

Abstract
Objective
Estrogen treatment limits the cytotoxic effects of chemotherapy in estrogen receptor-positive (ER+) breast cancer cell lines, suggesting that estrogen-pathway signaling may confer chemotherapeutic resistance. This study investigates the molecular responses of ER+ breast cancer cell lines to the chemotherapeutic agent, doxorubicin, in the presence or absence of estrogen.
Methods
ER+ MCF-7 and T47-D cells were cultured in hormone-starved or estrogen-containing media with or without doxorubicin at concentrations mimicking the low concentrations seen in plasma and tumor microenvironments in humans following typical bolus administration. Protein levels, phosphorylations, and interactions of estrogen-signaling molecules were assessed following these treatments, as well the effects of ER signaling inhibitors on cell proliferation.
Results
Surprisingly, estrogen and doxorubicin co-treatment markedly induced pro-growth alterations compared to doxorubicin alone and modestly enhanced estrogen alone-induced changes. Several inhibitors suppressed cell proliferation in the presence of doxorubicin and estrogen.
Conclusions
These findings demonstrate that molecular changes caused by doxorubicin in ER+ breast cancer cells can be reversed by estrogen, providing molecular evidence for the poorer responses of ER+ tumors to doxorubicin in the presence of physiologic estrogen levels. Our results also suggest that the addition of drugs targeting the ER, EGFR, the SFKs, MEK, PI3K, and/or the MMP proteins to a conventional chemotherapy regimen may improve chemosensitivity.
Keywords: doxorubicin, breast cancer, estrogen, estrogen receptor, EGFR
Introduction
Breast cancer is the most commonly diagnosed and the second most deadly cancer in women [1]. Clinically, breast tumors can be classified by hormone receptor status, with estrogen receptor-positive (ER+) cancers occurring three times more often than ER- cancers [2]. Patients with ER+ tumors are often treated with hormonal therapies to reduce estrogen responses within the tumor and/or with chemotherapy. However, several clinical studies have reported that patients with ER+ tumors respond less well to chemotherapy than their counterparts with ER- tumors [3-5]. Laboratory studies using ER+ breast cancer cell lines have also demonstrated that the presence of physiologic estrogen levels counter the effects of chemotherapy, which may explain the clinical observation of mitigated doxorubicin sensitivity in ER+ tumors [6, 7].
Doxorubicin hydrochloride (Adriamycin, Rubex) is one of several commonly used chemotherapeutic agents in the treatment of ER+ breast cancer. It is typically administered to cancer patients as a bolus of 40-70 mg/m2. Within about two hours of administration the serum concentration rapidly decreases to levels of 10-100 nM, which are maintained for days [8-12]. Similarly, a study of twelve breast cancer patients given a 90 mg/m2 bolus of epirubicin, an analog of doxorubicin, demonstrated that the average intratumoral concentration paralleled the mean plasma epirubicin level of 52 nM at 25 hr following administration [13]. Investigations of breast cancer cell lines have shown that 1 μM or greater concentrations of doxorubicin decrease cell viability, induce apoptosis, and cause cell cycle arrest [14-16]. However, little is known about whether and how the drug (especially at concentrations of 10-100 nM) affects cell proliferation, cell cycle progression, estrogen/estrogen receptor signaling, and whether estrogen effectors may mediate resistance to doxorubicin. Likewise, few studies explain how estrogen-mediated resistance may occur in estrogen-suppressed patients (i.e. post-menopausal patients, aromatase inhibitor [AI]-treated patients, oophorectomy patients, etc.). The concept of estrogen-mediated resistance is supported by findings of persistent mediators of estrogen signaling in both endocrine and chemotherapy treated patients [17-19].
Estrogen regulates the cellular proliferation, survival, and differentiation of many tissues [20-24] and acts by binding to its canonical receptors, ER-α and/or ER-β in the cytoplasm or nucleus of treated cells. These receptors form homo- and heterodimers that, in turn, activate classical and rapid signaling cascades [25, 26]. Transcription resulting from classical signaling is measurable within hours of estrogen stimulation and results from either direct binding of dimerized ER proteins to estrogen response elements (EREs) upstream of estrogen-responsive genes or participation in non-ERE-dependent transcriptional complexes in the nucleus. Rapid estrogen actions occur in the cytoplasm within minutes of estrogen exposure and are characterized by increased phosphorylation and activation of many signaling molecules including ER-α, epidermal growth factor receptor (EGFR) family members, extracellular regulated kinase (ERK) 1/2, c-Src, and phosphoinositide 3-kinase (PI3K) [27, 28]. Short stimulations with estrogen also induce the formation of complexes between ER-α, c-Src, PI3K, and/or MNAR (Modulator of Nongenomic Actions of the estrogen Receptor, a scaffolding protein also known as PELP-1) as well as between the androgen receptor (AR) and ER-α or c-Src. Some of these complexes result in post-translational modifications that enhance downstream signaling and ultimately gene transcription [29-31]. Phosphorylation of ERK 1/2, c-Src and PI3K can result in the activation of signaling cascades that regulate proliferation, survival, adhesion, and migration independently of and together with ER [28, 32]. Because of their involvement in estrogen action, these molecules are prime candidates for mediating resistance of ER+ breast cancer cells to chemotherapy.
Little is understood about the molecular effects of doxorubicin in the presence of estrogen on ER+ breast cancer despite extensive work with each chemical alone. This report tests the hypothesis that the doxorubicin resistance is mediated in part by estrogen pathway signaling. We seek to test the effects of doxorubicin and estrogen on various downstream components of estrogen signaling pathway. Our model was designed to mimic the prolonged estrogen and doxorubicin exposures observed clinically. We show that prolonged estrogen treatment alone modulated total and phosphorylated levels of specific signaling molecules similar to short estrogen treatments. Doxorubicin (25 nM) alone modestly increased levels of several hormone and growth factor signaling molecules that are downregulated by estrogen alone, suggesting mechanisms by which doxorubicin counteracts the pro-survival effects of estrogen. Surprisingly, treatment with both estrogen and doxorubicin modestly enhanced estrogen-induced changes and markedly induced pro-growth alterations compared to doxorubicin alone, providing molecular evidence for the poorer responses of ER+ tumors to doxorubicin in the presence of physiologic estrogen levels. We also tested inhibitors of several proteins involved in estrogen signaling to identify molecules whose inhibition might decrease intrinsic doxorubicin resistance. We show that inhibitors of the ERK pathway and of MMPs (matrix metalloproteinases that activate growth factor receptor pathways) cooperate with doxorubicin to reduce cell proliferation, while inhibitors of ER(s), Src-family kinase (SFK) protein(s), AR, and PI3K function independently of doxorubicin. Both classes of inhibitors are potential candidates for overcoming doxorubicin resistance in ER+ breast cancers.
Materials and Methods
Reagents
17-β estradiol was purchased from Sigma-Aldrich Corporation (Saint Louis, MO) and resuspended in ethanol for storage. The final media concentration used in experiments (10 nM) contained no more than 0.001% ethanol. Doxorubicin hydrochloride was obtained from Calbiochem (San Diego, CA). For inhibitor experiments PD-98059 (final concentration, 50 μM) was obtained from Biomol International (Enzo Life Sciences, Plymouth Meeting, PA), fulvestrant (1 μM) from AstraZeneca (Wilmington, DE), GM 6001 (10 μM) from Biomol International, gefitinib (10 μM) from LC Laboratories (Woburn, MA), SU6656 (1 μM) from Calbiochem, and LY 295002 (25 μM) from Cayman Chemical (Ann Arbor, MI). All inhibitors were suspended in dimethyl sulfoxide (DMSO; Fisher Scientific) except fulvestrant, which was mixed with ethanol. The final concentration of inhibitors contained no more than 1% DMSO or ethanol. Unless otherwise specified, all other chemical reagents were purchased from Fisher Scientific (Waltham, MA).
Cell Lines
The human ER+ breast cancer cell lines T47-D and MCF-7 were acquired from ATCC (Manassas, VA) and the A. Bouton lab (Univ. of Va.) and maintained at 37°C in a humidified 5% CO2 environment. T47-D and MCF-7 cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS, USA origin), 1 mM sodium pyruvate, and 1% penicillin/streptomycin. For hormone-starvation, cells were cultured in phenol red-free, low glucose DMEM supplemented with 5% charcoal-stripped FBS, 2 mM L-glutamine, 1 mM sodium pyruvate, and 0.76% D-(+)-glucose (45% w/v, Sigma), designated “CSSM.” Except where noted, all liquid cell culture reagents were from Gibco (Billings, MT).
Growth Assay
For each condition tested, 5 × 104 MCF-7 or 105 T47-D cells were seeded per well in a 6-well dish (Corning, Corning, NY) and incubated overnight. The next day, wells were washed three times with Dulbecco’s Phosphate-Buffered Saline (DPBS, Gibco) and replenished with fresh starvation medium. Inhibitors were added to the appropriate wells after 23 hrs, and 10 nM estrogen was added 1 hr later. In the absence of inhibitor, groups were incubated 24 hrs prior to estrogen addition. Cells were incubated an additional 24 hrs, and then the media was changed to fresh starvation medium with or without doxorubicin. Estrogen and inhibitors were added again to the appropriate groups, and cells were incubated another 48 hrs, trypsinized, and counted on a hemocytometer.
Cell Cycle Analysis
MCF-7 treatment groups of CSSM alone, CSSM + estrogen (E), CSSM + doxorubicin (Dox), and CSSM + estrogen + doxorubicin (Dox + E) were seeded in CSSM in triplicate at a density of 2 × 106 cells per 10 cm dish (Greiner bio-one, Frichenhausen, Germany), incubated overnight, and cultured as described for the growth assay. Following a PBS (1.4 M NaCl, 26.8 mM KCl, 40.6 mM Na2HPO4•7H2O) wash cells were detached with 5 ml/dish Accutase (Innovative Cell Technologies, San Diego, CA), and triplicate plates combined. All subsequent steps were at 4°C and utilized polypropylene tubes (Becton Dickenson, Franklin Lakes, NJ). Cell clumps were broken apart by pipetting repeatedly, centrifuged for 6 min at 200 × g, and resuspended in 0.5 ml PBS. Cells were then fixed in 4.5 ml cold 70% ethanol (Fisher Scientific) in ddH2O and stored at -20°C until analyzed. Just prior to analysis, ethanol-suspended cells were centrifuged 5 min at 200 × g, suspended in 1 ml cold PBS and counted by hemacytometer. One million cells were washed in cold PBS, pelleted, resuspended in 1 ml DAPI/Triton X-100 staining solution (0.1% w/v Triton-X-100 [LabChem Inc., Pittsburg, PA] and 1 μg/ml DAPI [Sigma-Aldrich] in ddH2O) for at least 30 min in the dark. DAPI fluorescence was detected by a CyAn™ ADP LX 9 Color flow cytometer (Beckman Coulter, Brea, CA). The pulse width–pulse area signal was used to discriminate between G2 cells and cell doublets and gate out the latter. Data were analyzed with the ModFit LT program, version 3.2.1 (Verity Software House, Topsham, ME).
Western Blot Analysis
For each of the four treatment groups, cells were scraped, pelleted, and lysed in NP-40 lysis buffer (1% (v/v) NP-40, 20 mM Tris HCl pH 8.0, 137 mM NaCl, 10% (v/v) glycerol, 2 mM ethylenediaminetetraacetic acid [EDTA]) supplemented with Protease Inhibitor Cocktail Set I (Calbiochem) and 100 μM Na3VO4, and clarified by centrifugation (9300 × g, 5 min). The protein concentration of the lysates was quantitated with BioRad Protein Assay reagent (Hercules, CA) and the Multiskan MCC plate reader (Fisher Scientific).
