; Herrmann and Rice, 1995; Laspia et al., 1989; Nabel and Baltimore, 1987; Peng et al., 1998; Rice and Mathews, 1988; Sobhian et al., 2010; Wei et al., 1998). This unique interaction of HIV-1 Tat with TAR it provides an ideal target for pharmaceutical intervention. In vitro a variety of compounds have been demonstrated to inhibit HIV-1 replication. Some of these compounds have been described to specifically interfere with Tat-TAR interaction. The Novartis compound CGP 40336A was described to selectively bind to the AU base pair above the trinucleotide bulge with additional stacking interactions to the bulge (Hamy et al., 1998). The multicyclic dyes, Hoechst 33258, DAPI and berenil bind to the cavity created by the trinucleotide bulge (Bailly et al., 1996; Dassonneville et al., 1997; Edwards and Sigurdsson, 2002; Mestre et al., 1999). Neomycin binds to the minor groove of the lower helix, whereas argininamide was found to bind to the U23-A27-U38 base triple (Aboul-ela et al., 1995; Brodsky et al., 1998; Brodsky and Williamson, 1997; Faber et al., 2000; Nifosi et al., 2000). Other compounds inhibit HIV-1 transcription without interfering with Tat-TAR interaction (K-12, Ro24-7429) (Baba et al., 1998; Baba et al., 1997; Hsu et al., 1993). However, none of these compounds have proven clinically useful. Ro24-7429 was even advanced into early clinical trials (Hsu et al., 1993), but no inhibitory effect on HIV-1 replication in patients was observed during a Phase I/II clinical trial, despite sufficiently high drug plasma levels and no development of viral resistance (Haubrich et al., 1995). While some drug targets, such as the Tat-TAR interaction are clearly defined, the more recent discoveries that WP631, Temacrazine or CDK inhibitors (Agbottah et al., 2005; Galons et al., 2010; Guendel et al., 2010; Kashanchi and Kehn-Hall, 2009; Malumbres et al., 2008; Van Duyne et al., 2008), would also inhibit HIV-1 transcription hint at the possibility that there are potential features of HIV-1 transcription that are not appreciated as drug targets yet.
Tat is released by infected lymphoid (Ensoli et al., 1993) and glial cells (Tardieu et al., 1992). Both forms of Tat are released (i.e. Tat formed by the first exon only, and that formed by both first and second exons) (Malim and Cullen, 1991) and are cytotoxic to neurons (Magnuson et al., 1995; Maragos et al., 2003; Sabatier et al., 1991; Weeks et al., 1995). Tat effects on neurons involve excitotoxic mechanisms, and this is similarly true for gp120. αv integrin subunit-containing receptors (Barillari et al., 1993; Etienne-Manneville and Hall, 2001; Noonan and Albini, 2000), vascular endothelial growth factor-1 receptor (VEGF-1 receptor or flt-1) (Krum and Rosenstein, 1998), low-density lipoprotein receptor-related protein (LPR) (Liu et al., 2000), and NMDA receptors (Haughey et al., 2001) (NMDA receptor activation may be secondary to GPCR activation) (Haughey and Mattson, 2002; Nath et al., 1996) have all been proposed as targets for Tat (Noonan and Albini, 2000; Rusnati and Presta, 2002). Interactions with excitatory amino acid receptors (Haughey and Mattson, 2002; Magnuson et al., 1995; Nath et al., 1996), with accompanying increases in Ca2+ and reactive oxygen species (Bonavia et al., 2001; Kruman et al., 1998; Nath et al., 1996), may be especially detrimental. Tat injection into the brain (Jones et al., 1998; Philippon et al., 1994; Sabatier et al., 1991), including the striatum (Bansal et al., 2000), causes gliosis and infiltration of macrophages, production of cytotoxic cytokines, and chemokines such as MCP-1 (Conant et al., 1998; Weiss et al., 1999). Intrastriatal Tat injections induce neurodegenerative changes (Aksenov et al., 2003; Philippon et al., 1994), which precedes peak increases in macrophages/microglia at 24 h (Aksenov et al., 2003). Dying neurons are no longer seen at 7 days following Tat exposure during peak periods of astrogliosis suggesting the neuronal losses are not secondary to reactive astroglial changes. There is also evidence from culture studies that Tat is directly neurotoxic because toxicity occurs in highly enriched cultures of striatal neurons (Bonavia et al., 2001). Very brief exposures to Tat can cause neuronal death (Magnuson et al., 1995; Nath et al., 1999). The core domain of Tat, amino acids 21-40 can induce cytopathic effects in monocytes and angiogenesis (Boykins et al., 1999).
Here we present data on small chemical molecules that can inhibit both Tat dependent transcription and Tat induced neurotoxicity. These small chemical molecules are inhibitors of GSK-3β, which is known to have important implications in HAND. We reasoned that signaling molecules downstream of Tat that may be common to its effects on HIV-1 replication and neurotoxicity may be important therapeutic targets.
RESULTS
HIV-1 transcriptional inhibitor screening system
We have previously developed a HTS compatible reporter cell line in which a stably integrated chronically active HIV-1 derived from a patient isolate drives eGFP expression controlled by a HIV-1 LTR (CUCY cells-Jurkat based) (Kempf et al., 2006). eGFP expression thus serves as a direct and quantitative marker of HIV-1 expression. By the inclusion of a second fluorescent protein that can be spectrally separated from GFP (DsRedExpress) and which is controlled by an activation-independent promoter (MSCV-LTR), we obtained a reporter cell line in which we can simultaneously measure the influence of compounds on HIV-1 transcription (on-target) and general transcription (off-target). By this means, false positive hits, in which the compound would non-specifically inhibit global transcription, could be minimized.
However, the requirement for a BSL2+/BSL3 facility to perform the actual drug screen is very restrictive and also significantly increases the costs of drug screening. To overcome these limitations, we had earlier attempted to establish a non-infectious drug screening system similar to CUCY cells, in which the integrated LTR-eGFP promoter construct in the parental JLTRG cells would be activated through stably integrated HIV-1 Tat expression plasmids. However, initial attempts to obtain high eGFP expressing cells through either stable transfection or retroviral transduction with Tat expression vectors failed. For example, retroviral vectors expressing HIV-1 Tat under the control of the murine leukemia promoter were found to efficiently transduce JLTRG cells, as indicated by the initially high measurable levels of eGFP expression, but then gradually lost eGFP expression, probably due to promoter methylation. We solved this problem by using retroviral vectors that express HIV-1 Tat under the control of the murine stem cell virus promoter (Swindle et al., 2004). Through an iterative process of retrovirally transducing JLTRG cells with the Tat vectors, sorting the transduced cells for high eGFP expression, supertransducing this eGFP-positive population, etc., we were able to obtain a cell population that generates a HIV-1 Tat driven eGFP fluorescence intensity similar to that observed in CUCY cells (Figure 1A). We were then able to transduce these cells with retroviral vectors expressing high levels of RFP, which similarly as in CUCY cells can serve as a simultaneously accessible marker for drug toxicities (Figure 1B). Final single cell cloning for high eGFP/RFP expressing cells resulted in the establishment of the clonal TiGR cell line. eGFP and DsRed expression in TiGR cells is stable. Over six months of continuous culture, we only observed a 10% decrease of eGFP expression at the population basis. However, at this time-point, a 100% double-positive cell population can be easily regenerated by cell sorting.
Figure 1. HIV-1 transcriptional inhibitor screening system.

Comparison of eGFP (A) and RFP (B) expression in CUCY cells and the TIGR reporter cells used in this application. C) Z-test results for TiGR cells. 2×105 TiGR cells were loaded into 48 wells of a 384 well plate and eGFP fluorescence intensity was measured (black circles) compared to eGFP fluorescence of the parental JLTRG cells (gray squares) loaded into 48 wells of the same plate.
We initially established the ideal per well cell density by titrating TiGR cells at various cell densities into 384 well plates. The eGFP/RFP signals increase in a linear manner from a cell density of 5 × 103 cells/well, with 2 × 105 cells (15-fold signal increase over background) being optimal for cell screening. At this cell density the Z’ was defined as 0.89, indicating that the assay is very robust (Figure 1C). The experimentally determined Z’ factor is a dimensionless statistical value designed to reflect the dynamic range as well as the variation of the assay. It calculates as Z’= 1 - (3σp + 3σn)/|μp - μn). With 3σn representing the standard deviation of the negative control samples, 3σp representing the standard deviation of the positive samples, μn the mean of the negative control samples and μp the mean of the positive samples. Z’ = 1 would be an ideal assay and 1 > Z’ > 0.5 are considered very good to excellent assays.
We next verified the TiGR based HTS assay in a small manually performed 1,000 compound screen and included some known inhibitors of HIV-1 transcription (Ro24-7429, WP631) as controls, which were efficiently detected (80-90% eGFP signal reduction on day 7; data not shown). As TiGR cells use fluorescence intensities as quantitative markers for HIV-1 expression and drug toxicities, drug effects can be assessed intervention free. As such, the assay not only determines cumulative inhibition at a defined time point, but also the inhibitory/toxic onset kinetics of a respective compound, which gives additional insights into the possible drug efficiency. Therefore, this assay system serves as a standardized platform to screen for Tat LTR transcriptional inhibitors.
Identification of Tat dependent transcription inhibitors
LOPAC-Sigma-Aldrich (1280 compounds) and Spectrum–Microsource (2000 compounds) small compound libraries were screened using the TiGR cells described above to identify novel HIV-1 transcription inhibitors. These libraries were chosen as they contain pharmacologically active compounds, known FDA-approved drugs, experimental bioactives, and pure natural products which provided a wide range of compounds with potential biological activity. Compounds were diluted in DMSO and screened at 10 μM for Tat transcriptional inhibition. TiGR cells were analyzed at Days 0, 1, 2, 3, 4 and 7 post drug treatments to determine the GPF and RFP signals and those compounds that exhibited greater than 30% inhibition of GFP expression without affecting the RFP expression were selected for further analysis. We identified five candidates based on these criteria. The top five candidates that were identified through the TiGR cell system were tested in TZM-bl cells, which contain an integrated LTR-luciferase reporter. As the TZM-bl system was utilized as our secondary screen, we chose 1 uM as a more stringent cut-off for selection of our lead candidate. TZM-bl cells were transfected with Tat followed by compound treatment the next day. Results shown in Table 1 indicate the % inhibition observed at 1 μM as compared to the DMSO control. The compound with the greatest inhibition was the chemotherapeutic agent Dactinomycin, which is a transcriptional inhibitor and anti-proliferation compound (Ho et al., 2008). Its mechanism of action is non-specific as it binds to multiple DNA structures such as GC-rich duplexes, single-stranded forms, or hairpin forms, as well as interfering with RNA polymerase (Kang and Park, 2009). Although toxicity was not observed in the TIGR cells, the above characteristics suggest the limited HIV-1 therapeutic potential of this compound; therefore we chose not to further pursue Dactinomycin. Out of the remaining compounds, the GSK-3 inhibitor 6BIO (6-bromoindirubin-3′-oxime) (Meijer et al., 2003; Polychronopoulos et al., 2004) was ranked the second most potent LTR transcriptional inhibitor (Table 1). Therefore, these results indicate that 6BIO reproducibly inhibits Tat dependent LTR transcription.
