

Abstract
Obesity is associated with elevated risk of heart disease. A solid understanding of the safety and potential adverse effects of high fat, low carbohydrate diet (HFLCD) similar to that used by humans for weight loss on the heart is crucial. High fat intake is known to promote increases in ROS and mitochondrial damage. We hypothesized that there would be adverse effects of HFLCD on myocardial ischemia-reperfusion injury through enhancing oxidative stress injury and impairing mitochondrial biogenesis in a non-genetic, diet-induced rat model of obesity. To test the hypothesis, 250g male Sprague-Dawley rats were fed an obesity-promoting diet for 7 weeks to induce obesity, then switched to HFLCD or a low fat control diet for 2 weeks. Isolated hearts underwent global low flow ischemia for 60 min and reperfusion for 60 min. HFLCD resulted in greater weight gain and lower myocardial glycogen, plasma adiponectin and insulin. Myocardial antioxidant genes transcript and protein expression of superoxide dismutase and catalase were reduced in HFLCD, along with increased oxidative gene NADPH oxidase-4 transcript and xanthine oxidase activity, and a 37% increase in nitrated protein (nitrotyrosine) in HFLCD hearts. The cardiac expression of key mitochondrial regulatory factors such as nuclear respiratory factor-1 and transcription factor A-mitochondrial were inhibited and myocardial mitochondrial DNA copy number decreased. The cardiac expression of adiponectin and its receptors were downregulated in HFLCD. HFLCD impaired recovery of left ventricular rate-pressure product after ischemia-reperfusion, and led to 3.5-fold increased injury as measured by LDH release. In conclusion, HFLCD leads to increased ischemic myocardial injury and impaired recovery of function following reperfusion and was associated with attenuation of mitochondrial biogenesis and enhanced oxidative stress in obese rats. These findings may have important implications for diet selection in obese patients with ischemic heart disease.
Keywords: Diet, Rat, Myocardial ischemia, Obesity, mitochondria, oxidative stress
1. Introduction
Obesity is a major public health problem in the developing world, and it is clear that obesity is associated with elevated risk of diabetes, hypertension, stroke, and ischemic heart disease, making dietary interventions to lose weight of paramount importance. There is great interest in specialty diets for weight loss, with manipulation of the macronutrient composition between fat, protein, and carbohydrate (CHO). Some specialists have advanced the concept that restriction of CHO with more allowance for protein and fat (a high-fat, low-carbohydrate diet, or HFLCD) is effective for weight loss. Consequently, a solid understanding of the safety and potential adverse effects of HFLCD is crucial. Almost a decade ago, a comprehensive review of the efficacy and safety of low-carbohydrate diets found inconclusive evidence to recommend either for or against HFLCD. Clinical outcomes studies examining the long term safety of these diets have yielded conflicting results, with some studies reporting an increase in adverse cardiovascular events in those on such diets, and others study finding no strong link between diet and cardiovascular events or mortality.
While effects of these diets on cardiovascular risk factors such as lipoprotein-associated cholesterol levels and diabetes or glucose tolerance are important, there are potentially other adverse consequences of these diets, including possible direct effects on the heart. Indeed as we have previously shown, rats undergoing short-term HFLCD feeding exhibited significantly increased ischemia-reperfusion injury, which was due to direct diet effects on the myocardium including impairment of insulin signaling and, possibly, derangements in glycogen stores. A limitation of this prior work in applying its results to the study of HFLCD in humans was that we used relatively young, normal weight rats, rather than an older, more obese animal, the typical human choosing such diets is typically older and overweight. Several models of obesity are commercially available and include the db/db mouse and Zucker diabetic fatty (ZDF; fa/fa), however, the obesity, insulin resistance, and diabetes seen in these inbred models are typically the result of gene mutations: the systemic leptin receptor defect seen in db/db mice and fa/fa rats is not considered to be a typical characteristic of human obesity and type 2 diabetes. For these reasons, a diet-induced, non-genetic model of obesity, due to its similarity with human obesity, is more clinically relevant to investigate the effects of HFLCD in obesity.