Whole cell lysates were analyzed by Western blot (100 μg/lane). Proteins were separated on 8% SDS-PAGE gels and transferred to polyvinylidene fluoride membranes (Immobilon-P, Millipore, Temecula, CA). Membranes were blocked in 5% milk or BSA (w/v) in Tris-buffered saline Tween-20 buffer (TBST; 100 μM Tris base, 1.5 mM NaCl, 1% (v/v) Tween-20) for 1-2 hrs at room temperature and then incubated in a primary antibody solution (identical to blocking buffer) overnight at 4°C (see Table 1 for details). Membranes were then thoroughly washed in TBST and incubated at room temperature with horseradish peroxidase-conjugated secondary antibody (goat anti-mouse or sheep anti-rabbit, GE Healthcare, Waukesha, WI, both 1:2000) for 1 hr. Membranes were again thoroughly washed and incubated with SuperSignal West Pico Chemiluminescent Substrate (Thermoscientific, Waltham, MA) for 2 min in the dark. Membranes were exposed to film (Blue Lite Autorad film, ISC Bioexpress, Kaysville, UT) and processed.
Table 1.
| Protein Target | Block & Ab Solution | Primary Ab Source, Clone/Catalog Number | Primary Ab Dilution | Primary Ab Host Species |
|---|---|---|---|---|
| AR | 5% milk | M. Weber lab (Univ. of VA), AR-21 | 1:10,000 | Rabbit |
| AR pS210/S213 | 5% BSA | Imgenex (San Diego), 156C125.2 | 1:1000 | Mouse |
| AR pY534 | 5% BSA | † | 1:1000 | Rabbit |
| β-actin | 5% milk | Sigma, AC-15 | 1:10,000 | Mouse |
| c-Src | 5% milk | Cell Signaling, #2108 | 1:1000 | Rabbit |
| c-Src | 5% milk | S. Parsons lab, 2-17 | 1:1000 | Mouse |
| c-Src pY418 | 5% BSA | Biosource (Camarillo, TX), #44-660G | 1:5000 | Rabbit |
| EGFR | 5% BSA | Cell Signaling, #2232 | 1:1000 | Rabbit |
| ER-α | 5% milk | Vector Labs, 1D5 | 1:1000 | Mouse |
| ER-α | 5% milk | Santa Cruz, HC-20 | 1:1000 | Rabbit |
| ER-α pS118 | 5% BSA | Cell Signaling, #2515 | 1:2000 | Rabbit |
| ER-α pS167 | 5% BSA | Cell Signaling, #2514 | 1:1000 | Rabbit |
| ER-α pY537 | 5% BSA | Abcam (Cambridge, MA), ab59177 | 1:1000 | Rabbit |
| ER-β | 5% milk | Millipore, 68-4 | 1:1000 | Rabbit |
| ER-β | 5% milk | Abcam, 9.88 | 1:1000 | Mouse |
| ERK 1/2 | 5% milk | B3B9* | 1:1000 | Mouse |
| ERK pT183/Y185 | 5% BSA | Sigma, MAPK-YT | 1:10,000 | Mouse |
| GAPDH | 5% milk | Millipore, 6C5 | 1:10,000 | Mouse |
| HER2 | 5% milk | Santa Cruz, C-18 | 1:1000 | Rabbit |
| HER2 pY877 | 5% BSA | Cell Signaling, #2241 | 1:1000 | Rabbit |
| MNAR | 5% milk | Bethyl (Montgomery, TX), #A300-180A-1 | 1:5000 | Rabbit |
| PI3K p85 | 5% milk | Millipore, AB6 | 1:125 | Mouse |
(Reuter, Catling et al. 1995)
Waller AS, Sharrard RM, Berthon P, Maitland NJ: Androgen receptor localisation and turnover in human prostate epithelium treated with the antiandrogen, casodex. J Mol Endocrinol 2000;24:339-351.
Reuter CW, Catling AD, Weber MJ: Immune complex kinase assays for mitogen-activated protein kinase and mek. Methods Enzymol 1995;255:245-256.
Quantitation and Statistical Analysis
AlphaEaseFC version 3.1.2 (Alpha Innotech Corp., San Leandro, CA) was used to quantitate scanned protein bands on film. All total protein levels were normalized to the loading control. Specific phosphorylations were calculated by first normalizing the phospho- and total protein bands to the loading control and then dividing the normalized phosphorylated band by the normalized total protein. All conditions were divided by the value for the untreated control and expressed as the fold change of the control. Comparisons between treatment groups were carried out using Student’s t-test or ANalysis Of VAriance between groups (ANOVA) corresponding to factorial experiments done in randomized blocks after transforming values to the log scale (to account for experiment-to-experiment variation in growth). All error bars indicate standard errors of the mean (SEM).
Results
Doxorubicin Impairs Estrogen-Induced Proliferation
It has previously been shown that at the level of the tumor microenvironment, estrogen is present at levels likely sufficient for signaling even in post-menopausal or ovarian suppressed patients {Lonning, 2011 #671}. In current standard of care, most patients receiving chemotherapy do not receive concurrent anti-estrogen therapy (AIs or SERMs), thus the hypothesis that estrogen signaling may counteract the effects of chemotherapy is clinically relevant.
In order to develop a cell-culture system that would reflect the clinical treatment paradigm for ER+ breast cancers and allow us to address the biological and molecular questions regarding ER signaling, two ER+ breast cancer cell lines (MCF-7 and T47-D), which are dependent on estrogen for growth and survival, were treated as depicted in Figure 1. Cells were hormone-starved and exposed to 10 nM estrogen alone, doxorubicin alone, estrogen and doxorubicin together, or maintained in hormone depleted conditions for a five day period. Following treatment, cells were assessed for protein levels and phosphorylation status of molecules involved in estrogen signaling by immunoprecipitation/Western blot or for biological responses, such as proliferation, cell death, and cell cycle progression. Figures 2-6 depict representative results of these studies and are described in more detail below.
Figure 1. Schematic depiction of the cell culture treatment protocol.
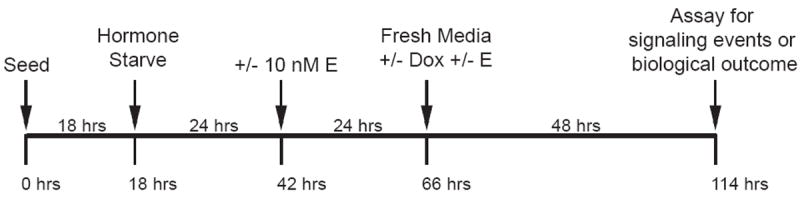
ER+ breast cancer cells were treated with estrogen (E) alone, doxorubicin (Dox) alone, or co-treated as depicted. Cells were seeded in growth medium, incubated overnight, and then hormone-starved. A day later, 10 nM estrogen was added to the appropriate groups, and cells were incubated for an additional 24 hrs. At that time, spent media was replaced with fresh starvation media supplemented with or without doxorubicin and estrogen. Forty-eight hours later, cells were lysed for biochemical analysis by immunoprecipitation or Western blotting, counted, or fixed and stained for cell cycle analysis by flow cytometry.
Figure 2. Doxorubicin impairs estrogen-induced proliferation.
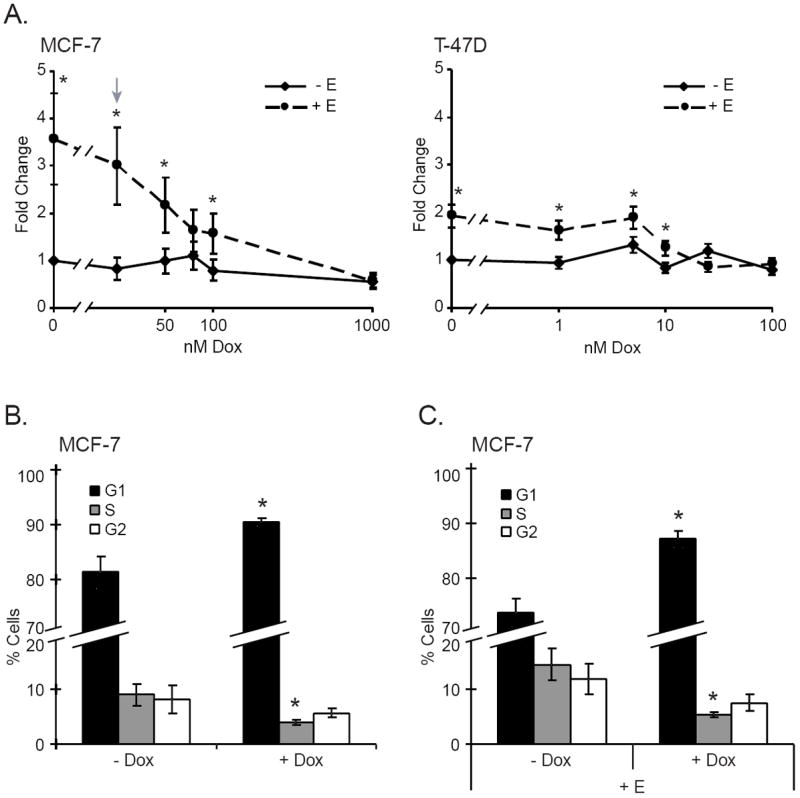
Cells were treated as described in Figure 1 and analyzed by cell counting or by cell cycle distribution. A. MCF-7 (left panel) and T47-D (right panel) cells were treated with increasing doxorubicin concentrations in the presence (dashed line) or absence (solid line) of 10 nM estrogen as described in Materials and Methods. The arrow indicates 25 nM doxorubicin in MCF-7 cells. *Indicates statistical significance between groups treated with and without estrogen (p ≤ 0.05). The results are expressed as the mean fold change in cell number ± SEM for five experiments. B. MCF-7 cells treated with or without 25 nM doxorubicin were stained with DAPI and subjected to cell cycle analysis, as described in Materials and Methods. Results are expressed as the mean percent cells in G1 (black bars), S (grey bars), and G2 (white bars) ± SEM for ten experiments. *Indicates statistical significance between groups treated with and without doxorubicin (p ≤ 0.05). C. MCF-7 cells treated with 10 nM estrogen were left untreated or exposed to 25 nM doxorubicin and analyzed as in 2B. *Indicates statistical significance between groups treated with and without estrogen (p ≤ 0.05).
Figure 6. Estrogen signaling persists in the presence of doxorubicin.
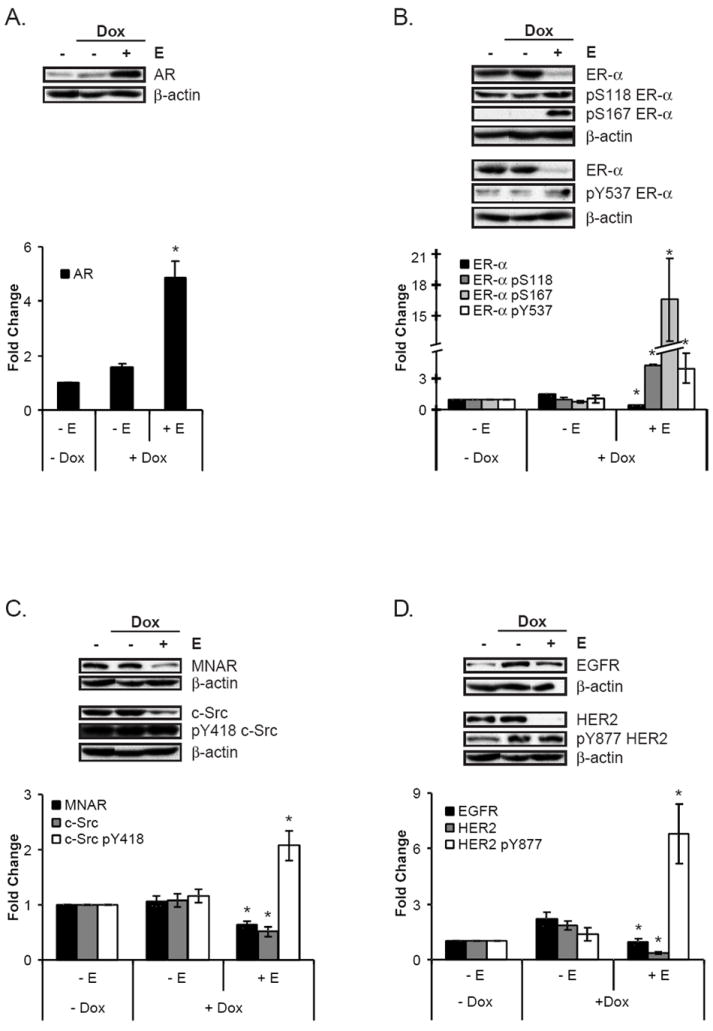
MCF-7 cells were treated according to the scheme depicted in Figure 1. Following lysis, proteins were analyzed by Western blotting, as represented in the upper portion of each panel. Graphs depict the quantitations of the eight or more experiments. The doxorubicin-treated groups with and without estrogen were expressed as the mean fold change of the untreated control ± SEM, as described in Materials and Methods. *Indicate statistical significance between the estrogen-treated groups with and without doxorubicin (p ≤ 0.05). A. As compared to doxorubicin alone, co-treatment with estrogen and doxorubicin increased AR levels. B. The addition of estrogen to doxorubicin decreased ER-α protein (black bars) but increased specific phosphorylation at S118 (dark grey bars), S167 (light grey bars), and Y537 (white bars). C. MNAR (black bars) and c-Src (grey bars) protein levels were decreased, while specific phosphorylation at c-Src Y418 (white bars) was increased from doxorubicin alone by co-treatment with doxorubicin and estrogen. D. As compared to doxorubicin alone, EGFR (black bars) and HER2 (grey bars) protein levels were reduced, but specific phosphorylation of HER2 Y877 (white bars) was increased by the addition of estrogen to doxorubicin treatment.