Table 1.
| Inhibitor | Description & Target | % Inhibition at 1μM |
|---|---|---|
| Dactiniomycin | Antineoplastic, intercalating agent | 98 |
| 6BIO | Glycogen synthase kinase 3α/β inhibitor | 62 |
| Quinine ethyl carbonate | Antimalarial | 34 |
| Indirubin-3′-oxime | CDK inhibitor | 25 |
| Epirubicin hydrochloride | Antineoplastic | 17 |
6BIO inhibits HIV-1 transcription without inducing cellular toxicity
The best candidate identified in our screen, 6BIO, was further tested using TZM-bl cells to determine its IC50. TZM-bl cells were transfected with Tat followed by treatment with various concentrations of 6BIO the next day. Luciferase assays performed at two days post 6BIO treatment indicated that 6BIO inhibited HIV-1 LTR Tat dependent transcription in a dose dependent manner (Figure 2A), with an IC50 of 40 nM. MTT assays were performed to determine the influence of 6BIO on cell viability. As can be seen in Figure 2B, at the concentrations used in our transcriptional assays, 6BIO did not affect cell viability. MTT assays were also performed with 6BIO on multiple other cell types, including uninfected and infected T-cell lines (CEM and ACH2), uninfected and infected monocytic cell lines (U937 and U1) and an astrocytoma cell line (U87MG). No cellular toxicity was observed upon treatment with 6BIO (1 uM) as compared to the DMSO control (data not shown). These results indicate that 6BIO can inhibit HIV-1 transcription without inhibiting cellular viability.
Figure 2. 6BIO inhibits HIV-1 transcription and replication without inducing cellular toxicity.

A) TZM-bl cells were transfected with 1.0 μg of Tat and treated the next day with DMSO, 6BIO (0.025, 0.05, 0.1, and 1.0 μM). Cells were processed 48 hours post drug treatment for luciferase assays. Assays were performed in triplicate and an average value is shown plus standard deviation. B) TZM-bl cells were treated with DMSO or 6BIO (0.025, 0.05, 0.1, and 1.0 μM). Cell proliferation/viability was determined by MTT assays. Treatments were performed in triplicate and samples analyzed at 48 hours. C) PHA and IL-2 activated PBMCs were kept in culture for 2 days prior to infection. Isolation and treatment of PBMCs were performed by following the guidelines of the Centers for Disease Control. Approximately 2.5×107 PBMCs were infected with 89.6 (35,520 RT units). Cells were resuspended in 6.5 ml of complete media and plated in a 96 well plate at 200 μl/well. 6BIO treatment (0, 0.1, 0.5 and 1.0 μM) was performed (only once) the day following infection. Samples were collected on days 7 and 14 and stored at −20°C for RT assay. Treatments were performed in triplicate and the average plus the standard deviation is displayed. D) PBMCs were processed as described for C. Cells were collected on days 7 and 14, washed 2X with PBS w/o Ca and Mg, resuspended in 70% ethanol, and stained with propidium iodine prior to cell cycle analysis by flow cytometry to determine apoptosis (sub-G1 peak). Triplicate wells were pooled for this analysis.
To determine whether 6BIO has an effect on HIV-1 replication in primary cells, we infected activated PBMCs with the dual tropic 89.6 virus and subsequently treated cells with vehicle (DMSO) or various concentrations of 6BIO (0.1, 0.5, and 1.0 μM). Treatment was performed only one time. Cells were maintained for 14 days and supernatants were collected at days 7 and 14 for RT analysis (Figure 2C). Results indicate that 1.0 μM of 6BIO inhibited virus replication by more than 50% after 7 and 14 days. Cells were also collected to determine the influence of 6BIO treatment on cell viability using PI staining/FACS analysis. Apoptosis was determined through cell cycle analysis (sub G1 peak). Uninfected PBMCs displayed low levels of apoptosis at both days 7 and 14 (Figure 2D). Infected PBMCs also demonstrated low levels of apoptosis at day 7. However, at day 14 an increase in apoptosis was observed in the infected cells probably due to viral induced cell death as the DMSO control cells were also beginning to die and due to the fact that BIO treatment was unable to completely suppress viral replication. Collectively these results indicate that the IC50 of the 6BIO inhibition is ~ 0.75 μM in HIV-1 infected PBMCs and that 6BIO can inhibit HIV-1 replication without inhibiting cellular viability.
Identification of a more potent 6BIO analog
Hit2Lead (Hit2Lead.com) was utilized to identify thirty-eight commercially available 6BIO analogs. These analogs were tested at 1 μM in the TZM-bl cells to test their ability to inhibit Tat-dependent LTR transcription (Figure 3A). Many of the analogs actually increased viral transcription as compared to the DMSO control, especially compounds 16 and 31 possibly due to removal of inhibitors from the promoter. In contrast, a few analogs tested displayed LTR transcriptional inhibition with different potencies (compounds 4, 6, 18). Derivative #6 showed particularly strong inhibition of LTR transcription. MTT assays were performed with derivative #6 on multiple cell lines, including uninfected and infected T-cells (CEM and ACH2), uninfected and infected monocytes (U937 and U1) and astrocytoma cells (U87MG). Results indicated that the transcriptional inhibition was not due to cellular toxicity (Figure 3B) as very little change in viability was observed upon treatment with derivative #6 as compared to the DMSO control. Derivative #6 was termed 6BIOder and selected for follow-up analysis.
Figure 3. 6BIO derivatives inhibit HIV-1 transcription without inducing cellular toxicity.

A) TZM-bl cells were transfected with 1.0 μg of Tat and treated the next day with DMSO, 6BIO, derivatives 1-38 at 1 μM. Cells were processed 48 hours post drug treatment for luciferase assays. Assays were performed in triplicate and an average value is shown plus standard deviation. B) CEM, ACH2, U937, U1, and U87MG cells were treated with DMSO and derivative #6 (1 μM). Cell proliferation/viability was determined by MTT assays. Treatments were performed in triplicate and samples analyzed at 48 hours. Percent viability is expressed as compared to the DMSO control.
Effect of 6BIO analogs on inhibition of HIV-1 in monocytes and astrocytoma cells
To further test the effect of 6BIOder, we assayed its effect in primary monocyte/macrophage infection along with the parent 6BIO drug. Figure 4A shows the general structure of 6BIO and 6BIOder. Both compounds are commercially available, however 6BIOder has not been studied in the context of HAND. Data in Figure 4B utilized monocyte/macrophages from a healthy donor infected with HIV-1 dual tropic 89.6 for 7 days. Both 6BIO and 6BIOder were added to the media during the course of infection (once only). Lane 1 shows normal replication of HIV-1 as evidence by RT activity in supernatant and Lane 2 is with DMSO control. Lane 3 used 6BIO (10 nM) and 4-7 utilized 6BIOder (0.1, 1, 10, 100 nM). There was considerable inhibition with 6BIOder at 10 nM. The IC50 for 6BIOder in these cells is 4 nM. Next, we performed a similar assay in U87MG cells with 6BIOder at 0.1, 1 and 10 nM (left hand graph). Again, inhibition was mostly observed at 1 nM with IC50 = 0.5 nM. Finally, we performed MTT assays with monocyte/macrophage from two healthy donors and U87MG at 10, 100, 1000 and 10,000 nM (lanes 2-5) and observed no apparent toxicity in these cells (Figure 4C). Collectively, these data indicate that when searching for 6BIO related compounds to inhibit HIV-1, we were able to identify 1 out of 38 compounds that had IC50 of 0.5-4 nM (depending on the cell type) and were not toxic to cells at 10 μM. This is roughly a 3 log difference between HIV-1 inhibitory activity and possible cell toxicity.
Figure 4. Effect of 6BIO and 6BIOder on HIV-1 replication.

A) Structure of 6BIO: 2-{[[2-(5-bromo-2-oxo-1,2-dihydro-3H-indol-3 ylidene)hydrazino](oxo) acetyl]amino} benzoic acid and 6BIOder: 6-bromo-5-methyl-1H-indole-2,3-dione 3-[(6-bromo-5-methyl-2-oxo-1,2-dihydro-3H-indol-3-ylidene)hydrazone]. B) PMBCs were obtained from the blood of healthy donor (YW), and purified by centrifugation through a layer of Lymphocyte Isolation Medium. Cells were resuspended in serum-free RPMI and plated on culture dish for 1 hour at 37°C. Non-adherent lymphocytes were removed and the adherent monocytes were cultured in RPMI plus 10% heat-inactivated FBS. Macrophages were further differentiated by incubating in 10 ng/ml M-CSF for 1 week with medium change every 2 days. Both macrophages and U87MG cells were infected with 89.6 (MOI:1). 6BIO (lane 3, 10 nM) 6BIOder (lanes 4-7: 0.1, 1, 10, 100 nM) were added to cells six hours after infection. U87MG cells were treated with 6BIOder at 0.1, 1 and 10 nm (left side of panel B). Samples were collected on day 7 for RT assay. Treatments were performed in triplicate and the average plus the standard deviation is displayed. C) MTT assays in two monocyte/macrophage healthy donors and U87MG were performed after treatment with 6BIOder at 10, 100, 1000 and 10,000 nM (lanes 2-5).
6BIOder does not inhibit cellular gene expression in the absence of Tat and is specific to GSK-3β
We next asked whether 6BIOder was inhibitory to genes that require CDK9/cyclin T. HIV-1 Tat utilizes CDK9/cyclin T for its activation of transcription. We therefore asked if cellular gene expression would be sensitive to 6BIOder in treated cells. Results in Figure 5A show RT/PCR results of 3 genes that require CDK9/cyclin T for their transcription. None of these genes showed a dramatic decrease after 6BIOder treatment in U937 or U87MG cells in the absence of Tat. Interestingly, we did observe an increase in expression for MCL-1, IL-8, and cyclin D1 following 6BIOder treatment of U937 cells. Next we asked if 6BIO or 6BIOder were able to inhibit GSK-3β kinase activity. U937 cells were treated with 6BIO or 6BIOder, followed by immunoprecipitation with anti-GSK-3β. The IPed material was utilized for in vitro kinase assays with glycogen synthase peptide 2 as the substrate. Results in Figure 5B indicate that BIOder and not 6BIO was able to inhibit 90% of the GSK-3β kinase activity at 1 nM. The estimated in vitro IC50 for 6BIOder in these kinase assays for GSK-3β is 0.03 nM. Collectively, these results indicate that 6BIOder is an effective GSK-3β inhibitor.
Figure 5. 6BIOder does not inhibit cellular gene expression in the absence of Tat and is specific to GSK-3β.
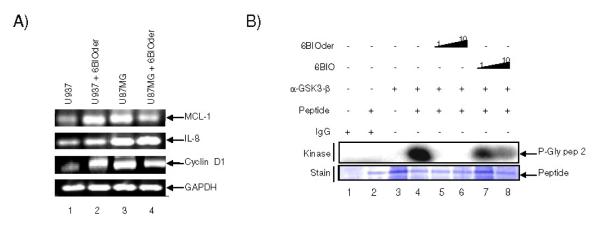
A) Total RNA was isolated from cells treated with 6BIOder (1μM) using Trizol. RNA was treated with 0.25 mg/ml DNase I for 1 hour, followed by heat inactivation at 65°C for 15 minutes. A total of 1 μg of total RNA was used to generate cDNA with the iScript cDNA Synthesis kit using oligo-dT reverse primers. Primers for PCR were MCL-1, IL-8, cyclin D1 and GAPDH as control. B) Two mg of U937 extract was IPed at 4°C overnight with GSK-3β antibody. The next day complexes were precipitated with A/G beads for two hours at 4°C. IPs were washed twice with TNE buffer and kinase buffer. Phosphorylation reactions were performed with IP material and 200 ng of glycogen synthase peptide 2 (Millipore) as substrate. Following incubation, samples were run on a 4-20% SDS-PAGE, dried and subjected to analyzed using Molecular Dynamics Phosphor Imager software.
Knockdown of GSK-3β decreases viral transcription in cells
We next asked whether downregulation of GSK-3β in cells could potentially decrease viral gene expression and/or viral load in infected cells. For that we used TZM-bl cells that were transfected with siRNA against GSK-3β or luciferase in the presence or absence of Tat and assayed for luciferase expression 48 hours post-transfection. Results of such an experiment are shown in Figure 6A where siLuc or siGSK-3β did not control much of basal transcription in the Hela TZM-bl cells (lanes 1 and 2). However, siGSK-3β did significantly reduce Tat activated transcription in these cells (compare lane 3 and 4). To confirm the knockdown, whole cell extract of TZM-bl transfected with siRNAs were run on a 4-20% SDS-PAGE and western blotted against GSK-3β and β-actin as control. More than 90% knockdown was observed with siGSK-3β (lower insert, lane 4).
Figure 6. Downregulation of GSK-3β decreases viral transcription in cells.

A) TZM-bl cells were transfected with siRNA against GSK-3β or luciferase (100 nM) in the presence or absence of Tat (1 μg) and assayed for luciferase expression 48 hours post-transfection. To confirm knockdown, 50 μg of whole cell extract of 293T cells (positive control), TZM-bl, TZM-bl transfected with luciferase siRNA (siLuc), and TZM-bl transfected with GSK-3β (siGSK-3β), were run on a 4-20% SDS-PAGE and western blotted against GSK-3β and β-actin. *p<0.01 is related to the comparison between siLuc and siGSK-3β. B) J1-1 cells were used for electroporation with siLuc or siGSK-3β. Log phase growing cells (5×105/ml) were electroporated with either siLuc or siGSK-3β (200 nm) and re-plated in complete media. Supernatants were processed for presence of RT at days 2 and 4. The effect of siGSK-3β treatment was abolished by day 6 (data not shown). *p<0.01 is related to the comparison between siLuc and siGSK-3β.
We next asked whether knockdown of GSK-3β could potentially decrease virus release from HIV-1 infected cells. For that we used J1-1 cells which are Jurkat derived, contain single copy integrated wild type virus, and release virus into the supernatant without addition of any external stimuli (i.e. TNF or HDAC inhibitors). We performed the experiment with either siLuc as control or siGSK-3β using electroporation. Results of such an experiment are shown in Figure 6B, where there was a marked decrease of RT from cells treated with siGSK-3β at days 2 and 4. Collectively these data imply that knockdown of GSK-3β in either HeLa or Jurkat (J1-1) based cells down regulated HIV gene expression and viral production.
Effect of 6BIOder on the dox-dependent HIV-rtTA viruses (Tat/TAR specificity)
We next asked if the effect of 6BIOder was specific to Tat function in HIV-1 expressing cells. For that we obtained two sets of constructs from the Berkhout lab which have mutation in Tat/TAR sequence. These viruses can be induced with dox and full particles are recovered in the supernatant (Verhoef et al., 2001). Briefly, the full-length, infectious HIV-1 molecular clone pLAI was used for construction of an HIV-rtTA virus genome, the transcription of which is controlled by dox. The viral transcriptional elements TAR and Tat were replaced by the prokaryotic tetO-rtTA elements (Figure 7A). TAR was inactivated by mutation of multiple nucleotides in the single-stranded bulge and loop domains, the binding sites for Tat and cyclin T, respectively. Also, the inactive TAR motif was inserted in both LTRs to minimize the chance of reversion to the wild-type virus by a recombination event. Inactivation of the Tat protein was accomplished by introduction of the Tyr26Ala point mutation. This single amino acid change resulted in a severe loss of Tat transcriptional activity and virus replication. Thus, both LTRs were modified, and this was done in the wild-type (W) and mutant (Y) Tat backgrounds, resulting in four HIV-rtTA constructs: KWK, KYK, SWS, and SYS. In the current study we used the KWK and KYK sets. The virus variant KWK is most wild type-like because it maintained the NF-B sites, SP1 sites, and a wild-type Tat protein, but it has a mutation in TAR. The KYK clone has similar promoter elements; however the Tat and TAR are both mutated. These HIV-rtTA replicate in a dox-dependent manner, when transfected into either cell lines or primary PBMCs.
Figure 7. Effect of BIOder in dox-dependent HIV-1 variants in macrophages.

A) HIV-1 genome and modifications that were introduced to construct HIV-rtTA. In brief, TAR-Tat transcriptional axis was replaced by the tetracycline-inducible tetO-rtTA system. Inactivation of TAR and Tat is indicated by crosses through the motifs (Verhoef et al., 2001). B) The pLAI chimera plasmids, KWK and KYK (20 μg), were individually transfected (electroporation) into the differentiated macrophages (~4 × 106) using 250 nM PMA for three days. The culture was maintained with dox (1,000 ng/ml) and BIOder (50 nM), and virus replication was monitored by measuring the amount of RT produced in the culture medium at day 10. No virus replication was observed in the absence of dox, indicating that replication is strictly dependent on the inserted Tet system. C) Macrophages were treated and transfected as described above in B. On day 10, cell pellets were lysed in Buffer RLT and RNA extracted by Qiagen’s RNeasy kit. RNA was treated with 0.25 mg/ml DNase I at 37°C for 1 hour, followed by heat inactivation at 65°C for 15 minutes. A total of 30 ng of total RNA was used to generate cDNA with the iScript cDNA Synthesis kit using random primers. PCR was performed with GAPDH and MCl-1 specific primers.
We performed our experiments with the KWK and KYK clones in primary monocytes that had been differentiated into macrophages with PMA (Figure 7B). Differentiated cells (3 days) were electroporated with 20 μg of either KWK or KYK molecular clones, and were cultured without or with dox (1,000 ng/ml). Virus production was measured by RT on culture supernatant samples. Cells treated with dox showed viral production from both KWK and KYK clones. When cells were treated with 6BIOder, viral replication was inhibited in the KWK (Tat+) and not KYK (Tat−) clone. We were also interested in determining the effect of Tat and BIOder treatment on cdk9 responsive genes in primary macrophages. For this analysis we used MCL-1 as an example, because we had observed an increase in expression following BIOder treatment in the monocytic cell line U937. Results in Figure 7C indicate that MCL-1 expression did not change following treatment with BIOder in the presence of KWK (Tat+); however a modest decrease (less than 2-fold) in expression was observed with BIOder treatment in the presence of KYK (Tat−) clone. These results further reinforce the notion that a functional Tat is required for the effect of 6BIOder in cells.
6BIOder protects neuronal cultures from the HIV-1 Tat protein
It has been previously shown that GSK-3 inhibitors such as lithium have neuroprotective effects (Cross et al., 2001). Therefore, we were interested in determining if 6BIO could protect against Tat induced death. To this end, rat mixed hippocampal cultures were preincubated with 6BIO prior to exposure to Tat. Cell death was analyzed 18 hours after Tat exposure by MTT assay. As expected, Tat treatment reduced cell viability (Figure 8A). Importantly, 6BIO was protective against Tat mediated neurodegeneration, with significant neuroprotective effects at 1.0 and 3.0 μM (p<0.05). There was neurotoxicity observed at 5.0 and 10.0 μM of 6BIO, with a LD50 of ~4 μM. 6BIOder had even more promising results, having a protective effect at 1.0 and 3.0 μM (p<0.05) (Figure 8B). Importantly, there was no neurotoxicity observed at higher concentrations of 6BIOder (5.0 and 10.0 μM). These results indicate that 6BIO and 6BIOder may be able to protect neuronal cultures from Tat induced cell death.
Figure 8. 6BIO protects neuronal cultures from the HIV-1 Tat protein.

Rat mixed hippocampal cultures were preincubated with various concentrations of (A) 6BIO (0.05-10 μM) or (B) 6BIOder (0.05-10 μM) for 1 hour at 37°C prior to an 18 hour exposure to 500 nM Tat1-72. After 18 hours, cell survival was measured by MTT assay. Statistical significance was determined by ANOVA, followed by Newman-Keuls post hoc pair-wise comparisons.
DISCUSSION
In this study, we identified 6BIO as the most potent inhibitor of Tat-dependent transcription out of 3280 compounds screened. 6BIO, a synthetic derivative of the natural product, 6-bromoindirubin, is a potent and specific GSK-3 inhibitor with an IC50 of 5 nM (Meijer et al., 2003). 6BIO can also inhibit CDK5/p35, CDK2/cyclin A, and CDK1/cyclin B complexes with higher IC50s of 0.08, 0.30, and 0.32 μM respectively (Meijer et al., 2003). Co-crystallization experiments indicated that 6BIO binds to the ATP pocket of GSK-3β, forming a van der Waals contact with Leu132, which is replaced by a phenylalanine in CDK2 and CDK5, providing one explanation for the preference of 6BIO for GSK-3β (Meijer et al., 2003). 6BIO was found to be an effective GSK-3 inhibitor both in vitro and in vivo, through the accumulation of unphosphorylated β-catenin and through modulating Wnt signaling in Xenopus embryos (Meijer et al., 2003). Along these lines, there are other GSK-3 inhibitors reported including lithium, SB-216763, and SB-415286. Lithium is active in the 10-20 mM range and is known to inhibit other molecules including polyphosphate-1-phosphate, inositol monophosphatase, casein kinase-II, MAP kinase-activated protein kinase-2, and p38-regulated/activated kinase (Berridge et al., 1989; Davies et al., 2000). SB-216763 and SB-415286 were originally identified as GSK-3α inhibitors through a high throughput screen of the SmithKline Beecham compound library against rabbit GSK-3α and subsequently were shown to inhibit human GSK-3 with IC50s of 34 nM and 78 nM respectively (Coghlan et al., 2000). 6BIO has the lowest IC50 of all these GSK-3 inhibitors (5 nM) and therefore has the most therapeutic potential. In addition, in this study we identified a second generation 6BIO analog, 6BIOder, which has an in vitro IC50 of 0.03 nM and demonstrated neuronal protection with less toxicity than 6BIO. At this stage it cannot be ruled out that 6BIOder acts through another unidentified kinase (i.e., CK1, CLK1 or DYRK from infected cells), or by inhibition of a collection of various kinases, which jointly results in selective inhibition of Tat-dependent transcription. Another complicating factor at this point is that cell type difference that was also observed when 6BIOder inhibited HIV-1 better in U87MG as compared to monocyte/macrophages cells.