Previous studies have shown that ER+ breast cancer cells treated with less than 1 μM doxorubicin can survive in the presence of estrogen [6, 7], suggesting that in the presence of the sub-lethal doxorubicin concentrations achievable in human tumors, ER+ breast cancers must be able to employ resistance mechanisms (e.g. estrogen-mediated, SFK-mediated, cell cycle arrest, IGFR-mediated, etc.). Given this, we sought to establish the biological effect of nanomolar concentrations of doxorubicin in our system. MCF-7 and T47-D cells were exposed to doxorubicin in the presence or absence of estrogen (according to the scheme in Fig. 1) and counted after five days as a measure of the cumulative net growth and survival of cells in an estrogenic environment prior to and during doxorubicin treatment. Figure 2A shows that estrogen-induced proliferation was diminished in both cell lines with increasing doxorubicin concentrations, with an approximate 50% decrease at 25 nM in MCF-7 cells. Since the estrogen response in MCF-7 cells in the absence of doxorubicin was more robust than in T47-D cells, and the 25 nM drug concentration permitted an analysis of both estrogen and doxorubicin effects, this doxorubicin concentration and MCF-7 cells were chosen for all subsequent experiments. Within the doxorubicin range tested (0 – 1 μM) and in the absence of estrogen, little change in cell number was observed, consistent with the finding that doxorubicin is most cytotoxic for proliferating cells, and ER+ cells require estrogen for proliferation [33, 34]. Apoptosis was also not detected above background by the terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) or Poly ADP-Ribose Polymerase (PARP) cleavage assays nor was a change in cell death observed by the trypan blue assay in either cell line (data not shown). In contrast, ER- BT-20 breast cancer cells were not dependent upon estrogen for proliferation and demonstrated significant reductions in cell number upon doxorubicin treatment, even in the absence of estrogen (Supplemental Data 1). These results raised the question of what effect, if any, doxorubicin might have on cell cycle progression in ER+ breast cancer cells, in both the absence and presence of estrogen. To address this question, MCF-7 cells were either left untreated or treated with 25 nM doxorubicin alone or together with estrogen for five days and subjected to cell cycle analysis by flow cytometry as a point-in-time measure of the effect of doxorubicin. Figure 2B shows that doxorubicin alone-treated cells arrested in G1, and fewer progressed to S phase compared to non-treated cells, consistent with prior reports and further affirming the validity of our model system [16, 35]. Cells treated with doxorubicin and estrogen together exhibited a similar G1 accumulation and decrease in S phase entry (Fig. 2C). A slight, but statistically insignificant, trend towards reduced G1 and elevated S phases upon the addition of estrogen to doxorubicin treatment (Figs. 2B and 2C, + Dox bars) and the differences in the temporal nature of the assays may explain the apparent differences in proliferation/cell cycle progression between the growth and cell cycle assays. These data indicate that a sub-apoptotic doxorubicin concentration can cause cell cycle blocks that estrogen cannot fully overcome.
Prolonged Estrogen and/or Doxorubicin Treatment Cause Changes in Levels and/or Phosphorylation Status of Estrogen Signaling Molecules
To gain insight into the effects of the various doxorubicin/estrogen treatments on molecules known to be involved in estrogen signaling, lysates from treated cells were analyzed by Western blotting for total protein levels or specific phosphorylations. Our first analyses were carried out on cells stimulated long-term with estrogen alone and compared to those maintained under hormone-deprived, or non-treated, conditions. Figure 3 shows that when compared to hormone-deprived controls, prolonged estrogen treatment alone caused accumulation of AR (Fig. 3A) and PI3K (Fig. 3B) proteins, both of which have been shown to mediate estrogen-induced proliferation and/or survival [36, 37]. Reduction of ER-α (Fig. 3C), c-Src (Fig. 3D), MNAR (Fig. 3D), EGFR (Fig. 3E), and HER2 (Fig. 3E) protein levels was also observed. Changes to these levels may be the result of rapid protein turnover, a hallmark of active signaling, and/or reduced gene expression [38, 39]. In addition, specific phosphorylations associated with increased activity on ER-α (Fig. 3C), c-Src (Fig. 3D), and HER2 (Fig. 3E) were elevated, similar to that seen with acute estrogen action [40-43]. Supplemental Data 2 (-Dox columns) provides the quantification of these and other effects on ER-β and ERK 1/2 proteins and on AR and ERK 1/2 phosphorylations, which were modestly or insignificantly altered by prolonged estrogen treatment.
Figure 3. Prolonged levels of estrogen alter the protein levels and increase specific phosphorylation of estrogen signaling molecules.
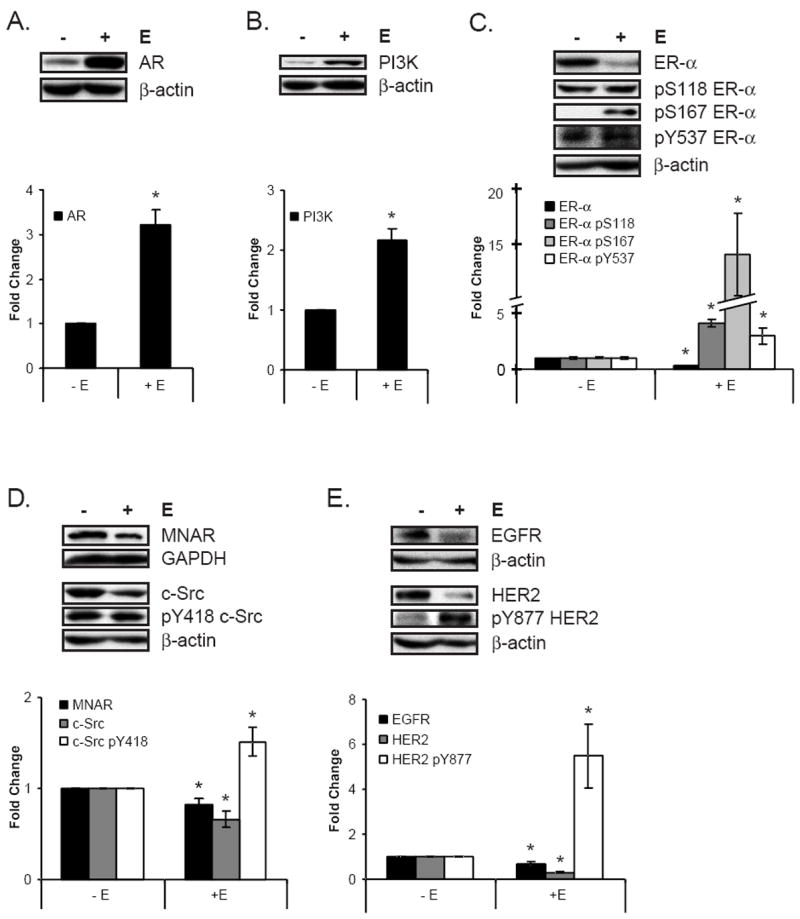
MCF-7 cells, cultured for 72 hrs in the presence or absence of estrogen, were lysed and analyzed by Western blotting. Within each panel, the Western blots are representative of nine or more experiments that were quantitated and shown in the graph. For each graph, the untreated control was set to 1, and the estrogen-treated group was expressed as the mean fold change of the untreated control ± SEM, as described in Materials and Methods. *Indicates statistical significance between groups treated with and without estrogen (p ≤ 0.05). A. Androgen receptor protein levels increased with estrogen treatment. B. Estrogen enhanced PI3K protein levels. C. ER-α levels (black bars) were reduced by estrogen treatment, even as specific phosphorylations at ER-α S118 (dark grey bars), ER-α S167 (light grey bars), and ER-α Y537 (white bars) were elevated. D. MNAR (black bars) and c-Src (grey bars) protein levels decreased with estrogen treatment, but the specific phosphorylation of c-Src’s activating tyrosine (c-Src pY418, white bars) was increased. E. EGFR (black bars) and HER2 (grey bars) levels were reduced, but HER2 specific phosphorylation at Y877 (white bars) was enhanced with estrogen exposure.
The finding that 25 nM doxorubicin could induce a G1 block in the presence of estrogen suggested that extended doxorubicin treatment could affect estrogen downstream signaling (Fig. 2C). Given that low levels of doxorubicin alone also caused cell cycle arrest (Fig. 2B), we asked whether molecules involved in estrogen signaling were affected by doxorubicin treatment in the absence of estrogen. Cell lysates were analyzed by Western blotting following treatment with 25 nM doxorubicin to study these molecular effects. Figure 4 shows that in the absence of estrogen, prolonged doxorubicin treatment resulted in increases in AR (Fig. 4A), ER-α (Fig. 4A), EGFR (Fig. 4B), and HER2 (Fig. 4B) protein levels, an effect that is opposite that of prolonged estrogen alone treatment (with the exception of AR, whose protein levels were elevated by both treatments) (Fig. 3). See also Supplemental Data 2 (-E, compare −Dox to +Dox), which additionally indicates a small but significant reduction in ER-α pS167 levels. These results indicate that doxorubicin alone can have effects on proteins involved in estrogen signaling that are antagonistic to the effects of estrogen alone.
Figure 4. Doxorubicin treatment enhances the levels of pro-proliferative molecules.
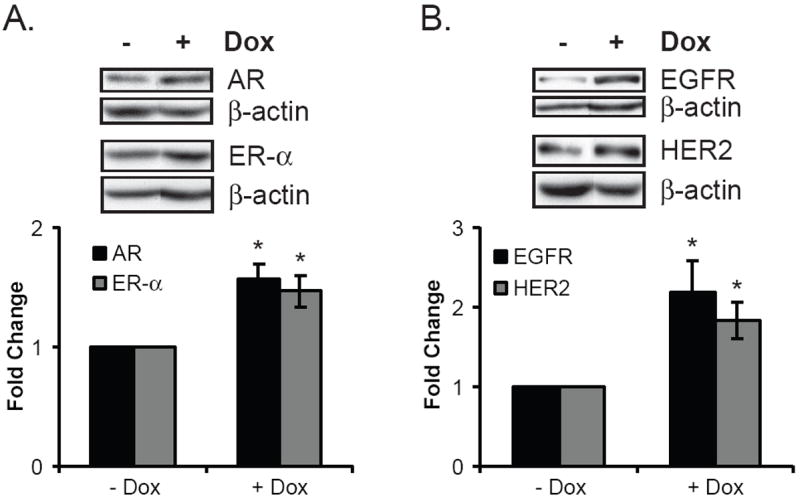
MCF-7 cells were treated as depicted in Figure 1, lysed, and analyzed by Western blotting. Within each panel, the Western blots are representative of the ten or more experiments quantitated in the graph. For each graph the untreated control was set to 1, and the doxorubicin-treated group is expressed as the mean fold change of the untreated control ± SEM, as described in Materials and Methods. *Indicates statistical significance between groups treated with and without doxorubicin (p ≤ 0.05). A. Doxorubicin modestly increased the protein levels of the hormone receptors AR (black bar) and ER-α (grey bar). B. Levels of the growth factor receptor signaling molecules EGFR (black bar) and HER2 (grey bar) were also elevated with doxorubicin treatment.
Results depicted in Figures 3 and 4 indicate that estrogen and doxorubicin individually affect estrogen signaling molecules, but little is known about the signaling changes that occur when an estrogen-responsive tumor is treated with doxorubicin in the absence of estrogen-suppression. To determine the molecular effects of doxorubicin on breast cancer cells cultured in conditions reflective of the human microenvironment, levels and phosphorylation states of proteins from estrogen-exposed cells were compared to those treated with both estrogen and doxorubicin. Surprisingly, doxorubicin and estrogen co-treatment augmented several estrogen-alone responses (Figs. 3 and 5). In comparison to estrogen alone, doxorubicin plus estrogen modestly but significantly increased AR protein levels (Fig. 5A), increased HER2 specific phosphorylation (Fig. 5D), and reduced MNAR protein (Fig. 5B). In contrast, dual doxorubicin/estrogen treatment modestly reduced estrogen alone-induced increases in PI3K levels (Fig. 5C). However, doxorubicin did not affect many estrogen alone-stimulated changes, including those to ER-α, c-Src and EGFR protein levels and ER-α and c-Src phosphorylations. (See Supplemental Data 2 for quantitative comparisons of protein and phosphorylation levels between groups treated with and without doxorubicin on an estrogenic background [-Dox + E vs. +Dox + E].) Contrary to expectations, these results suggest that although doxorubicin had a slight detrimental effect on estrogen signaling (with respect to PI3K), it also left unaffected and even enhanced other aspects of the estrogen signaling pathway.
Figure 5. A low concentration of doxorubicin has both positive and negative effects on estrogen signaling.
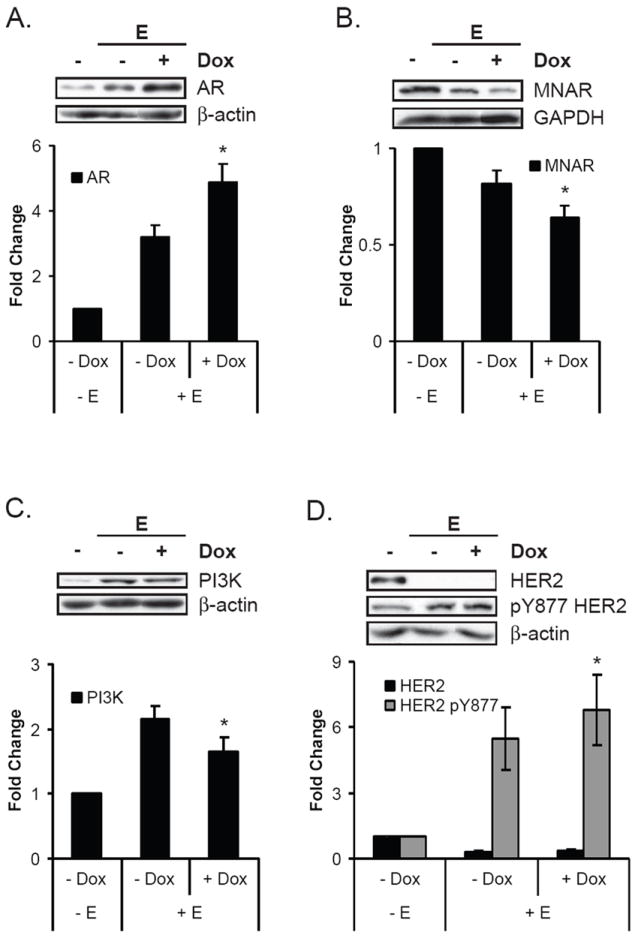
MCF-7 cells were treated as depicted in Figure 1, lysed, and analyzed by Western blotting. Within each panel, the Western blots are representative of the nine or more experiments that were quantitated in the graph. Estrogen-treated groups are expressed as the mean fold change of the untreated control ± SEM, as described in Materials and Methods. *Indicates statistical significance between groups treated with and without estrogen (p ≤ 0.05). A. The addition of doxorubicin to estrogen treatment further increased estrogen-induced AR protein levels. B. MNAR levels were further decreased when doxorubicin was added to estrogen. C. Estrogen-induced elevation of PI3K protein levels was partially reversed by the addition of doxorubicin. D. Though the co-treatment of doxorubicin and estrogen did not significantly change HER2 levels (black bars) from estrogen-alone levels, it caused a slight increase in HER2 specific phosphorylation at Y877 (grey bars).
In addition to chemotherapy, some patients with ER+ tumors are candidates for adjuvant estrogen-ablation therapy. To better understand the molecular changes that occur when ER+ breast tumors are treated with chemotherapy in the presence or absence of estrogen suppression, we compared cells exposed to doxorubicin with and without estrogen treatment. Figure 6 and Supplemental Data 2 show that compared to doxorubicin alone, co-treatment with estrogen resulted in increased AR (Fig. 6A) and decreased ER-α (Fig. 6B), MNAR (Fig. 6C), c-Src (Fig. 6C), EGFR (Fig. 6D) and HER2 (Fig. 6D) protein levels, consistent with changes seen with estrogen alone (Fig. 3). We also observed increased specific phosphorylation at residues correlated with ER-α (Fig. 6B), c-Src (Fig. 6C), and HER2 (Fig. 6D) activation, indicating active estrogen signaling in the presence of doxorubicin. Together, these data indicated that a low dose of doxorubicin cannot completely suppress estrogen signaling in estrogen-responsive breast cancer cells.
Inhibitors of Estrogen Signaling Molecules Reduce MCF-7 Cell Growth in Doxorubicin-Dependent and -Independent Manners
The biochemical studies depicted in Figs. 3-6 and summarized in Supplemental Data 2 clearly demonstrated that doxorubicin treatment resulted in changes to proteins involved in estrogen signaling, whether doxorubicin was administered with or without estrogen. Given these alterations, we sought to determine if targeted inhibitors of estrogen effectors would improve the cytotoxic effects of doxorubicin in the presence of estrogen. To this end, various combinations of doxorubicin, estrogen, and inhibitors of the ER(s), SFK family, EGFR, PI3K, MEK, AR, and MMP proteins were tested for their net effects on MCF-7 cell proliferation. Figure 7 shows that neither PD-98059, a MEK inhibitor, nor GM 6001/Galardin, a pan-MMP inhibitor, significantly affected cell number in the absence of estrogen whether doxorubicin was present or not (Figs. 7A & B, bars 1 vs. 3, and 5). However, in the presence of estrogen, the MEK inhibitor alone reduced cell number (Fig. 7A, bar 2 vs. 4), an effect that was significantly augmented by doxorubicin (Fig. 7A, bar 4 vs. 8). The MMP inhibitor, on the other hand, had little effect on cell number in the absence of doxorubicin and presence of estrogen (Fig. 7B, bar 2 vs. 4) but significantly enhanced the cytotoxic effect of doxorubicin when estrogen was present (Fig. 7B, bar 6 vs. 8) or not (Fig. 7B, bar 5 vs. 7). When administered in the presence of estrogen, the addition of doxorubicin to the MEK and MMP inhibitors resulted in a statistically significant reduction in net proliferation (35.6% and 37.8% reduction, respectively; bars 4 vs. 8). Of all the tested inhibitors, only PD-98059 and GM 6001 exhibited this cooperativity with doxorubicin to reduce cell growth. These results suggest that therapeutically targeting MEK and MMP molecules in doxorubicin-resistant ER+ breast cancers is worthy of further investigation.
Figure 7. MEK and MMP inhibitors cooperate with doxorubicin to inhibit growth of MCF-7 cells.
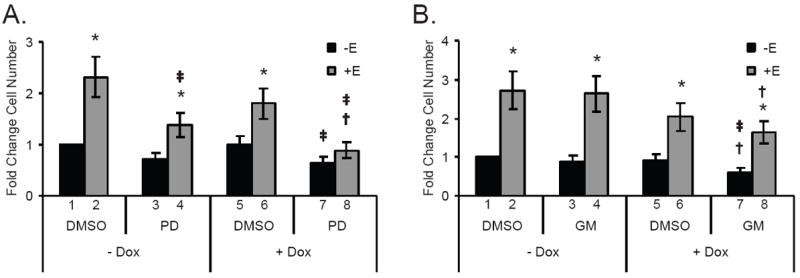
MCF-7 cells treated according to the protocol depicted in Figure 1 were also treated with targeted inhibitor or DMSO a 1 hr prior to estrogen addition. The inhibitor or DMSO remained present in the media until cells were counted. Graphs depict the results of three or more experiments as the mean fold change in cell number ± SEM for each treatment group compared to the - Dox, - E, DMSO control, which was set to 1. Black bars represent groups treated in the absence of estrogen whereas grey bars indicate estrogen-stimulated groups. *Indicates that the addition of estrogen has significantly altered (p ≤ 0.05) the level from that of its non-estrogen treated counterpart (e.g. DMSO - Dox - E vs. DMSO - Dox + E), †denotes a significant change with doxorubicin-treatment from the level of its non-doxorubicin treated counterpart (e.g. DMSO - Dox - E vs. DMSO + Dox - E), and ‡ signifies a difference between groups treated with DMSO and an inhibitor (e.g. DMSO - Dox + E vs. inhibitor - Dox + E). A. The MEK inhibitor, PD-98059 (PD, 50 μM), impaired estrogen-induced proliferation and cooperated with doxorubicin in the presence of estrogen to further reduce cell numbers to basal levels. B. GM 6001 (GM, 10 μM), a pan-MMP inhibitor, demonstrated doxorubicin-dependent decreases in cell number.
In contrast to the MEK and MMP inhibitors, the remainder of the tested targeted drugs acted largely independently of doxorubicin. Figure 8 shows that fulvestrant/Faslodex (an ER-α and -β targeted-drug, Fig. 8A) and gefitinib/Iressa (an EGFR inhibitor, Fig. 8B) inhibited estrogen responses to nearly the same extent in the presence or absence of doxorubicin (Bars 4 vs. 8 in each panel). Similar responses were seen with SU6656 (a SFK inhibitor, Supplemental Data 4A) and LY 294002 (a PI3K inhibitor, Supplemental Data 4B) as with fulvestrant; these three drugs showed the strongest inhibition of estrogen-responses. Bicalutamide/Casodex (an AR-targeted drug, Supplemental Data 4C) additionally demonstrated doxorubicin-independent inhibition, though its effects and mechanism of action are less well understood (see Discussion and Supplemental Data 5). While it was known that the ER, EGFR, SFKs, PI3K and AR mediated estrogen-induced proliferation/survival of ER+ breast cancer cells [34, 36, 44, 45], the efficacy of their inhibitors in the presence of doxorubicin and estrogen had not previously been tested. These findings indicate that the ER protein(s), SFK protein(s), and PI3K are critical to tumor cell growth in the presence or absence of doxorubicin and could be exploited as therapeutic targets in treating doxorubicin-resistant breast cancer cells.
Figure 8. ER and EGFR-targeted drugs are effective inhibitors of estrogen-induced MCF-7 cell growth that act independently of doxorubicin.
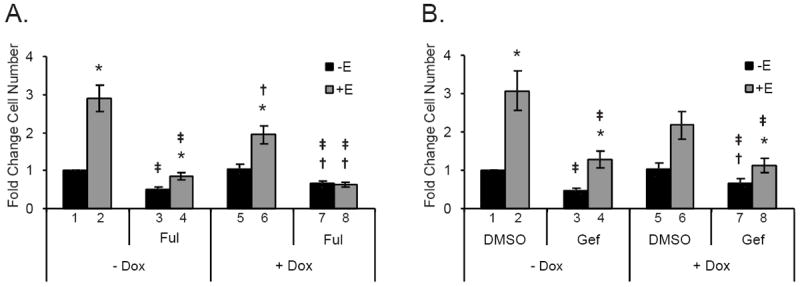
MCF-7 cells were treated and analyzed as in Figure 7. *Indicates that the addition of estrogen has significantly altered (p ≤ 0.05) a level from that of its non-estrogen treated counterpart (e.g. DMSO - Dox - E vs. DMSO - Dox + E), †denotes a significant change with doxorubicin-treatment from the level of its non-doxorubicin treated counterpart (e.g. DMSO - Dox - E vs. DMSO + Dox - E), and ‡ signifies a difference between groups treated with DMSO and an inhibitor (e.g. DMSO - Dox + E vs. inhibitor - Dox + E). Cells treated with 1 μM fulvestrant (Ful, an ER-targeted drug, panel A) or 10 μM gefitinib (Gef, an EGFR inhibitor, panel B) demonstrated inhibitor-dependent decreases in estrogen-induced proliferation in both the absence and presence of doxorubicin.