Glycogen synthase kinase -3 (GSK-3) is a serine/threonine protein kinase that was originally described as a critical regulator of glycogen metabolism through the phoshorylation of glycogen synthase (Plyte et al., 1992). The human GSK-3 family comprises two isoforms, GSK-3α and GSK-3β, which share 97% sequence identity in their kinase domain, but differ in their N- and C-terminus regions. GSK-3α/GSK-3β are implicated in the regulation of glycogen synthesis, the Wnt / β-catenin signaling pathway, PI3K pathway, cell cycle control, transcriptional regulation, and apoptosis (Frame and Cohen, 2001). The ability of GSK-3α/GSK-3β to regulate this vast array of cellular processes may be related to its numerous substrates including glycogen synthase, axin, β-catenin, APC, cyclin D1, c-Jun, c-Myc, C/EBPα/β, NFATc, RelA and CREB to name just a few (Buss et al., 2004; Frame and Cohen, 2001). Interestingly, phosphorylation of some substrates such as glycogen synthase, but not of others such as β-catenin, by GSK-3 requires a “priming” phosphorylation of the substrate at a serine residue C-terminal to the GSK-3 phosphorylation site (Ali et al., 2001). GSK-3β is negatively regulated by PKB/AKT phosphorylation of Ser9 (Meijer et al., 2004; van Weeren et al., 1998). There has been much interest in inhibiting GSK-3β for the treatment of Alzheimer’s disease, and other neurological disorders. This is in large part due to its proapoptotic effects in neuronal cells (Hetman et al., 2000). Likewise, GSK-3β inhibitors lithium, SB 216763, and SB 415286 have been shown to protect cerebellar granule neurons from apoptosis (Cross et al., 2001).
Tat induces GSK-3β activity, which can be reversed by the addition of the GSK-3β inhibitor lithium (Maggirwar et al., 1999). Furthermore, the GSK-3β inhibitors lithium and valproic acid (VPA) can protect against Tat and gp120 mediated neurotoxicity (Dou et al., 2003; Dou et al., 2005; Everall et al., 2002). Rodent and human neurons exposed to culture fluids from HIV-1-infected monocyte-derived macrophages (MDMs) were protected from cell death in the presence GSK-3β inhibitors (lithium, AR-A014418 and 66BIO) (Dou et al., 2005; Nguyen et al., 2009). Importantly, lithium treatment also resulted in neuronal protection and neurogenesis in SCID HIV-1 encephalitis (HIVE) mice (Dou et al., 2005). Sui et al. investigated the role of GSK-3β in NF-kB regulated neuronal apoptosis (Sui et al., 2006). They found that neurons exposed to HIVADA-macrophage conditioned medium (MCM) displayed decreased NF-kB activity in a Tat dependent manner. GSK-3β inhibition through lithium or indirubin treatment blocked NF-kB inhibition, the suppressive binding of RelA to HDAC3, and neuronal apoptosis (Sui et al., 2006). Lithium treatment also inhibits HIV-1 replication of both T- and M-tropic viruses in PBMCs as well as TNF stimulated J1.1 cells (Kumar et al., 2008). Therefore, pharmacological inhibition of GSK-3β may have implications for the treatment of HAND as well as in the inhibition of HIV-1 replication in PBMCs.
The role of GSK-3β in HAND is not entirely clear, but it is well known that GSK-3β is important for both inflammation and cell migration (Jope et al., 2007). An upstream negative regulator of GSK-3β is PI3K, which limits the release of pro-inflammatory cytokines from monocytes and macrophages (Fukao and Koyasu, 2003). Likewise, several studies have demonstrated that the inhibition of GSK-3β is associated with the suppression of inflammation. Specifically, in response to stimulation of Toll-like receptors in both monocytes and peripheral blood mononuclear cells, GSK-3β activity is necessary for the production of pro-inflammatory cytokines, such as interleukin-6 (IL-6), IL-1β, and tumor necrosis factor (TNF), and reduction of the anti-inflammatory cytokine IL-10 (Martin et al., 2005). In terms of cell migration, GSK-3β inhibition prevents extension of lamellipodia in keratinocytes (Koivisto et al., 2004) and reduces axon elongation rates in neurons (Owen and Gordon-Weeks, 2003). Inhibition of GSK-3 through 6BIO treatment or RNAi also prevented migration of epithelial cells (Farooqui et al., 2006). Thus it would be expected that GSK-3β inhibition through 6BIO treatment would have a profound effect on both inflammatory responses and cellular migration in response to inflammatory signals.
Our studies demonstrate for the first time the ability of 6BIO to inhibit Tat-dependent transcription. GSK-3β regulates a number of transcription factors and co-factors including β-catenin, c-Jun, c-Myc, C/EBPα/β, NFATc, RelA and CREB (Buss et al., 2004; Frame and Cohen, 2001), most of which have also been implicated in Tat mediated transcription (Rohr et al., 2003). Studies are underway to identify factor occupancy changes at LTR following 6BIO treatment to elucidate the mechanism of 6BIO inhibition of Tat dependent transcription. Of particular interest is the β-catenin/ T-cell factor-4 (TCF-4) pathway, which has important connections in neuronal development and multiple neurological disorders (Cerpa et al., 2009). Interestingly, TCF-4 has been shown to inhibit HIV-1 transcription (Carroll-Anzinger et al., 2007; Rossi et al., 2006; Wortman et al., 2002). While initial studies indicated that the TCF-4 mediated inhibition of HIV-1 transcription was β-catenin independent (Wortman et al., 2002), later studies utilizing a TCF-4 dominant negative construct, which is mutated in the β-catenin binding site, suggest that β-catenin is important for the observed effects (Carroll-Anzinger et al., 2007). β-catenin binding to TCF-4 results in the release of TCF-4 repressors, such as transducin-like enhancer, allowing the TCF-4/β-catenin complex to bind to DNA and regulate transcription. β-catenin proteasomal degradation is induced by GSK-3β dependent phosphorylation (Amit et al., 2002; Rubinfeld et al., 1996) and thus stabilization of β-catenin would be expected following 6BIO treatment, as shown in [76].
In addition to the β-catenin/TCF-4 pathway, the NF-kB pathway is highly regulated by GSK-3β. Expression of a constitutively active GSK-3β mutant (S9A) and inhibition of the PI3K pathway (thus allowing GSK-3β to remain active) results in astrocyte apoptosis (Rao et al., 2004). This may be due at least in part to the inhibition of the NF-κB pathway (Bournat et al., 2000; Rao et al., 2004; Sanchez et al., 2003). In the presence of constitutively active GSK-3β, inhibition of NF-κB was observed along with stabilization of the NF-κB-inhibitory protein, IκBα and down-regulation of IκB kinase (IKK) activity. GSK-3β can directly inhibit NF-kB through phosphorylation of RelA at serine 468, resulting in an inactivate form of NF-kB (Buss et al., 2004). However, the reverse has also been demonstrated, where GSK-3β was shown to be essential for NF-κB mediated apoptosis in response to TNF-α treatment (Hoeflich et al., 2000). Thus the influence of GSK-3β on NF-κB activity may be stimulus, cell type and/or promoter specific.
MATERIALS AND METHODS
Cell Culture and Reagents
TZM-bl, U87MG, and 293T Cells were grown and cultured to confluency in Dulbecco’s modified Eagle’s medium supplemented with 10% heat-inactivated FBS, 1% L-glutamine, and 1% streptomycin/penicillin (Gibco/BRL, Gaithersburg, MD, USA). The latently infected promonocytic U1 cell line and the uninfected corresponding U937 cell line, as well as infected J1.1, ACH2 and their uninfected counterparts Jurkat and CEM (12D7) cells were cultured up to 1 × 105 cells per ml (early log phase of growth) in RPMI-1640 medium supplemented with 10% heat-inactivated FBS, 1% L-glutamine, and 1% streptomycin/penicillin. ACH2, J1-1 contain a single integrated copy of HIV-1 genome, whereas U1 cells harbor two copies (one wild type and one mutant) of the viral genome in parental U973 cells. All cells were incubated at 37°C and 5% CO2. The reporter T-cell lines JLTRG and TiGR were maintained at an average cell density of 0.5 × 106 cells/mL in RPMI 1640 (Mediatech, Herndon, VA, USA) supplemented with 2 mM l-glutamine, 100 U/mL penicillin, 100 μg/mL streptomycin, and 10% heat-inactivated fetal bovine serum (FBS; HyClone, Logan, UT, USA) (12). TiGR cells were derived by retroviral transduction of JLTRG cells with a retroviral MSCV-LTR driven Tat-expression vector. JC53BL-13 cells (TZM-bL) were cultured and infected as previously described (13). Briefly, the cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM; Mediatech) supplemented with 2 mM L-glutamine, 100 U/mL penicillin, 100 μg/mL streptomycin, and 10% heat-inactivated FBS.
PBMCs used to generate infectious viral supernatants were isolated from the blood of healthy donors by Ficoll-Paque™ density gradient centrifugation (Amersham 6BIOsciences, Uppsala, Sweden) and were cultured in RPMI 1640 supplemented with 10% heat-inactivated FBS, 2 mM l-glutamine, 100 U/mL penicillin, and 100 μg/mL streptomycin. PBMCs were initially PHA/interleukin-2 stimulated and infected with HIV-1 89.6 strain 4 days following stimulation. All antibodies were purchased from BD Pharmingen (San Diego, CA, USA). PHA-L was obtained from Sigma (St. Louis, MO, USA), and IL-2 was purchased from 6BIOsource International (Camarillo, CA, USA).
384-Well Plate-Based Fluorometry Assays
All plate-based experiments were performed in 384-well optical bottom black wall plates (Nalgen Nunc International, Rochester, NY, USA) and designed to obtain a final cell density of 1 × 106 cells/mL in a final volume of 90 μL phenol red-free RPMI 1640 per well. This optimal number was obtained by titrating TiGR cells over a range of cell numbers per well (1 × 103 to 1 × 106 cell/well). The phenol red-free RPMI 1640 used in all experiments was supplemented with 2 mM l-glutamine, 100 U/mL penicillin, 100 μg/mL streptomycin, and 2% heat-inactivated FBS. Analysis was performed using a Synergy™ HT Multi-Detection Microplate Reader (6BIO-Tek Instruments, Winooski, VT, USA), equipped with the following filter set: excitation, 488/20 nm; emission, 525/20 nm.