Discussion
In clinical practice, 75% of breast tumors are classified as ER+ by IHC (using a 1% positive staining cutoff), and many of the tumors with poor prognostic features are treated with a chemotherapy regimen likely to contain an anthracycline such as doxorubicin [46]. However, several studies have demonstrated that chemotherapy is less effective in patients with tumors that are ER+ than ER- [5]. This finding suggests that some mechanism(s) limit(s) chemotherapy-induced apoptosis or cytostasis, including perhaps estrogen signaling itself. However, many in the clinical community have avoided concomitant treatment of ER+ breast cancer with chemotherapy and inhibitors targeting estrogen-signaling molecules based on the rationale that estrogen promotes cell cycling and, therefore, chemotherapeutic efficacy [47]. This appeared to be confirmed by clinical findings in ER+ patients that the efficacy of tamoxifen (an ER-targeted drug) was not improved by concurrent exposure to chemotherapy [48, 49]. A closer examination of the seminal in vitro study supporting these conclusions raises questions as to its usefulness for informing clinical practice: the in vitro experiments demonstrating antagonism between estrogen treatment and chemotherapy involved super-physiological estrogen stimulation, as estrogen was added in the presence of serum [47], potentially resulting in abnormal proliferation properties and responses to chemotherapy. These findings are contrasted by those summarized in a meta-analysis of 194 breast cancer trials: there was a measurable trend towards increased disease-free and overall survival in ER+ and ER status unknown subjects given concurrent verses sequential treatment with chemotherapy and tamoxifen [50]. Additionally, a recent small, single-arm study in advanced breast cancer patients treated concurrently with anthracycline-based chemotherapy and an aromatase inhibitor, which blocks estrogen production [51]. The regimen was well tolerated and 87% of the patients experienced clinical benefit over the course of the trial, supporting the rationale that concomitant estrogen-signaling inhibition and chemotherapy may positively affect clinical outcomes. The limited scope of the clinical trials studying concomitant treatment (i.e. before the era of modern chemotherapy regimens, aromatase inhibitors or other targeted inhibitors) [48, 49] leaves the field of combined chemo-endocrine therapy ripe for the further investigation of modern combinatorial regimens and novel molecularly-targeted drugs.
Given that several studies working within a physiological range of estrogen have shown that doxorubicin is less effective in the presence of estrogen and given the availability of targeted inhibitors to mediators of estrogen signaling, we hypothesized that molecules involved in the estrogen response may mediate doxorubicin resistance and their inhibition during chemotherapy may improve patient outcomes. Therefore, we sought to define specific alterations in estrogen signaling molecules that occurred in response to chemotherapy (doxorubicin in this study) alone or together with estrogen, so as to identify druggable targets for the improvement of chemotherapeutic outcomes of patients with ER+ tumors. Our study demonstrated that in the presence of estrogen, sub-apoptotic levels of doxorubicin (similar to those found intratumorally and in sera of treated patients) permitted and in some cases enhanced changes in a subset of estrogen signaling molecules that estrogen alone induced. Furthermore, inhibiting many of these same molecules augmented the anti-proliferative effect of doxorubicin, suggesting that several mediators of estrogen signaling, including ER, c-Src, PI3K, MEK, and MMP proteins, limit the cytotoxic effects of doxorubicin and are thus promising therapeutic targets to be used in combination with the drug.
Although high levels of doxorubicin are administered to patients, the intratumoral concentration of the drug has not been reported. However, Hunz and colleagues have shown that intratumoral levels of epirubicin, an analog of doxorubicin with highly similar properties, to be in the range of 1-50 nM in breast cancer patients 25 hrs after being given a 90 mg/m2 dose [13]. Additional evidence suggests that intratumoral concentrations of doxorubicin may be quite low, including the rapid reduction in serum concentrations that occurs within hours of its administration [8-12] and the poor apoptotic responses of doxorubicin-treated ER+ breast tumors [3-5]. Furthermore, since anti-estrogen therapy is not administered concurrently with chemotherapy in clinical practice, most breast cancer patients receiving adjuvant chemotherapy are exposed to physiologic levels of estrogen at the time of its administration. (Post-menopausal patients have low, but measurable, serum estradiol levels and higher intratumoral estradiol levels). Based on these treatment paradigms, our cell culture assays initially tested estrogen responses to a range of doxorubicin concentrations from 0 - 1 μM. Our data demonstrated that in ER+ cell lines estrogen can induce proliferation in the presence of doxorubicin at concentrations of 100 nM or less (Fig. 2 and data not shown). Within this range, doxorubicin reduced the proliferative effect of estrogen alone by causing G1 accumulation and reduced S phase entry but did not induce apoptosis, which is consistent with clinical observations of estrogen-responsive tumors treated with chemotherapy. These cell culture studies provide rational support of the hypothesis that very low intratumoral concentrations of chemotherapy coupled with an estrogenic environment may explain the poor pathologic complete response rates of ER+ tumors treated with chemotherapy alone. We speculate that within a tumor, most cells may arrest in the presence of doxorubicin but some resume estrogen-induced proliferation as doxorubicin levels drop.
The results shown in Figure 3 and Supplemental Data 2, demonstrated that ER+ cancer cells subjected to prolonged estrogen elicited similar molecular responses to those described for brief estrogen treatments [28, 38, 52-55]. Though we chose to focus on changes in members of the rapid estrogen signaling pathways in this study, transcriptional changes have also been reported to occur within hours of estrogen stimulation [56]. Indeed, the ER-α, AR, EGFR, and HER2 protein level fluctuations that we observed under the various treatment paradigms are consistent with estrogen-regulated, ER-α– mediated transcriptional mechanisms that have been described previously following several days of estrogen exposure [38, 39, 57-59]. However, we speculate that regulation of protein turnover may also play a role in altering levels of the various signaling molecules, such as occurs when phosphorylation of Ser118 targets ER-α for proteasomal degradation [60]. Whether transcriptional or post-transcriptional/translational modification mechanisms are at play, overall, prolonged estrogen exposure mirrored pro-proliferation and pro-survival responses stimulated by shorter courses of estrogen.
Interestingly, doxorubicin was found to suppress estrogen signaling pathways when administered alone (Fig. 4). The upregulation of ER-α, EGFR, and HER2 levels that occurred in the presence of doxorubicin was opposite of estrogen’s effect on these molecules, suggesting that doxorubicin may antagonize ER pathway signaling. Furthermore, doxorubicin-induced upregulation of ER-α and AR correlated with cell cycle arrest (Figs. 2C and 4A), suggesting that the upregulation of the AR may have facilitated a doxorubicin response. These results mimic those in patients with ER+ breast cancers that co-express the AR and ER, as they are reported to have better survival outcomes [61] than AR-negative patients when treated with chemo- and endocrine therapies.
Doxorubicin-induced changes, however, were counteracted by the addition of estrogen. As shown by the increased phosphorylations and protein level modulations in Figure 6, doxorubicin was unable to prevent estrogen signaling, even enhancing it in some instances (Figs. 5 and 6). Furthermore, associations between c-Src and ER-α that are described as supporting estrogen signaling within minutes of stimulation [29] were altered in much the same way as with estrogen treatment alone (Supplemental Data 3). On a molecular level, these data demonstrated that low levels of doxorubicin do not completely ablate estrogen signaling.
When comparing estrogen effects to those resulting from dual treatment of estrogen and doxorubicin, doxorubicin was shown to have little effect on estrogen-alone signaling, as described above. For example, doxorubicin plus estrogen modestly enhanced AR and MNAR protein levels and HER2 specific phosphorylations over those seen with estrogen alone, while reducing increases in PI3K levels (Fig. 5). However, despite the negative impact of doxorubicin on proliferation (as seen in Figs. 2C, 2D, 7, 8 and Supplemental Data 4), most specific phosphorylation levels and pro-proliferative protein-protein interactions of estrogen signaling effectors were maintained at estrogen-alone levels (Supplemental Data 2 and data not shown). Based on these findings we speculate that within a physiologic system, cells treated with doxorubicin in an estrogenic environment are molecularly poised to participate in robust estrogen signaling upon the removal of doxorubicin.
Previous small molecule inhibitor studies had identified mediators of growth and survival in ER+ breast cancer cells but had not considered their effects in the context of estrogen and chemotherapy together [36, 37, 62]. Our findings (Figs. 7, 8, and Supplemental Data 4) provide rationale for targeting the canonical ER proteins themselves, the SFK proteins, PI3K, MEK, and/or EGFR in patients treated with doxorubicin. In our study the most robust inhibition of proliferation and survival were demonstrated by fulvestrant, SU6656, and LY 294002, respective inhibitors of ER(s), SFK proteins, and PI3K. These inhibitors have all shown efficacy as single agents in inhibiting growth and survival in ER+ cell lines [53, 62-64]; however, monotherapies (except AIs and fulvestrant) to date have all failed in preclinical xenograft models or clinical trials for ER+ tumors [65-68]. For this reason, we suggest that outcomes may be improved with combinatorial treatment of targeted therapies with one another or with chemotherapeutic agents. Fulvestrant, gefitinib, LY 294002, and dasatinib, a kinase inhibitor that targets the SFK proteins, have all shown promising results when combined with doxorubicin in ER+ breast cancer cell culture and/or xenograft studies, [6, 69-71]. Furthermore, a recent report demonstrating that 82% of advanced cancer patients treated with concurrent gefitinib and liposomal doxorubicin experienced clinical benefit over the course of the study [72]. Neither this study nor in vitro work on cardiomyocytes suggest that the addition of the tested inhibitors would increase the cardiotoxicity inherent to anthracycline-based chemotherapy [72, 73], though this has yet to be tested directly in large clinical trials. Together these data imply that ER, SFK proteins, PI3K, MEK, and/or EGFR inhibitors may improve clinical outcomes when combined with chemotherapy
Inhibition of MEK and MMP proteins showed less anti-proliferative efficacy when administered alone than did inhibitors of ER, SFKs and PI3K, but nevertheless were able to enhance the cytotoxic effect of doxorubicin in the presence of estrogen (Fig. 7), suggesting that these enzymes may also play important roles in MCF-7 cell resistance to doxorubicin. Work in sarcoma and rat neuronal cells showed that MMP-7 and MMP-3 inhibit doxorubicin action by negatively regulating Fas action, a mechanism that may also be operative in ER+ breast cancer [74, 75]. We speculate that the modest effect of the MMP inhibitor may be due to its broad spectrum of action and its ability to simultaneously inhibit pro-proliferative and anti-proliferative MMPs [76, 77]. Similarly, the multiple functions of the MEK/MAP kinase pathway can give rise to contrasting outcomes following its inhibition, which appear to depend on cellular context [78-80].
From our studies, the role of the AR in mediating estrogen-induced proliferation and survival was less clear. Inhibition of cell proliferation following treatment with bicalutamide indicated AR involvement in estrogen action (Supplemental Data 4C). Bicalutamide has been shown to downregulate the AR and inhibit its transcriptional activities in prostate cancer cell lines [81, 82]. However, we found that bicalutamide behaved differently in ER+ MCF-7 and T47-D cells than in LNCaP prostate cancer cells (Supplemental Data 5 and data not shown). For example, in ER+ cell lines cultured in the presence of estrogen, the drug had no effect on AR levels or phosphorylation, while in the absence of estrogen, bicalutamide increased both. We also demonstrated through AR knock-down that the AR was not necessary for estrogen- or doxorubicin-dependent changes in proliferation or growth. Together, these data suggested that in ER+ breast cancer the AR level may be enhanced, but appears not to be necessary for, estrogen-induced proliferation.
In summary, we describe doxorubicin-induced changes on estrogen signaling and show that estrogen signaling is indeed relevant to chemotherapeutic efficacy. We demonstrated the surprising finding that in the presence of estrogen the MCF-7 breast cancer cell line responded to sub-apoptotic levels of doxorubicin (which may occur intratumorally) by enhancing estrogen-stimulated changes in phosphorylation and protein levels of a subset of estrogen signaling molecules. Further, the negative effects of doxorubicin alone correlated with suppression of the estrogen signaling pathway (Fig. 9). These findings provide rationale for targeting estrogen/ER effector proteins to enhance chemosensitivity of ER+ tumors. Such dual therapy may ultimately improve outcomes for patients with ER+ breast tumors receiving chemotherapy. Only a few clinical trials have been conducted combining conventional chemotherapy with targeted therapy aimed at estrogen signaling related molecules in the ER+ setting, though the success of HER2 targeted therapy combined with chemotherapy validates such an approach [83, 84]. Our study indicates that the addition of drugs targeting the ER, EGFR, the SFKs, MEK, PI3K, and/or the MMP proteins to a conventional chemotherapy regimen may improve chemosensitivity. Additionally, it appears that in vitro doxorubicin treatment of ER+ breast cancer is superior when administered in the absence of estrogen. Clinically, this supports the design of combination chemoendocrine clinical trials, several of which are ongoing.
Figure 9. Model of doxorubicin action on an estrogen-stimulated ER+ breast cancer cell.
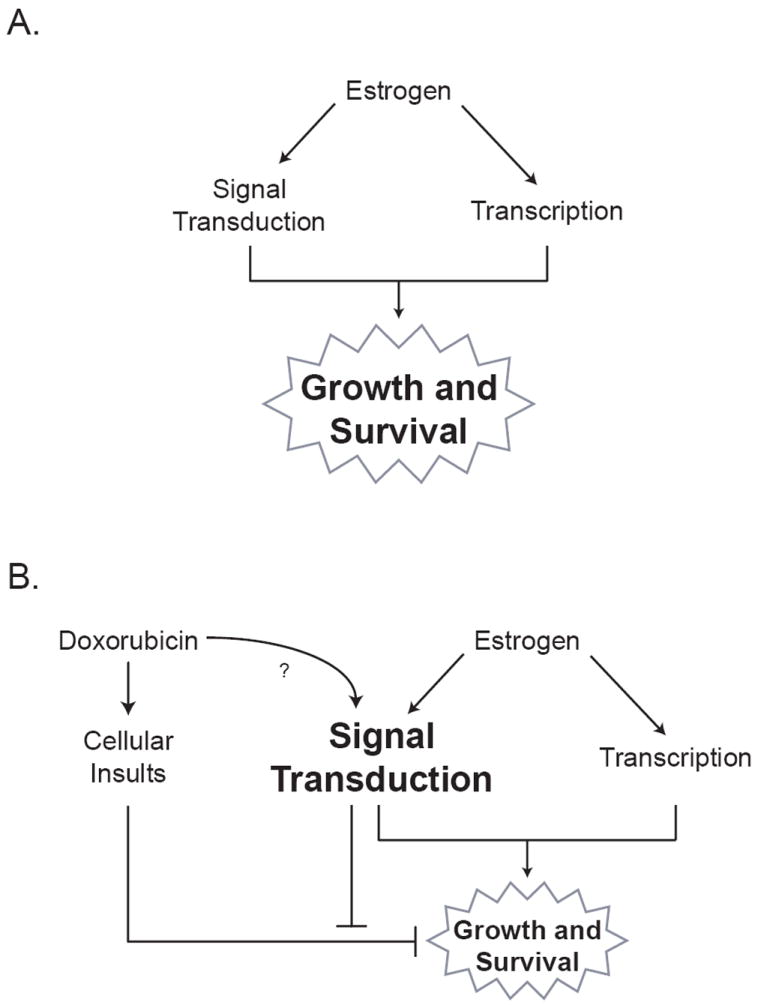
A. Estrogen increases growth and survival through intracellular signaling and transcriptional mechanisms. B. Doxorubicin induces cellular insults that impair estrogen-stimulated growth and survival responses. Through an unknown mechanism, doxorubicin also alters the protein levels and phosphorylation states of various ER effector molecules in the presence of estrogen in ways that are consistent with more robust estrogen signaling. Additionally, some estrogen signaling molecules (such as MEK and MMP proteins) counteract doxorubicin’s impairment of growth and survival, thereby lessening the cytotoxic effects of doxorubicin. The net effect of doxorubicin action in the presence of estrogen is reduced but persistent growth and survival of ER+ breast cancer cells.
Supplementary Material
Acknowledgments
We would like to thank W. Faust, Jr. for his technical contribution to this work as well as K. Hulse, Parsons-Weber-Parsons Research Group, the Women’s Oncology Group, and the past and present members of the S. Parsons lab for their helpful discussions. We are grateful to the M. Weber group for providing the anti-MAPK and anti-AR Western blotting antibodies.
Funding
This study was generously supported by the National Cancer Institute (RO1 CA 123037) (S.J.P), National Cancer Institute Institutional Training Grant (T32 CA009109) (J.E.P), and the Wagner Fellowship (J.E.P.).
List of Abbreviations
- ANOVA
ANalysis Of Variance
- AR
androgen receptor
- BSA
bovine serum albumin
- CSSM
charcoal-stripped serum medium
- DMEM
Dulbecco’s Modified Eagle Medium
- DMSO
dimethyl sulfoxide
- Dox
doxorubicin
- E
estrogen
- EDTA
ethylenediaminetetraacetic acid
- EGFR
epidermal growth factor receptor
- ER
estrogen receptor
- ERE
estrogen response element
- ERK
extracellular regulated kinase
- FBS
fetal bovine serum
- Ful
fulvestrant, ER inhibitor
- Gef
gefitinib, EGFR inhibitor
- GM
GM 6001, MMP inhibitor
- GPR30
G-protein coupled receptor 30
- HER2
human EGFR 2
- IGF-1
insulin-like growth factor-1
- PARP
poly ADP-ribose polymerase
- MAPK
mitogen-activated protein kinase
- MEK
mitogen-activated ERK-activating kinase
- MMP
matrix metalloproteinase
- PBS
phosphate-buffered saline
- PD
PD-98059, MEK inhibitor
- PI3K
phosphoinositide 3-kinase
- MNAR
modular of nongenomic actions of the estrogen receptor
- pS
phosphoserine
- pY
phosphotyrosine
- SEM
standard error of the mean
- SFK
c-Src family kinase
- TBST
Tris-buffered saline Tween-20 buffer
- TUNEL
terminal deoxynucleotidyl transferase dUTP nick end labeling
Footnotes
Declaration of Interests
The authors declare that they have no competing interests.
References
- 1.NCI. Seer cancer statistics review 1975-2007. National Cancer Institute; 2011. Tables 1.23-21.28. [Google Scholar]
- 2.Kocic B, Filipovic S, Petrovic B, Mijalkovic D, Rancic N, Poultsidi A. Clinical and biological characteristics of breast cancer. J Buon. 2010;15:660–667. [PubMed] [Google Scholar]
- 3.Conforti R, Boulet T, Tomasic G, Taranchon E, Arriagada R, Spielmann M, Ducourtieux M, Soria JC, Tursz T, Delaloge S, Michiels S, Andre F. Breast cancer molecular subclassification and estrogen receptor expression to predict efficacy of adjuvant anthracyclines-based chemotherapy: A biomarker study from two randomized trials. Ann Oncol. 2007;18:1477–1483. doi: 10.1093/annonc/mdm209. [DOI] [PubMed] [Google Scholar]
- 4.Kuerer HM, Newman LA, Smith TL, Ames FC, Hunt KK, Dhingra K, Theriault RL, Singh G, Binkley SM, Sneige N, Buchholz TA, Ross MI, McNeese MD, Buzdar AU, Hortobagyi GN, Singletary SE. Clinical course of breast cancer patients with complete pathologic primary tumor and axillary lymph node response to doxorubicin-based neoadjuvant chemotherapy. J Clin Oncol. 1999;17:460–469. doi: 10.1200/JCO.1999.17.2.460. [DOI] [PubMed] [Google Scholar]
- 5.Berry DA, Cirrincione C, Henderson IC, Citron ML, Budman DR, Goldstein LJ, Martino S, Perez EA, Muss HB, Norton L, Hudis C, Winer EP. Estrogen-receptor status and outcomes of modern chemotherapy for patients with node-positive breast cancer. JAMA. 2006;295:1658–1667. doi: 10.1001/jama.295.14.1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Teixeira C, Reed JC, Pratt MA. Estrogen promotes chemotherapeutic drug resistance by a mechanism involving bcl-2 proto-oncogene expression in human breast cancer cells. Cancer Res. 1995;55:3902–3907. [PubMed] [Google Scholar]
- 7.Leung LK, Wang TT. Differential effects of chemotherapeutic agents on the bcl-2/bax apoptosis pathway in human breast cancer cell line mcf-7. Breast Cancer Res Treat. 1999;55:73–83. doi: 10.1023/a:1006190802590. [DOI] [PubMed] [Google Scholar]
- 8.de Bruijn P, Verweij J, Loos WJ, Kolker HJ, Planting AS, Nooter K, Stoter G, Sparreboom A. Determination of doxorubicin and doxorubicinol in plasma of cancer patients by high-performance liquid chromatography. Anal Biochem. 1999;266:216–221. doi: 10.1006/abio.1998.2943. [DOI] [PubMed] [Google Scholar]
- 9.Benjamin RS, Riggs CE, Jr, Bachur NR. Plasma pharmacokinetics of adriamycin and its metabolites in humans with normal hepatic and renal function. Cancer Res. 1977;37:1416–1420. [PubMed] [Google Scholar]
- 10.Creasey WA, McIntosh LS, Brescia T, Odujinrin O, Aspnes GT, Murray E, Marsh JC. Clinical effects and pharmacokinetics of different dosage schedules of adriamycin. Cancer Res. 1976;36:216–221. [PubMed] [Google Scholar]
- 11.Wurz GT, Soc L, Emshoff VD, Cadman TB, DeGregorio MW. Pharmacokinetic analysis of high-dose toremifene in combination with doxorubicin. Cancer Chemother Pharmacol. 1998;42:363–366. doi: 10.1007/s002800050830. [DOI] [PubMed] [Google Scholar]
- 12.Rodvold KA, Rushing DA, Tewksbury DA. Doxorubicin clearance in the obese. J Clin Oncol. 1988;6:1321–1327. doi: 10.1200/JCO.1988.6.8.1321. [DOI] [PubMed] [Google Scholar]
- 13.Hunz M, Jetter A, Warm M, Pantke E, Tuscher M, Hempel G, Jaehde U, Untch M, Kurbacher C, Fuhr U. Plasma and tissue pharmacokinetics of epirubicin and paclitaxel in patients receiving neoadjuvant chemotherapy for locally advanced primary breast cancer. Clin Pharmacol Ther. 2007;81:659–668. doi: 10.1038/sj.clpt.6100067. [DOI] [PubMed] [Google Scholar]
- 14.Saunders DE, Lawrence WD, Christensen C, Wappler NL, Ruan H, Deppe G. Paclitaxel-induced apoptosis in mcf-7 breast-cancer cells. Int J Cancer. 1997;70:214–220. doi: 10.1002/(sici)1097-0215(19970117)70:2<214::aid-ijc13>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 15.Sharma G, Tyagi AK, Singh RP, Chan DC, Agarwal R. Synergistic anti-cancer effects of grape seed extract and conventional cytotoxic agent doxorubicin against human breast carcinoma cells. Breast Cancer Res Treat. 2004;85:1–12. doi: 10.1023/B:BREA.0000020991.55659.59. [DOI] [PubMed] [Google Scholar]
- 16.Rusetskaya NV, Lukyanova NY, Chekhun VF. Molecular profile and cell cycle in mcf-7 and mcf-7/dox cells exposed to conventional and liposomal forms of doxorubicin. Exp Oncol. 2009;31:140–143. [PubMed] [Google Scholar]
- 17.Yamashita H, Takahashi S, Ito Y, Yamashita T, Ando Y, Toyama T, Sugiura H, Yoshimoto N, Kobayashi S, Fujii Y, Iwase H. Predictors of response to exemestane as primary endocrine therapy in estrogen receptor-positive breast cancer. Cancer Sci. 2009;100:2028–2033. doi: 10.1111/j.1349-7006.2009.01274.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.McGlynn LM, Kirkegaard T, Edwards J, Tovey S, Cameron D, Twelves C, Bartlett JM, Cooke TG. Ras/raf-1/mapk pathway mediates response to tamoxifen but not chemotherapy in breast cancer patients. Clin Cancer Res. 2009;15:1487–1495. doi: 10.1158/1078-0432.CCR-07-4967. [DOI] [PubMed] [Google Scholar]
- 19.Yamashita H, Nishio M, Toyama T, Sugiura H, Kondo N, Kobayashi S, Fujii Y, Iwase H. Low phosphorylation of estrogen receptor alpha (eralpha) serine 118 and high phosphorylation of eralpha serine 167 improve survival in er-positive breast cancer. Endocr Relat Cancer. 2008;15:755–763. doi: 10.1677/ERC-08-0078. [DOI] [PubMed] [Google Scholar]