To determine the Z’-factor, JLTRG or TiGR cells were adjusted to a cell density of 2 × 106 cells/mL in phenol red-free RPMI 1640 supplemented with 2% FBS, of which 50 μL were loaded per well.
Compounds/inhibitors
The indirubin derivatives used in this study were: 1: 2-{[[2-(5-bromo-2-oxo-1,2-dihydro-3H-indol-3-ylidene)hydrazino](oxo)acetyl]amino}benzoic acid, 2: N’~1~,N’~4~-bis(5-methyl-2-oxo-1,2-dihydro-3H-indol-3-ylidene)terephthalohydrazide, 3: 5-bromo-3-({2-[(2-oxo-1,2-dihydro-3H-indol-3-ylidene)amino]phenyl}imino)-1,3-dihydro-2H-indol-2-one, 4: 4-bromo-5-methyl-1H-indole-2,3-dione 3-oxime, 5: 4-bromo-5-methyl-1H-indole-2,3-dione 3-(N-phenylsemicarbazone), 6: 6-bromo-5-methyl-1H-indole-2,3-dione 3-[(6-bromo-5-methyl-2-oxo-1,2-dihydro-3H-indol-3-ylidene)hydrazone], 7: N’-(5-bromo-7-methyl-2-oxo-1,2-dihydro-3H-indol-3-ylidene)-2-chlorobenzohydrazide, 8: N’-(5-bromo-2-oxo-1,2-dihydro-3H-indol-3-ylidene)-2-chlorobenzohydrazide, 9: 5,7-dibromo-1H-indole-2,3-dione 3-(phenylhydrazone), 10: 5,7-dibromo-1H-indole-2,3-dione 3-oxime, 11: 2-chloro-N’-(5,7-dibromo-2-oxo-1,2-dihydro-3H-indol-3-ylidene)benzohydrazide, 12: 2-bromo-N’-(5,7-dibromo-2-oxo-1,2-dihydro-3H-indol-3-ylidene)benzohydrazide, 13: N’-(4-bromo-5-methyl-2-oxo-1,2-dihydro-3H-indol-3-ylidene)-3,5-dihydroxybenzohydrazide, 14: N’-(5-bromo-2-oxo-1,2-dihydro-3H-indol-3-ylidene)-2-methyl-3-furohydrazide, 15: N-(1-{[2-(5-fluoro-2-oxo-1,2-dihydro-3H-indol-3-ylidene)hydrazino]carbonyl}-2-phenylvinyl)benzamide, 16: N’-(5-bromo-2-oxo-1,2-dihydro-3H-indol-3-ylidene)-2,4-dichlorobenzohydrazide, 17: 4-bromo-5-methyl-1H-indole-2,3-dione 3-(phenylhydrazone), 18: 6-chloro-7-methyl-1H-indole-2,3-dione 3-oxime, 19: 4-chloro-7-methyl-1H-indole-2,3-dione 3-oxime, 20: 3-[(1H-indazol-5-ylamino)methylene]-1,3-dihydro-2H-indol-2-one, 21: 2-(5-bromo-2-methyl-1H-indol-3-yl)-N’-(2-oxo-1,2-dihydro-3H-indol-3-ylidene)acetohydrazide, 22: N’-(2-oxo-1,2-dihydro-3H-indol-3-ylidene)-3-phenyl-1H-pyrazole-5-carbohydrazide, 23: N’-(5-bromo-2-oxo-1,2-dihydro-3H-indol-3-ylidene)-3-phenyl-1Hpyrazole-5-carbohydrazide, 24: N’-(5,7-dibromo-2-oxo-1,2-dihydro-3H-indol-3-ylidene)-3-phenyl-1Hpyrazole-5-carbohydrazide, 25: N-[1-{[2-(5-bromo-2-oxo-1,2-dihydro-3H-indol-3-ylidene)hydrazino]carbonyl}-2-(3,4-dimethoxyphenyl)vinyl]benzamide, 26: N-[1-{[2-(5-bromo-7-methyl-2-oxo-1,2-dihydro-3H-indol-3-ylidene)hydrazino]carbonyl}-2-(2,5-dimethoxyphenyl)vinyl]benzamide, 27: 3-(4-methoxyphenyl)-N’-(2-oxo-1,2-dihydro-3H-indol-3-ylidene)-1Hpyrazole-5-carbohydrazide, 28: N’-(5-bromo-2-oxo-1,2-dihydro-3H-indol-3-ylidene)-3-(4-methoxyphenyl)-1H-pyrazole-5-carbohydrazide, 29: 3-(4-ethoxyphenyl)-4-methyl-N’-(2-oxo-1,2-dihydro-3H-indol-3-ylidene)-1H-pyrazole-5-carbohydrazide, 30: 3-(2-naphthyl)-N’-(2-oxo-1,2-dihydro-3H-indol-3-ylidene)-1H-pyrazole-5-Carbohydrazide, 31: N-(2-{[2-(5-bromo-2-oxo-1,2-dihydro-3H-indol-3-ylidene)hydrazino]carbonyl}phenyl)benzamide, 32: N’-(5-bromo-2-oxo-1,2-dihydro-3H-indol-3-ylidene)-3-methyl-1Hpyrazole-5-carbohydrazide, 33: 5-(5-bromo-2-oxo-1,2-dihydro-3H-indol-3-ylidene)-2,4-imidazolidinedione, 34: 5-bromo-5′-chloro-3,3′-biindole-2,2′(1H,1′H)-dione, 35: 5-chloro-3,3′-biindole-2,2′(1H,1′H)-dione, 36: 5-fluoro-3,3′-biindole-2,2′(1H,1′H)-dione, 37: 5-bromo-7-methyl-3,3′-biindole-2,2′(1H,1′H)-dione, 38: 6-chloro-7-methyl-3,3′-biindole-2,2′(1H,1′H)-dione. All inhibitors were prepared in 10 mM stock solution dissolved in DMSO.
Derivative Screening and Luciferase Assay
TZM-bl cells were transfected with pc-Tat (1 μg) or silencing RNA (100 nM) against GSK-3β (Cell Signaling Technology, Danvers, MA, USA) or luciferase (Dharmacon, Lafayette, CO, USA) using the AttracteneLipofectamine reagent (InvitrogenQiagen, Chatsworth, CA, USA ) according to the manufacturer’s instructions. TZM-bl cells contain an integrated copy of the firefly luciferase gene under the control of the HIV-1 promoter (obtained through the NIH AIDS Research and Reference Reagent Program). The next day, cells were treated with DMSO or the indicated compound at 1 μM. Forty-eight hours post drug treatment, luciferase activity of the firefly luciferase was measured with the BrightGlo Luciferase Assay (Promega, Madison, WI, USA) and luminescence was read from a 96 well plate on an EG&G Berthold luminometeR (Berthold Technologies, Oak Ridge, TN, USA).
MTT Assay
Fifty thousand cells were plated per well in a 96-well plate and the next day cells were treated with 1 μM compound or DMSO. Forty-eight hours later, 10 μl MTT reagent (5 mg/ml) was added to each well and plates incubated at 37°C for 2 hours. Next, 100 μl of DMSO was added to each well and the plate was shaken for 15 minutes at room temperature. The assay was read at 570 nM using a SpectraMax 340 plate reader (Molecular Devices, Sunnyvale, CA, USA).
Protein extracts and immunoblotting
Whole cell extracts from were prepared. Cells were collected, washed once with PBS and pelleted. Cells were lysed in a buffer containing containing Tris-HCl pH 7.5, 120 mM NaCl, 5 mM EDTA, 0.5% NP-40, 50 mM NaF, 0.2 mM Na3VO4, 1 mM DTT and one tablet complete protease inhibitor cocktail per 50 ml. Lysis was performed under ice-cold conditions, incubated on ice for 30 minutes and spun at 4°C for 5 minutes at 14,000 rpm. The protein concentration for each preparation was determined with a Bio-Rad protein assay kit (Bio-Rad Laboratories, Hercules, CA, USA). Cell extracts were resolved by SDS PAGE on a 4-20% tris-glycine gel (Invitrogen, Carlsbad, CA, USA). Proteins were transferred to polyvinylidene difluoride microporous membranes using the iBlot dry blotting system as described by the manufacturer (Invitrogen ). Membranes were blocked with Dulbecco’s phosphate-buffered saline (PBS) 0.1% Tween-20 + 3% BSA. Primary antibody against specified proteins was incubated with the membrane in blocking solution overnight at 4°C. Antibodies against GSK-3β (1V001) and β-actin (C-11) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Membranes were washed twice with PBS + 0.1% Tween-20 and incubated with HRP-conjugated secondary antibody for one hour in blocking solution. Presence of secondary antibody was detected by SuperSignal West Dura Extended Duration Substrate (Pierce, Rockford, IL, USA). Luminescence was visualized on a Kodak 1D image station (Carestream Helath, Rochester, NY, USA).
RT assay
For RT assays, viral supernatants (10 μl) were incubated in a 96-well plate with RT reaction mixture containing 1× RT buffer (50 mM Tris-HCl, 1 mM DTT, 5 mM MgCl2, 20 mM KCl), 0.1% Triton, poly(A) (10-2 U), poly(dT) (10-2 U) and [3H]TTP. The mixture was incubated overnight at 37°C and 5 μl of the reaction mix was spotted on a DEAE Filter mat paper (PerkinElmer, Shelton, CT, USA) washed four times with 5% Na2HPO4 and three times with water, and then dried completely. RT activity was measured in a Betaplate counter (Wallac, Gaithersburg, MD).
RT-PCR and primers
For RNA analysis of CDK9/cyclin T-dependent genes (Mcl-1, IL-8, cyclin D1) following drug treatments, total RNA was isolated from U937 and U87MG cells using Trizol (Invitrogen) according to the manufacturer’s protocol. A total of 1μg of RNA from the RNA fraction was treated with 0.25mg/ml DNase I for 60min, followed by heat inactivation at 65°C for 15min. A total of 1μg of total RNA was used to generate cDNA with the iScript cDNA Synthesis kit (BIO-Rad) using oligo-dT reverse primers.