- 20.Critchley HO, Saunders PT. Hormone receptor dynamics in a receptive human endometrium. Reprod Sci. 2009;16:191–199. doi: 10.1177/1933719108331121. [DOI] [PubMed] [Google Scholar]
- 21.Ropero AB, Alonso-Magdalena P, Quesada I, Nadal A. The role of estrogen receptors in the control of energy and glucose homeostasis. Steroids. 2008;73:874–879. doi: 10.1016/j.steroids.2007.12.018. [DOI] [PubMed] [Google Scholar]
- 22.Belcher SM. Rapid signaling mechanisms of estrogens in the developing cerebellum. Brain Res Rev. 2008;57:481–492. doi: 10.1016/j.brainresrev.2007.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Alvaro D, Mancino MG, Onori P, Franchitto A, Alpini G, Francis H, Glaser S, Gaudio E. Estrogens and the pathophysiology of the biliary tree. World J Gastroenterol. 2006;12:3537–3545. doi: 10.3748/wjg.v12.i22.3537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Khosla S. Update on estrogens and the skeleton. J Clin Endocrinol Metab. 2010;95:3569–3577. doi: 10.1210/jc.2010-0856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Welboren WJ, Sweep FC, Span PN, Stunnenberg HG. Genomic actions of estrogen receptor alpha: What are the targets and how are they regulated? Endocr Relat Cancer. 2009;16:1073–1089. doi: 10.1677/ERC-09-0086. [DOI] [PubMed] [Google Scholar]
- 26.McDevitt MA, Glidewell-Kenney C, Jimenez MA, Ahearn PC, Weiss J, Jameson JL, Levine JE. New insights into the classical and non-classical actions of estrogen: Evidence from estrogen receptor knock-out and knock-in mice. Mol Cell Endocrinol. 2008;290:24–30. doi: 10.1016/j.mce.2008.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fox EM, Andrade J, Shupnik MA. Novel actions of estrogen to promote proliferation: Integration of cytoplasmic and nuclear pathways. Steroids. 2009;74:622–627. doi: 10.1016/j.steroids.2008.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lannigan DA. Estrogen receptor phosphorylation. Steroids. 2003;68:1–9. doi: 10.1016/s0039-128x(02)00110-1. [DOI] [PubMed] [Google Scholar]
- 29.Wong CW, McNally C, Nickbarg E, Komm BS, Cheskis BJ. Estrogen receptor-interacting protein that modulates its nongenomic activity-crosstalk with src/erk phosphorylation cascade. Proc Natl Acad Sci U S A. 2002;99:14783–14788. doi: 10.1073/pnas.192569699. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 30.Cheskis BJ, Greger J, Cooch N, McNally C, McLarney S, Lam HS, Rutledge S, Mekonnen B, Hauze D, Nagpal S, Freedman LP. Mnar plays an important role in era activation of src/mapk and pi3k/akt signaling pathways. Steroids. 2008;73:901–905. doi: 10.1016/j.steroids.2007.12.028. [DOI] [PubMed] [Google Scholar]
- 31.Chieffi P, Kisslinger A, Sinisi AA, Abbondanza C, Tramontano D. 17beta-estradiol-induced activation of erk1/2 through endogenous androgen receptor-estradiol receptor alpha-src complex in human prostate cells. Int J Oncol. 2003;23:797–801. [PubMed] [Google Scholar]
- 32.Thomas SM, Brugge JS. Cellular functions regulated by src family kinases. Annu Rev Cell Dev Biol. 1997;13:513–609. doi: 10.1146/annurev.cellbio.13.1.513. [DOI] [PubMed] [Google Scholar]
- 33.Barranco SC. Cellular and molecular effects of adriamycin on dividing and nondividing cells. Pharmacol Ther. 1984;24:303–319. doi: 10.1016/0163-7258(84)90007-x. [DOI] [PubMed] [Google Scholar]
- 34.Migliaccio A, Varricchio L, De Falco A, Castoria G, Arra C, Yamaguchi H, Ciociola A, Lombardi M, Di Stasio R, Barbieri A, Baldi A, Barone MV, Appella E, Auricchio F. Inhibition of the sh3 domain-mediated binding of src to the androgen receptor and its effect on tumor growth. Oncogene. 2007;26:6619–6629. doi: 10.1038/sj.onc.1210487. [DOI] [PubMed] [Google Scholar]
- 35.Yun C, Lee JH, Park H, Jin YM, Park S, Park K, Cho H. Chemotherapeutic drug, adriamycin, restores the function of p53 protein in hepatitis b virus × (hbx) protein-expressing liver cells. Oncogene. 2000;19:5163–5172. doi: 10.1038/sj.onc.1203896. [DOI] [PubMed] [Google Scholar]
- 36.Castoria G, Migliaccio A, Bilancio A, Di Domenico M, de Falco A, Lombardi M, Fiorentino R, Varricchio L, Barone MV, Auricchio F. Pi3-kinase in concert with src promotes the s-phase entry of oestradiol-stimulated mcf-7 cells. Embo J. 2001;20:6050–6059. doi: 10.1093/emboj/20.21.6050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lobenhofer EK, Huper G, Iglehart JD, Marks JR. Inhibition of mitogen-activated protein kinase and phosphatidylinositol 3-kinase activity in mcf-7 cells prevents estrogen-induced mitogenesis. Cell Growth Differ. 2000;11:99–110. [PubMed] [Google Scholar]
- 38.Read LD, Keith D, Jr, Slamon DJ, Katzenellenbogen BS. Hormonal modulation of her-2/neu protooncogene messenger ribonucleic acid and p185 protein expression in human breast cancer cell lines. Cancer Res. 1990;50:3947–3951. [PubMed] [Google Scholar]
- 39.Bourdeau V, Deschenes J, Metivier R, Nagai Y, Nguyen D, Bretschneider N, Gannon F, White JH, Mader S. Genome-wide identification of high-affinity estrogen response elements in human and mouse. Mol Endocrinol. 2004;18:1411–1427. doi: 10.1210/me.2003-0441. [DOI] [PubMed] [Google Scholar]
- 40.Arnold SF, Vorojeikina DP, Notides AC. Phosphorylation of tyrosine 537 on the human estrogen receptor is required for binding to an estrogen response element. J Biol Chem. 1995;270:30205–30212. doi: 10.1074/jbc.270.50.30205. [DOI] [PubMed] [Google Scholar]
- 41.Arnold SF, Obourn JD, Jaffe H, Notides AC. Serine 167 is the major estradiol-induced phosphorylation site on the human estrogen receptor. Mol Endocrinol. 1994;8:1208–1214. doi: 10.1210/mend.8.9.7838153. [DOI] [PubMed] [Google Scholar]
- 42.Murphy LC, Weitsman GE, Skliris GP, Teh EM, Li L, Peng B, Davie JR, Ung K, Niu YL, Troup S, Tomes L, Watson PH. Potential role of estrogen receptor alpha (eralpha) phosphorylated at serine118 in human breast cancer in vivo. J Steroid Biochem Mol Biol. 2006;102:139–146. doi: 10.1016/j.jsbmb.2006.09.021. [DOI] [PubMed] [Google Scholar]
- 43.Antoniotti S, Taverna D, Maggiora P, Sapei ML, Hynes NE, De Bortoli M. Oestrogen and epidermal growth factor down-regulate erbb-2 oncogene protein expression in breast cancer cells by different mechanisms. Br J Cancer. 1994;70:1095–1101. doi: 10.1038/bjc.1994.454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Song RX, Chen Y, Zhang Z, Bao Y, Yue W, Wang JP, Fan P, Santen RJ. Estrogen utilization of igf-1-r and egf-r to signal in breast cancer cells. J Steroid Biochem Mol Biol. 2010;118:219–230. doi: 10.1016/j.jsbmb.2009.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Castoria G, Barone MV, Di Domenico M, Bilancio A, Ametrano D, Migliaccio A, Auricchio F. Non-transcriptional action of oestradiol and progestin triggers DNA synthesis. Embo J. 1999;18:2500–2510. doi: 10.1093/emboj/18.9.2500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gianni L, Norton L, Wolmark N, Suter TM, Bonadonna G, Hortobagyi GN. Role of anthracyclines in the treatment of early breast cancer. J Clin Oncol. 2009;27:4798–4808. doi: 10.1200/JCO.2008.21.4791. [DOI] [PubMed] [Google Scholar]
- 47.Hug V, Hortobagyi GN, Drewinko B, Finders M. Tamoxifen-citrate counteracts the antitumor effects of cytotoxic drugs in vitro. J Clin Oncol. 1985;3:1672–1677. doi: 10.1200/JCO.1985.3.12.1672. [DOI] [PubMed] [Google Scholar]
- 48.Rivkin SE, Green S, Metch B, Cruz AB, Abeloff MD, Jewell WR, Costanzi JJ, Farrar WB, Minton JP, Osborne CK. Adjuvant cmfvp versus tamoxifen versus concurrent cmfvp and tamoxifen for postmenopausal, node-positive, and estrogen receptor-positive breast cancer patients: A southwest oncology group study. J Clin Oncol. 1994;12:2078–2085. doi: 10.1200/JCO.1994.12.10.2078. [DOI] [PubMed] [Google Scholar]
- 49.Pritchard KI, Paterson AH, Fine S, Paul NA, Zee B, Shepherd LE, Abu-Zahra H, Ragaz J, Knowling M, Levine MN, Verma S, Perrault D, Walde PL, Bramwell VH, Poljicak M, Boyd N, Warr D, Norris BD, Bowman D, Armitage GR, Weizel H, Buckman RA. Randomized trial of cyclophosphamide, methotrexate, and fluorouracil chemotherapy added to tamoxifen as adjuvant therapy in postmenopausal women with node-positive estrogen and/or progesterone receptor-positive breast cancer: A report of the national cancer institute of canada clinical trials group. Breast cancer site group. J Clin Oncol. 1997;15:2302–2311. doi: 10.1200/JCO.1997.15.6.2302. [DOI] [PubMed] [Google Scholar]
- 50.EBCTCG EBCTCG. Effects of chemotherapy and hormonal therapy for early breast cancer on recurrence and 15-year survival: An overview of the randomised trials. Lancet. 2005;365:1687–1717. doi: 10.1016/S0140-6736(05)66544-0. [DOI] [PubMed] [Google Scholar]
- 51.de la Haba-Rodriguez J, Mancha RG, Manga GP, Aguilar EA, Baena Canada JM, Rovira PS, Conejo EA. Exemestane and chemotherapy as first-line treatment of metastatic breast cancer: Results of a phase ii study. Clin Breast Cancer. 2010;10:313–317. doi: 10.3816/CBC.2010.n.041. [DOI] [PubMed] [Google Scholar]
- 52.Migliaccio A, Castoria G, Di Domenico M, de Falco A, Bilancio A, Lombardi M, Bottero D, Varricchio L, Nanayakkara M, Rotondi A, Auricchio F. Sex steroid hormones act as growth factors. J Steroid Biochem Mol Biol. 2002;83:31–35. doi: 10.1016/s0960-0760(02)00264-9. [DOI] [PubMed] [Google Scholar]