Immunoprecipitation and in vitro kinase assay
For immunoprecipitation (IP) 2 mg of extract from 6BIO or 6BIOder-treated (1, 10 μM) U937 cells were immunoprecipitated at 4°C overnight with GSK-3β antibody. The next day complexes were precipitated with A/G beads (Calbiochem) for two hours at 4°C. IPs were washed twice with appropriate TNE buffer and kinase buffer. Reaction mixtures (20 μl) contained final concentrations: 40 mM β-glycerophosphate pH 7.4, 7.5 mM MgCl2, 7.5 mM EGTA, 5% glycerol, [γ-32P]ATP (0.2 mM, 1 μCi), 50 mM NaF, 1 mM orthovanadate, and 0.1% (v/v) β-mercaptoethanol. Phosphorylation reactions were performed with IP material and 200 ng of glycogen synthase peptide 2 (Millipore) as substrate in TTK kinase buffer containing 50 mM HEPES (pH 7.9), 10 mM MgCl2, 6 mM EGTA, and 2.5 mM dithiothreitol. Reactions were incubated at 37°C for 1 hour and stopped by the addition of 1 volume of Laemmli sample buffer containing 5% β-mercaptoethanol and ran on a 4-20% SDS-PAGE. Gels were subjected to autoradiography and quantitation using a Molecular Dynamics PhosphorImager software (Amersham 6BIOsciences, Piscataway, NJ, USA).
CONCLUSIONS
Our studies indicated that 6BIO and 6BIOder can inhibit both Tat-dependent transcription and neuronal cell death. The combined anti-proliferative and anti-inflammatory properties of 6BIO and 6BIOder make them an attractive treatment towards the control of HAND. While the mechanism of neuroprotection is currently unknown and likely to be multifaceted; it is clear that 6BIO and 6BIOder have great potential to be used as a supplement to current HIV-1 therapies.
ACKNOWLEGEMENTS
We would like to thank Ben Berkhout for the Dox Tat/TAR viruses. This work was supported by NIH grants AI0078859, AI0074410 and NS070740 to Fatah Kashanchi and NIH grant NS060632 to Lena Al-Harthi. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aboul-ela F, Karn J, Varani G. The structure of the human immunodeficiency virus type-1 TAR RNA reveals principles of RNA recognition by Tat protein. J Mol Biol. 1995;253:313–332. doi: 10.1006/jmbi.1995.0555. [DOI] [PubMed] [Google Scholar]
- Agbottah E, de La Fuente C, Nekhai S, Barnett A, Gianella-Borradori A, Pumfery A, Kashanchi F. Antiviral activity of CYC202 in HIV-1-infected cells. J Biol Chem. 2005;280:3029–3042. doi: 10.1074/jbc.M406435200. [DOI] [PubMed] [Google Scholar]
- Aksenov MY, Hasselrot U, Wu G, Nath A, Anderson C, Mactutus CF, Booze RM. Temporal relationships between HIV-1 Tat-induced neuronal degeneration, OX-42 immunoreactivity, reactive astrocytosis, and protein oxidation in the rat striatum. Brain Res. 2003;987:1–9. doi: 10.1016/s0006-8993(03)03194-9. [DOI] [PubMed] [Google Scholar]
- Ali A, Hoeflich KP, Woodgett JR. Glycogen synthase kinase-3: properties, functions, and regulation. Chem Rev. 2001;101:2527–2540. doi: 10.1021/cr000110o. [DOI] [PubMed] [Google Scholar]
- Amit S, Hatzubai A, Birman Y, Andersen JS, Ben-Shushan E, Mann M, Ben-Neriah Y, Alkalay I. Axin-mediated CKI phosphorylation of beta-catenin at Ser 45: a molecular switch for the Wnt pathway. Gene Dev. 2002;16:1066–1076. doi: 10.1101/gad.230302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baba M, Okamoto M, Kawamura M, Makino M, Higashida T, Takashi T, Kimura Y, Ikeuchi T, Tetsuka T, Okamoto T. Inhibition of human immunodeficiency virus type 1 replication and cytokine production by fluoroquinoline derivatives. Mol Pharmacol. 1998;53:1097–1103. [PubMed] [Google Scholar]
- Baba M, Okamoto M, Makino M, Kimura Y, Ikeuchi T, Sakaguchi T, Okamoto T. Potent and selective inhibition of human immunodeficiency virus type 1 transcription by piperazinyloxoquinoline derivatives. Antimicrob Agents Chemother. 1997;41:1250–1255. doi: 10.1128/aac.41.6.1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailly C, Colson P, Houssier C, Hamy F. The binding mode of drugs to the TAR RNA of HIV-1 studied by electric linear dichroism. Nucleic Acids Res. 1996;24:1460–1464. doi: 10.1093/nar/24.8.1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bansal AK, Mactutus CF, Nath A, Maragos W, Hauser KF, Booze RM. Neurotoxicity of HIV-1 proteins gp120 and Tat in the rat striatum. Brain Res. 2000;879:42–49. doi: 10.1016/s0006-8993(00)02725-6. [DOI] [PubMed] [Google Scholar]
- Barillari G, Gendelman R, Gallo RC, Ensoli B. The Tat protein of human immunodeficiency virus type 1, a growth factor for AIDS Kaposi sarcoma and cytokine-activated vascular cells, induces adhesion of the same cell types by using integrin receptors recognizing the RGD amino acid sequence. Proc Natl Acad Sci U S A. 1993;90:7941–7945. doi: 10.1073/pnas.90.17.7941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge MJ, Downes CP, Hanley MR. Neural and developmental actions of lithium: a unifying hypothesis. Cell. 1989;59:411–419. doi: 10.1016/0092-8674(89)90026-3. [DOI] [PubMed] [Google Scholar]
- Bonavia R, Bajetto A, Barbero S, Albini A, Noonan DM, Schettini G. HIV-1 Tat causes apoptotic death and calcium homeostasis alterations in rat neurons. Biochem Biophys Res Commun. 2001;288:301–308. doi: 10.1006/bbrc.2001.5743. [DOI] [PubMed] [Google Scholar]
- Bournat JC, Brown AM, Soler AP. Wnt-1 dependent activation of the survival factor NF-kappaB in PC12 cells. J Neurosci Res. 2000;61:21–32. doi: 10.1002/1097-4547(20000701)61:1<21::AID-JNR3>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Boykins RA, Mahieux R, Shankavaram UT, Gho YS, Lee SF, Hewlett IK, Wahl LM, Kleinman HK, Brady JN, Yamada KM, Dhawan S. Cutting edge: a short polypeptide domain of HIV-1-Tat protein mediates pathogenesis. J Immunol. 1999;163:15–20. [PubMed] [Google Scholar]
- Brodsky AS, Erlacher HA, Williamson JR. NMR evidence for a base triple in the HIV-2 TAR C-G.C+ mutant-argininamide complex. Nucleic Acids Res. 1998;26:1991–1995. doi: 10.1093/nar/26.8.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodsky AS, Williamson JR. Solution structure of the HIV-2 TAR-argininamide complex. J Mol Biol. 1997;267:624–639. doi: 10.1006/jmbi.1996.0879. [DOI] [PubMed] [Google Scholar]
- Buss H, Dorrie A, Schmitz ML, Frank R, Livingstone M, Resch K, Kracht M. Phosphorylation of serine 468 by GSK-3beta negatively regulates basal p65 NF-kappaB activity. J Biol Chem. 2004;279:49571–49574. doi: 10.1074/jbc.C400442200. [DOI] [PubMed] [Google Scholar]
- Carroll-Anzinger D, Kumar A, Adarichev V, Kashanchi F, Al-Harthi L. Human immunodeficiency virus-restricted replication in astrocytes and the ability of gamma interferon to modulate this restriction are regulated by a downstream effector of the Wnt signaling pathway. J Virol. 2007;81:5864–5871. doi: 10.1128/JVI.02234-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerpa W, Toledo EM, Varela-Nallar L, Inestrosa NC. The role of Wnt signaling in neuroprotection. Drug News Perspect. 2009;22:579–591. doi: 10.1358/dnp.2009.10.1436817. [DOI] [PubMed] [Google Scholar]
- Coghlan MP, Culbert AA, Cross DA, Corcoran SL, Yates JW, Pearce NJ, Rausch OL, Murphy GJ, Carter PS, Roxbee Cox L, Mills D, Brown MJ, Haigh D, Ward RW, Smith DG, Murray KJ, Reith AD, Holder JC. Selective small molecule inhibitors of glycogen synthase kinase-3 modulate glycogen metabolism and gene transcription. Chem Biol. 2000;7:793–803. doi: 10.1016/s1074-5521(00)00025-9. [DOI] [PubMed] [Google Scholar]
- Conant K, Garzino-Demo A, Nath A, McArthur JC, Halliday W, Power C, Gallo RC, Major EO. Induction of monocyte chemoattractant protein-1 in HIV-1 Tat-stimulated astrocytes and elevation in AIDS dementia. Proc Natl Acad Sci U S A. 1998;95:3117–3121. doi: 10.1073/pnas.95.6.3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cross DA, Culbert AA, Chalmers KA, Facci L, Skaper SD, Reith AD. Selective small-molecule inhibitors of glycogen synthase kinase-3 activity protect primary neurones from death. J Neurochem. 2001;77:94–102. doi: 10.1046/j.1471-4159.2001.t01-1-00251.x. [DOI] [PubMed] [Google Scholar]
- Dassonneville L, Hamy F, Colson P, Houssier C, Bailly C. Binding of Hoechst 33258 to the TAR RNA of HIV-1. Recognition of a pyrimidine bulge-dependent structure. Nucleic Acids Res. 1997;25:4487–4492. doi: 10.1093/nar/25.22.4487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies SP, Reddy H, Caivano M, Cohen P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J. 2000;351:95–105. doi: 10.1042/0264-6021:3510095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou H, Birusingh K, Faraci J, Gorantla S, Poluektova LY, Maggirwar SB, Dewhurst S, Gelbard HA, Gendelman HE. Neuroprotective activities of sodium valproate in a murine model of human immunodeficiency virus-1 encephalitis. J Neurosci. 2003;23:9162–9170. doi: 10.1523/JNEUROSCI.23-27-09162.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dou H, Ellison B, Bradley J, Kasiyanov A, Poluektova LY, Xiong H, Maggirwar S, Dewhurst S, Gelbard HA, Gendelman HE. Neuroprotective mechanisms of lithium in murine human immunodeficiency virus-1 encephalitis. J Neurosci. 2005;25:8375–8385. doi: 10.1523/JNEUROSCI.2164-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards TE, Sigurdsson ST. Electron paramagnetic resonance dynamic signatures of TAR RNA-small molecule complexes provide insight into RNA structure and recognition. Biochemistry. 2002;41:14843–14847. doi: 10.1021/bi026299a. [DOI] [PubMed] [Google Scholar]