- 53.Chen Y, Alvarez EA, Azzam D, Wander SA, Guggisberg N, Jorda M, Ju Z, Hennessy BT, Slingerland JM. Combined src and er blockade impairs human breast cancer proliferation in vitro and in vivo. Breast Cancer Res Treat. 2010 doi: 10.1007/s10549-010-1024-7. [DOI] [PubMed] [Google Scholar]
- 54.Crowder RJ, Phommaly C, Tao Y, Hoog J, Luo J, Perou CM, Parker JS, Miller MA, Huntsman DG, Lin L, Snider J, Davies SR, Olson JA, Jr, Watson MA, Saporita A, Weber JD, Ellis MJ. Pik3ca and pik3cb inhibition produce synthetic lethality when combined with estrogen deprivation in estrogen receptor-positive breast cancer. Cancer Res. 2009;69:3955–3962. doi: 10.1158/0008-5472.CAN-08-4450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hitosugi T, Sasaki K, Sato M, Suzuki Y, Umezawa Y. Epidermal growth factor directs sex-specific steroid signaling through src activation. J Biol Chem. 2007;282:10697–10706. doi: 10.1074/jbc.M610444200. [DOI] [PubMed] [Google Scholar]
- 56.Hewitt SC, Li Y, Li L, Korach KS. Estrogen-mediated regulation of igf1 transcription and uterine growth involves direct binding of estrogen receptor alpha to estrogen-responsive elements. J Biol Chem. 2010;285:2676–2685. doi: 10.1074/jbc.M109.043471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lee YR, Park J, Yu HN, Kim JS, Youn HJ, Jung SH. Up-regulation of pi3k/akt signaling by 17beta-estradiol through activation of estrogen receptor-alpha, but not estrogen receptor-beta, and stimulates cell growth in breast cancer cells. Biochem Biophys Res Commun. 2005;336:1221–1226. doi: 10.1016/j.bbrc.2005.08.256. [DOI] [PubMed] [Google Scholar]
- 58.Apparao KB, Lovely LP, Gui Y, Lininger RA, Lessey BA. Elevated endometrial androgen receptor expression in women with polycystic ovarian syndrome. Biol Reprod. 2002;66:297–304. doi: 10.1095/biolreprod66.2.297. [DOI] [PubMed] [Google Scholar]
- 59.Yarden RI, Wilson MA, Chrysogelos SA. Estrogen suppression of egfr expression in breast cancer cells: A possible mechanism to modulate growth. J Cell Biochem Suppl. 2001;(Suppl 36):232–246. doi: 10.1002/jcb.1142. [DOI] [PubMed] [Google Scholar]
- 60.Valley CC, Metivier R, Solodin NM, Fowler AM, Mashek MT, Hill L, Alarid ET. Differential regulation of estrogen-inducible proteolysis and transcription by the estrogen receptor alpha n terminus. Mol Cell Biol. 2005;25:5417–5428. doi: 10.1128/MCB.25.13.5417-5428.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Castellano I, Allia E, Accortanzo V, Vandone AM, Chiusa L, Arisio R, Durando A, Donadio M, Bussolati G, Coates AS, Viale G, Sapino A. Androgen receptor expression is a significant prognostic factor in estrogen receptor positive breast cancers. Breast Cancer Res Treat. 2010;124:607–617. doi: 10.1007/s10549-010-0761-y. [DOI] [PubMed] [Google Scholar]
- 62.Okubo S, Kurebayashi J, Otsuki T, Yamamoto Y, Tanaka K, Sonoo H. Additive antitumour effect of the epidermal growth factor receptor tyrosine kinase inhibitor gefitinib (iressa, zd1839) and the antioestrogen fulvestrant (faslodex, ici 182,780) in breast cancer cells. Br J Cancer. 2004;90:236–244. doi: 10.1038/sj.bjc.6601504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Creighton CJ, Fu X, Hennessy BT, Casa AJ, Zhang Y, Gonzalez-Angulo AM, Lluch A, Gray JW, Brown PH, Hilsenbeck SG, Osborne CK, Mills GB, Lee AV, Schiff R. Proteomic and transcriptomic profiling reveals a link between the pi3k pathway and lower estrogen-receptor (er) levels and activity in er+ breast cancer. Breast Cancer Res. 2010;12:R40. doi: 10.1186/bcr2594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Reddy KB, Glaros S. Inhibition of the map kinase activity suppresses estrogen-induced breast tumor growth both in vitro and in vivo. Int J Oncol. 2007;30:971–975. [PubMed] [Google Scholar]
- 65.Creighton CJ, Massarweh S, Huang S, Tsimelzon A, Hilsenbeck SG, Osborne CK, Shou J, Malorni L, Schiff R. Development of resistance to targeted therapies transforms the clinically associated molecular profile subtype of breast tumor xenografts. Cancer Res. 2008;68:7493–7501. doi: 10.1158/0008-5472.CAN-08-1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Untch M, Gelber RD, Jackisch C, Procter M, Baselga J, Bell R, Cameron D, Bari M, Smith I, Leyland-Jones B, de Azambuja E, Wermuth P, Khasanov R, Feng-Yi F, Constantin C, Mayordomo JI, Su CH, Yu SY, Lluch A, Senkus-Konefka E, Price C, Haslbauer F, Suarez Sahui T, Srimuninnimit V, Colleoni M, Coates AS, Piccart-Gebhart MJ, Goldhirsch A. Estimating the magnitude of trastuzumab effects within patient subgroups in the hera trial. Ann Oncol. 2008;19:1090–1096. doi: 10.1093/annonc/mdn005. [DOI] [PubMed] [Google Scholar]
- 67.Regan MM, Gelber RD. Using clinical trial data to tailor adjuvant treatments for individual patients. Breast. 2007;16(Suppl 2):S98–104. doi: 10.1016/j.breast.2007.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Perrault DJ, Logan DM, Stewart DJ, Bramwell VH, Paterson AH, Eisenhauer EA. Phase ii study of flutamide in patients with metastatic breast cancer. A national cancer institute of canada clinical trials group study. Invest New Drugs. 1988;6:207–210. doi: 10.1007/BF00175399. [DOI] [PubMed] [Google Scholar]
- 69.Pichot CS, Hartig SM, Xia L, Arvanitis C, Monisvais D, Lee FY, Frost JA, Corey SJ. Dasatinib synergizes with doxorubicin to block growth, migration, and invasion of breast cancer cells. Br J Cancer. 2009;101:38–47. doi: 10.1038/sj.bjc.6605101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ren W, Korchin B, Zhu QS, Wei C, Dicker A, Heymach J, Lazar A, Pollock RE, Lev D. Epidermal growth factor receptor blockade in combination with conventional chemotherapy inhibits soft tissue sarcoma cell growth in vitro and in vivo. Clin Cancer Res. 2008;14:2785–2795. doi: 10.1158/1078-0432.CCR-07-4471. [DOI] [PubMed] [Google Scholar]
- 71.Wang YA, Johnson SK, Brown BL, McCarragher LM, Al-Sakkaf K, Royds JA, Dobson PR. Enhanced anti-cancer effect of a phosphatidylinositol-3 kinase inhibitor and doxorubicin on human breast epithelial cell lines with different p53 and oestrogen receptor status. Int J Cancer. 2008;123:1536–1544. doi: 10.1002/ijc.23671. [DOI] [PubMed] [Google Scholar]
- 72.Campos SM, Berlin ST, Parker LM, Chen WY, Bunnell CA, Atkinson T, Lee J, Matulonis U, Hirsch MS, Harris L, Krasner CN. Phase i trial of liposomal doxorubicin and zd1839 in patients with refractory gynecological malignancies or metastatic breast cancer. Int J Clin Oncol. 2010;15:390–398. doi: 10.1007/s10147-010-0073-6. [DOI] [PubMed] [Google Scholar]
- 73.Patten RD, Pourati I, Aronovitz MJ, Baur J, Celestin F, Chen X, Michael A, Haq S, Nuedling S, Grohe C, Force T, Mendelsohn ME, Karas RH. 17beta-estradiol reduces cardiomyocyte apoptosis in vivo and in vitro via activation of phospho-inositide-3 kinase/akt signaling. Circ Res. 2004;95:692–699. doi: 10.1161/01.RES.0000144126.57786.89. [DOI] [PubMed] [Google Scholar]
- 74.Mitsiades N, Yu WH, Poulaki V, Tsokos M, Stamenkovic I. Matrix metalloproteinase-7-mediated cleavage of fas ligand protects tumor cells from chemotherapeutic drug cytotoxicity. Cancer Res. 2001;61:577–581. [PubMed] [Google Scholar]
- 75.Wetzel M, Rosenberg GA, Cunningham LA. Tissue inhibitor of metalloproteinases-3 and matrix metalloproteinase-3 regulate neuronal sensitivity to doxorubicin-induced apoptosis. Eur J Neurosci. 2003;18:1050–1060. doi: 10.1046/j.1460-9568.2003.02838.x. [DOI] [PubMed] [Google Scholar]
- 76.Nilsson UW, Garvin S, Dabrosin C. Mmp-2 and mmp-9 activity is regulated by estradiol and tamoxifen in cultured human breast cancer cells. Breast Cancer Res Treat. 2007;102:253–261. doi: 10.1007/s10549-006-9335-4. [DOI] [PubMed] [Google Scholar]
- 77.Philips N, McFadden K. Inhibition of transforming growth factor-beta and matrix metalloproteinases by estrogen and prolactin in breast cancer cells. Cancer Lett. 2004;206:63–68. doi: 10.1016/j.canlet.2003.10.019. [DOI] [PubMed] [Google Scholar]
- 78.Zhao Y, Shen S, Guo J, Chen H, Greenblatt DY, Kleeff J, Liao Q, Chen G, Friess H, Leung PS. Mitogen-activated protein kinases and chemoresistance in pancreatic cancer cells. J Surg Res. 2006;136:325–335. doi: 10.1016/j.jss.2006.06.031. [DOI] [PubMed] [Google Scholar]
- 79.Choi J, Yip-Schneider M, Albertin F, Wiesenauer C, Wang Y, Schmidt CM. The effect of doxorubicin on mek-erk signaling predicts its efficacy in hcc. J Surg Res. 2008;150:219–226. doi: 10.1016/j.jss.2008.01.029. [DOI] [PubMed] [Google Scholar]
- 80.Brantley-Finley C, Lyle CS, Du L, Goodwin ME, Hall T, Szwedo D, Kaushal GP, Chambers TC. The jnk, erk and p53 pathways play distinct roles in apoptosis mediated by the antitumor agents vinblastine, doxorubicin, and etoposide. Biochem Pharmacol. 2003;66:459–469. doi: 10.1016/s0006-2952(03)00255-7. [DOI] [PubMed] [Google Scholar]
- 81.Waller AS, Sharrard RM, Berthon P, Maitland NJ. Androgen receptor localisation and turnover in human prostate epithelium treated with the antiandrogen, casodex. J Mol Endocrinol. 2000;24:339–351. doi: 10.1677/jme.0.0240339. [DOI] [PubMed] [Google Scholar]
- 82.Masiello D, Cheng S, Bubley GJ, Lu ML, Balk SP. Bicalutamide functions as an androgen receptor antagonist by assembly of a transcriptionally inactive receptor. J Biol Chem. 2002;277:26321–26326. doi: 10.1074/jbc.M203310200. [DOI] [PubMed] [Google Scholar]
- 83.NCI. Clinical trials database. National Cancer Institute; 2011. [Google Scholar]
- 84.Buzdar AU. Role of biologic therapy and chemotherapy in hormone receptor- and her2-positive breast cancer. Ann Oncol. 2009;20:993–999. doi: 10.1093/annonc/mdn739. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.