- Ensoli B, Buonaguro L, Barillari G, Fiorelli V, Gendelman R, Morgan RA, Wingfield P, Gallo RC. Release, uptake, and effects of extracellular human immunodeficiency virus type 1 Tat protein on cell growth and viral transactivation. J Virol. 1993;67:277–287. doi: 10.1128/jvi.67.1.277-287.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etienne-Manneville S, Hall A. Integrin-mediated activation of Cdc42 controls cell polarity in migrating astrocytes through PKCzeta. Cell. 2001;106:489–498. doi: 10.1016/s0092-8674(01)00471-8. [DOI] [PubMed] [Google Scholar]
- Everall IP, Bell C, Mallory M, Langford D, Adame A, Rockestein E, Masliah E. Lithium ameliorates HIV-gp120-mediated neurotoxicity. Mol Cell Neurosci. 2002;21:493–501. doi: 10.1006/mcne.2002.1196. [DOI] [PubMed] [Google Scholar]
- Faber C, Sticht H, Schweimer K, Rosch P. Structural rearrangements of HIV-1 Tatresponsive RNA upon binding of neomycin B. J Biol Chem. 2000;275:20660–20666. doi: 10.1074/jbc.M000920200. [DOI] [PubMed] [Google Scholar]
- Farooqui R, Zhu S, Fenteany G. Glycogen synthase kinase-3 acts upstream of ADPribosylation factor 6 and Rac1 to regulate epithelial cell migration. Exp Cell Res. 2006;312:1514–1525. doi: 10.1016/j.yexcr.2006.01.018. [DOI] [PubMed] [Google Scholar]
- Frame S, Cohen P. GSK3 takes centre stage more than 20 years after its discovery. Biochem J. 2001;359:1–16. doi: 10.1042/0264-6021:3590001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujinaga K, Cujec TP, Peng J, Garriga J, Price DH, Grana X, Peterlin BM. The ability of positive transcription elongation factor B to transactivate human immunodeficiency virus transcription depends on a functional kinase domain, cyclin T1, and Tat. J Virol. 1998;72:7154–7159. doi: 10.1128/jvi.72.9.7154-7159.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukao T, Koyasu S. PI3K and negative regulation of TLR signaling. Trends Immunol. 2003;24:358–363. doi: 10.1016/s1471-4906(03)00139-x. [DOI] [PubMed] [Google Scholar]
- Galons H, Oumata N, Meijer L. Cyclin-dependent kinase inhibitors: a survey of recent patent literature. Expert Opin Ther Pat. 2010;20:377–404. doi: 10.1517/13543770903524284. [DOI] [PubMed] [Google Scholar]
- Guendel I, Agbottah ET, Kehn-Hall K, Kashanchi F. Inhibition of human immunodeficiency virus type-1 by cdk inhibitors. AIDS Res Ther. 2010;7:7. doi: 10.1186/1742-6405-7-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamy F, Brondani V, Florsheimer A, Stark W, Blommers MJ, Klimkait T. A new class of HIV-1 Tat antagonist acting through Tat-TAR inhibition. Biochemistry. 1998;37:5086–5095. doi: 10.1021/bi972947s. [DOI] [PubMed] [Google Scholar]
- Haubrich RH, Flexner C, Lederman MM, Hirsch M, Pettinelli CP, Ginsberg R, Lietman P, Hamzeh FM, Spector SA, Richman DD. A randomized trial of the activity and safety of Ro 24-7429 (Tat antagonist) versus nucleoside for human immunodeficiency virus infection. The AIDS Clinical Trials Group 213 Team. J Infect Dis. 1995;172:1246–1252. doi: 10.1093/infdis/172.5.1246. [DOI] [PubMed] [Google Scholar]
- Haughey NJ, Mattson MP. Calcium dysregulation and neuronal apoptosis by the HIV-1 proteins Tat and gp120. J Acquir Immune Defic Syndr. 2002;31(Suppl 2):S55–61. doi: 10.1097/00126334-200210012-00005. [DOI] [PubMed] [Google Scholar]
- Haughey NJ, Nath A, Mattson MP, Slevin JT, Geiger JD. HIV-1 Tat through phosphorylation of NMDA receptors potentiates glutamate excitotoxicity. J Neurochem. 2001;78:457–467. doi: 10.1046/j.1471-4159.2001.00396.x. [DOI] [PubMed] [Google Scholar]
- Herrmann CH, Rice AP. Lentivirus Tat proteins specifically associate with a cellular protein kinase, TAK, that hyperphosphorylates the carboxyl-terminal domain of the large subunit of RNA polymerase II: candidate for a Tat cofactor. J Virol. 1995;69:1612–1620. doi: 10.1128/jvi.69.3.1612-1620.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetman M, Cavanaugh JE, Kimelman D, Xia Z. Role of glycogen synthase kinase-3beta in neuronal apoptosis induced by trophic withdrawal. J Neurosci. 2000;20:2567–2574. doi: 10.1523/JNEUROSCI.20-07-02567.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho CY, Wong CH, Li HY. Perturbation of the chromosomal binding of RCC1, Mad2 and survivin causes spindle assembly defects and mitotic catastrophe. J Cell Biochem. 2008;105:835–846. doi: 10.1002/jcb.21879. [DOI] [PubMed] [Google Scholar]
- Hoeflich KP, Luo J, Rubie EA, Tsao MS, Jin O, Woodgett JR. Requirement for glycogen synthase kinase-3beta in cell survival and NF-kappaB activation. Nature. 2000;406:86–90. doi: 10.1038/35017574. [DOI] [PubMed] [Google Scholar]
- Hsu MC, Dhingra U, Earley JV, Holly M, Keith D, Nalin CM, Richou AR, Schutt AD, Tam SY, Potash MJ, et al. Inhibition of type 1 human immunodeficiency virus replication by a tat antagonist to which the virus remains sensitive after prolonged exposure in vitro. Proc Natl Acad Sci U S A. 1993;90:6395–6399. doi: 10.1073/pnas.90.14.6395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones M, Olafson K, Del Bigio MR, Peeling J, Nath A. Intraventricular injection of human immunodeficiency virus type 1 (HIV-1) tat protein causes inflammation, gliosis, apoptosis, and ventricular enlargement. J Neuropathol Exp Neurol. 1998;57:563–570. doi: 10.1097/00005072-199806000-00004. [DOI] [PubMed] [Google Scholar]
- Jope RS, Yuskaitis CJ, Beurel E. Glycogen synthase kinase-3 (GSK3): inflammation, diseases, and therapeutics. Neurochem Res. 2007;32:577–595. doi: 10.1007/s11064-006-9128-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang HJ, Park HJ. Novel molecular mechanism for actinomycin d activity as an oncogenic promoter g-quadruplex binder. Biochemistry. 2009;48:7392–7398. doi: 10.1021/bi9006836. [DOI] [PubMed] [Google Scholar]
- Kashanchi F, Kehn-Hall K. Cyclin Dependent Kinases as Attractive Targets to Prevent Transcription from Viral Genomes. Curr Pharm Design. 2009;15:2520–2532. doi: 10.2174/138161209788682280. [DOI] [PubMed] [Google Scholar]
- Kempf MC, Jones J, Heil ML, Kutsch O. A high-throughput drug screening system for HIV-1 transcription inhibitors. J Biomol Screen. 2006;11:807–815. doi: 10.1177/1087057106290292. [DOI] [PubMed] [Google Scholar]
- Koivisto L, Hakkinen L, Matsumoto K, McCulloch CA, Yamada KM, Larjava H. Glycogen synthase kinase-3 regulates cytoskeleton and translocation of Rac1 in long cellular extensions of human keratinocytes. Exp Cell Res. 2004;293:68–80. doi: 10.1016/j.yexcr.2003.09.026. [DOI] [PubMed] [Google Scholar]
- Krum JM, Rosenstein JM. VEGF mRNA and its receptor flt-1 are expressed in reactive astrocytes following neural grafting and tumor cell implantation in the adult CNS. Exp Neurol. 1998;154:57–65. doi: 10.1006/exnr.1998.6930. [DOI] [PubMed] [Google Scholar]
- Kruman II, Nath A, Mattson MP. HIV-1 protein Tat induces apoptosis of hippocampal neurons by a mechanism involving caspase activation, calcium overload, and oxidative stress. Exp Neurol. 1998;154:276–288. doi: 10.1006/exnr.1998.6958. [DOI] [PubMed] [Google Scholar]
- Kumar A, Zloza A, Moon RT, Watts J, Tenorio AR, Al-Harthi L. Active betacatenin signaling is an inhibitory pathway for human immunodeficiency virus replication in peripheral blood mononuclear cells. J Virol. 2008;82:2813–2820. doi: 10.1128/JVI.02498-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laspia MF, Rice AP, Mathews MB. HIV-1 Tat protein increases transcriptional initiation and stabilizes elongation. Cell. 1989;59:283–292. doi: 10.1016/0092-8674(89)90290-0. [DOI] [PubMed] [Google Scholar]
- Liu Y, Jones M, Hingtgen CM, Bu G, Laribee N, Tanzi RE, Moir RD, Nath A, He JJ. Uptake of HIV-1 tat protein mediated by low-density lipoprotein receptor-related protein disrupts the neuronal metabolic balance of the receptor ligands. Nat Med. 2000;6:1380–1387. doi: 10.1038/82199. [DOI] [PubMed] [Google Scholar]
- Maggirwar SB, Tong N, Ramirez S, Gelbard HA, Dewhurst S. HIV-1 Tat-mediated activation of glycogen synthase kinase-3beta contributes to Tat-mediated neurotoxicity. J Neurochem. 1999;73:578–586. doi: 10.1046/j.1471-4159.1999.0730578.x. [DOI] [PubMed] [Google Scholar]
- Magnuson DS, Knudsen BE, Geiger JD, Brownstone RM, Nath A. Human immunodeficiency virus type 1 tat activates non-N-methyl-D-aspartate excitatory amino acid receptors and causes neurotoxicity. Ann Neurol. 1995;37:373–380. doi: 10.1002/ana.410370314. [DOI] [PubMed] [Google Scholar]
- Malim MH, Cullen BR. HIV-1 structural gene expression requires the binding of multiple Rev monomers to the viral RRE: implications for HIV-1 latency. Cell. 1991;65:241–248. doi: 10.1016/0092-8674(91)90158-u. [DOI] [PubMed] [Google Scholar]
- Malumbres M, Pevarello P, Barbacid M, Bischoff JR. CDK inhibitors in cancer therapy: what is next? Trends Pharmacol Sci. 2008;29:16–21. doi: 10.1016/j.tips.2007.10.012. [DOI] [PubMed] [Google Scholar]
- Maragos WF, Tillman P, Jones M, Bruce-Keller AJ, Roth S, Bell JE, Nath A. Neuronal injury in hippocampus with human immunodeficiency virus transactivating protein, Tat. Neuroscience. 2003;117:43–53. doi: 10.1016/s0306-4522(02)00713-3. [DOI] [PubMed] [Google Scholar]
- Martin M, Rehani K, Jope RS, Michalek SM. Toll-like receptor-mediated cytokine production is differentially regulated by glycogen synthase kinase 3. Nat Immunol. 2005;6:777–784. doi: 10.1038/ni1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meijer L, Flajolet M, Greengard P. Pharmacological inhibitors of glycogen synthase kinase 3. Trends Pharmacol Sci. 2004;25:471–480. doi: 10.1016/j.tips.2004.07.006. [DOI] [PubMed] [Google Scholar]
- Meijer L, Skaltsounis AL, Magiatis P, Polychronopoulos P, Knockaert M, Leost M, Ryan XP, Vonica CA, Brivanlou A, Dajani R, Crovace C, Tarricone C, Musacchio A, Roe SM, Pearl L, Greengard P. GSK-3-selective inhibitors derived from Tyrian purple indirubins. Chem Biol. 2003;10:1255–1266. doi: 10.1016/j.chembiol.2003.11.010. [DOI] [PubMed] [Google Scholar]
- Mestre B, Arzumanov A, Singh M, Boulme F, Litvak S, Gait MJ. Oligonucleotide inhibition of the interaction of HIV-1 Tat protein with the trans-activation responsive region (TAR) of HIV RNA. Biochim Biophys Acta. 1999;1445:86–98. doi: 10.1016/s0167-4781(99)00019-6. [DOI] [PubMed] [Google Scholar]
- Nabel G, Baltimore D. An inducible transcription factor activates expression of human immunodeficiency virus in T cells. Nature. 1987;326:711–713. doi: 10.1038/326711a0. [DOI] [PubMed] [Google Scholar]
- Nath A, Conant K, Chen P, Scott C, Major EO. Transient exposure to HIV-1 Tat protein results in cytokine production in macrophages and astrocytes. A hit and run phenomenon. J Biol Chem. 1999;274:17098–17102. doi: 10.1074/jbc.274.24.17098. [DOI] [PubMed] [Google Scholar]
- Nath A, Psooy K, Martin C, Knudsen B, Magnuson DS, Haughey N, Geiger JD. Identification of a human immunodeficiency virus type 1 Tat epitope that is neuroexcitatory and neurotoxic. J Virol. 1996;70:1475–1480. doi: 10.1128/jvi.70.3.1475-1480.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen TB, Lucero GR, Chana G, Hult BJ, Tatro ET, Masliah E, Grant I, Achim CL, Everall IP, Grp HNR. Glycogen synthase kinase-3 beta (GSK-3 beta) inhibitors AR-A014418 and B6B3O prevent human immunodeficiency virus-mediated neurotoxicity in primary human neurons. J Neurovirol. 2009;15:434–438. doi: 10.1080/13550280903168131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nifosi R, Reyes CM, Kollman PA. Molecular dynamics studies of the HIV-1 TAR and its complex with argininamide. Nucleic Acids Res. 2000;28:4944–4955. doi: 10.1093/nar/28.24.4944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noonan D, Albini A. From the outside in: extracellular activities of HIV Tat. Adv Pharmacol. 2000;48:229–250. doi: 10.1016/s1054-3589(00)48008-7. [DOI] [PubMed] [Google Scholar]
- Owen R, Gordon-Weeks PR. Inhibition of glycogen synthase kinase 3beta in sensory neurons in culture alters filopodia dynamics and microtubule distribution in growth cones. Mol Cell Neurosci. 2003;23:626–637. doi: 10.1016/s1044-7431(03)00095-2. [DOI] [PubMed] [Google Scholar]
- Peng J, Zhu Y, Milton JT, Price DH. Identification of multiple cyclin subunits of human P-TEFb. Genes Dev. 1998;12:755–762. doi: 10.1101/gad.12.5.755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Philippon V, Vellutini C, Gambarelli D, Harkiss G, Arbuthnott G, Metzger D, Roubin R, Filippi P. The basic domain of the lentiviral Tat protein is responsible for damages in mouse brain: involvement of cytokines. Virology. 1994;205:519–529. doi: 10.1006/viro.1994.1673. [DOI] [PubMed] [Google Scholar]
- Plyte SE, Hughes K, Nikolakaki E, Pulverer BJ, Woodgett JR. Glycogen synthase kinase-3: functions in oncogenesis and development. Biochim Biophys Acta. 1992;1114:147–162. doi: 10.1016/0304-419x(92)90012-n. [DOI] [PubMed] [Google Scholar]
- Polychronopoulos P, Magiatis P, Skaltsounis AL, Myrianthopoulos V, Mikros E, Tarricone A, Musacchio A, Roe SM, Pearl L, Leost M, Greengard P, Meijer L. Structural basis for the synthesis of indirubins as potent and selective inhibitors of glycogen synthase kinase-3 and cyclin-dependent kinases. J Med Chem. 2004;47:935–946. doi: 10.1021/jm031016d. [DOI] [PubMed] [Google Scholar]
- Rao R, Hao CM, Breyer MD. Hypertonic stress activates glycogen synthase kinase 3beta-mediated apoptosis of renal medullary interstitial cells, suppressing an NFkappaB-driven cyclooxygenase-2-dependent survival pathway. J Biol Chem. 2004;279:3949–3955. doi: 10.1074/jbc.M309325200. [DOI] [PubMed] [Google Scholar]
- Rice AP, Mathews MB. Transcriptional but not translational regulation of HIV-1 by the tat gene product. Nature. 1988;332:551–553. doi: 10.1038/332551a0. [DOI] [PubMed] [Google Scholar]
- Rohr O, Marban C, Aunis D, Schaeffer E. Regulation of HIV-1 gene transcription: from lymphocytes to microglial cells. J Leukoc Biol. 2003;74:736–749. doi: 10.1189/jlb.0403180. [DOI] [PubMed] [Google Scholar]
- Rossi A, Mukerjee R, Ferrante P, Khalili K, Amini S, Sawaya BE. Human immunodeficiency virus type 1 Tat prevents dephosphorylation of Sp1 by TCF-4 in astrocytes. J Gen Virol. 2006;87:1613–1623. doi: 10.1099/vir.0.81691-0. [DOI] [PubMed] [Google Scholar]
- Rubinfeld B, Albert I, Porfiri E, Fiol C, Munemitsu S, Polakis P. Binding of GSK3beta to the APC-beta-catenin complex and regulation of complex assembly. Science. 1996;272:1023–1026. doi: 10.1126/science.272.5264.1023. [DOI] [PubMed] [Google Scholar]
- Rusnati M, Presta M. HIV-1 Tat protein and endothelium: from protein/cell interaction to AIDS-associated pathologies. Angiogenesis. 2002;5:141–151. doi: 10.1023/a:1023892223074. [DOI] [PubMed] [Google Scholar]
- Sabatier JM, Vives E, Mabrouk K, Benjouad A, Rochat H, Duval A, Hue B, Bahraoui E. Evidence for neurotoxic activity of tat from human immunodeficiency virus type 1. J Virol. 1991;65:961–967. doi: 10.1128/jvi.65.2.961-967.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez JF, Sniderhan LF, Williamson AL, Fan S, Chakraborty-Sett S, Maggirwar SB. Glycogen synthase kinase 3beta-mediated apoptosis of primary cortical astrocytes involves inhibition of nuclear factor kappaB signaling. Mol Cell Biol. 2003;23:4649–4662. doi: 10.1128/MCB.23.13.4649-4662.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobhian B, Laguette N, Yatim A, Nakamura M, Levy Y, Kiernan R, Benkirane M. HIV-1 Tat assembles a multifunctional transcription elongation complex and stably associates with the 7SK snRNP. Mol Cell. 2010;38:439–451. doi: 10.1016/j.molcel.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sui Z, Sniderhan LF, Fan S, Kazmierczak K, Reisinger E, Kovacs AD, Potash MJ, Dewhurst S, Gelbard HA, Maggirwar SB. Human immunodeficiency virus-encoded Tat activates glycogen synthase kinase-3beta to antagonize nuclear factor-kappaB survival pathway in neurons. Eur J Neurosci. 2006;23:2623–2634. doi: 10.1111/j.1460-9568.2006.04813.x. [DOI] [PubMed] [Google Scholar]
- Swindle CS, Kim HG, Klug CA. Mutation of CpGs in the murine stem cell virus retroviral vector long terminal repeat represses silencing in embryonic stem cells. J Biol Chem. 2004;279:34–41. doi: 10.1074/jbc.M309128200. [DOI] [PubMed] [Google Scholar]
- Tardieu M, Hery C, Peudenier S, Boespflug O, Montagnier L. Human immunodeficiency virus type 1-infected monocytic cells can destroy human neural cells after cell-to-cell adhesion. Ann Neurol. 1992;32:11–17. doi: 10.1002/ana.410320104. [DOI] [PubMed] [Google Scholar]
- Van Duyne R, Cardenas J, Easley R, Wu W, Kehn-Hall K, Klase Z, Mendez S, Zeng C, Chen H, Saifuddin M, Kashanchi F. Effect of transcription peptide inhibitors on HIV-1 replication. Virology. 2008;376:308–322. doi: 10.1016/j.virol.2008.02.036. [DOI] [PubMed] [Google Scholar]
- van Weeren PC, de Bruyn KM, de Vries-Smits AM, van Lint J, Burgering BM. Essential role for protein kinase B (PKB) in insulin-induced glycogen synthase kinase 3 inactivation. Characterization of dominant-negative mutant of PKB. J Biol Chem. 1998;273:13150–13156. doi: 10.1074/jbc.273.21.13150. [DOI] [PubMed] [Google Scholar]
- Verhoef K, Marzio G, Hillen W, Bujard H, Berkhout B. Strict control of human immunodeficiency virus type 1 replication by a genetic switch: Tet for Tat. J Virol. 2001;75:979–987. doi: 10.1128/JVI.75.2.979-987.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weeks BS, Lieberman DM, Johnson B, Roque E, Green M, Loewenstein P, Oldfield EH, Kleinman HK. Neurotoxicity of the human immunodeficiency virus type 1 tat transactivator to PC12 cells requires the Tat amino acid 49-58 basic domain. J Neurosci Res. 1995;42:34–40. doi: 10.1002/jnr.490420105. [DOI] [PubMed] [Google Scholar]
- Wei P, Garber ME, Fang SM, Fischer WH, Jones KA. A novel CDK9-associated C-type cyclin interacts directly with HIV-1 Tat and mediates its high-affinity, loop-specific binding to TAR RNA. Cell. 1998;92:451–462. doi: 10.1016/s0092-8674(00)80939-3. [DOI] [PubMed] [Google Scholar]
- Weiss JM, Nath A, Major EO, Berman JW. HIV-1 Tat induces monocyte chemoattractant protein-1-mediated monocyte transmigration across a model of the human blood-brain barrier and up-regulates CCR5 expression on human monocytes. J Immunol. 1999;163:2953–2959. [PubMed] [Google Scholar]
- Wortman B, Darbinian N, Sawaya BE, Khalili K, Amini S. Evidence for regulation of long terminal repeat transcription by Wnt transcription factor TCF-4 in human astrocytic cells. J Virol. 2002;76:11159–11165. doi: 10.1128/JVI.76.21.11159-11165.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]