Reactive oxygen species (ROS) are implicated in a wide range of pathological conditions including ischemia-reperfusion injury, heart failure progression and aging. High fat diets increase fat-mediated oxidative stress and decrease antioxidative enzyme gene expression. Mitochondria (mt) are both major producers of ROS, as well as vulnerable targets of oxidative damage. Alterations of mitochondrial structure and function as a result of increased oxidative stress have been identified as an important contributor to the pathogenesis of heart disease. Typically, homeostasis is maintained by a balance between ROS formation and endogenous antioxidant defenses. ROS can be produced by several mechanisms including mitochondrial electron transport, NADPH oxidase, and xanthine dehydrogenase/xanthine oxidase. It has been well established that multiple endogenous antioxidant mechanisms such as the superoxide dismutases (SOD1, SOD2), glutathione peroxidase (GPX) and catalase are all crucial in maintaining cellular redox balance. However, if this balance is disturbed, oxidative stress increases, leading to damage of essential cellular components, including the mitochondria. On the other hand, mitochondrial biogenesis is a continuous renewal process marked by regular mitochondrial turnover. The control mechanisms involved include nuclear regulatory factor (NRF1), and mitochondrial transcription factor A (TFAM). They function as transcription factors which not only regulate mitochondrial DNA transcription and replication, but also maintain cellular mitochondrial density, and activate the expression of some key metabolic genes such as cytochrome c oxidase (COX).
Given these considerations, the present study investigated the hypothesis that HFLCD increases oxidative stress and impairs mitochondrial biogenesis, associated with an increase in myocardial ischemia-reperfusion (I/R) injury. A non-genetic, diet-induced model of obesity in the rat was studied to provide additional relevance to the use of HFLCD in obese humans for weight loss. An isolated heart ischemia-reperfusion procedure was used for determining the diets effect on cardiac function and ischemic injury, which may provide important implications for diet selection in obese patients with ischemic heart disease.
2. Methods and materials
2.1. Ethics Statement
Animal experiments were approved by the Institutional Animal Care and Use Committee of the University of Alabama at Birmingham and followed the Guide for the Care and Use of Laboratory Animals (National Academy of Sciences, 1996).
2.2. Animal and diets
Eight-week-old 250 gram male Sprague-Dawley rats (Taconic Farms) were fed standard chow diet and an obesity-promoting Western Diet (WD; 40% kcal from fat, 15% protein, 45% carbohydrate; TestDiet 5TTC, Richmond, IN) for 7 weeks to induce obesity. The diets were then switched to an alternate diet similar to certain types used by humans for weight loss: HFLCD (60%/30%/10%; TestDiet 5TSY) or a low fat control diet (CONT; 16%/19%/65%; TestDiet 5TJM) for 2 weeks (Table 1). The rats were housed three per cage at 22 °C on a 12-hour light–dark cycle. Animals were allowed ad libitum access to food. Water was available to the animals at all times. Weight gain and food intake were monitored once a week. To summarize, the overall experimental diet and measurement scheme is shown in Figure 1.
Table 1.
Compositions of diets (WD, CONT and HFLCD).
| Component | WD | CONT | HFLCD | HFLCD/CONT Ratio |
|---|---|---|---|---|
| Overview and macronutrients | ||||
| Total fat (% kcal) | 40 | 16 | 60 | 3.75 |
| Saturated fat (% kcal) | 11 | 8 | 30 | 3.75 |
| Protein (% kcal) | 15 | 19 | 30 | 1.58 |
| Carbohydrate (% kcal) | 45 | 65 | 10 | 0.15 |
| Energy density (kcal/g) | 4.62 | 3.87 | 5.01 | 1.29 |
|
| ||||
| Individual components | ||||
| Fat (%total weight) | ||||
| Milkfat | 0 | 2.1 | 10.3 | 4.9 |
| Lard | 10 | 2.1 | 10.3 | 4.9 |
| Soybean oil | 0 | 0.4 | 2.0 | 5 |
| Corn oil | 10.2 | 0.2 | 0.8 | 4 |
| Vegetable oil | 0 | 2.1 | 10.3 | 4.9 |
| Protein (%total weight) | ||||
| Casein | 19.4 | 20 | 41.2 | 2 |
| Cystine | 0.06 | 0.3 | 0.6 | 2 |
| Carbohydrate (%total weight) | ||||
| Corn starch | 0 | 39.7 | 8.0 | 0.2 |
| Dextrin | 20 | 13.2 | 2.7 | 0.2 |
| Sucrose | 30.9 | 10 | 2.0 | 0.2 |
Ratios of HFLCD/CONT are listed to demonstrate that the relative proportions of each type of fat, protein, and carbohydrate additive are identical between HFLCD and CONT diets.
Figure 1.
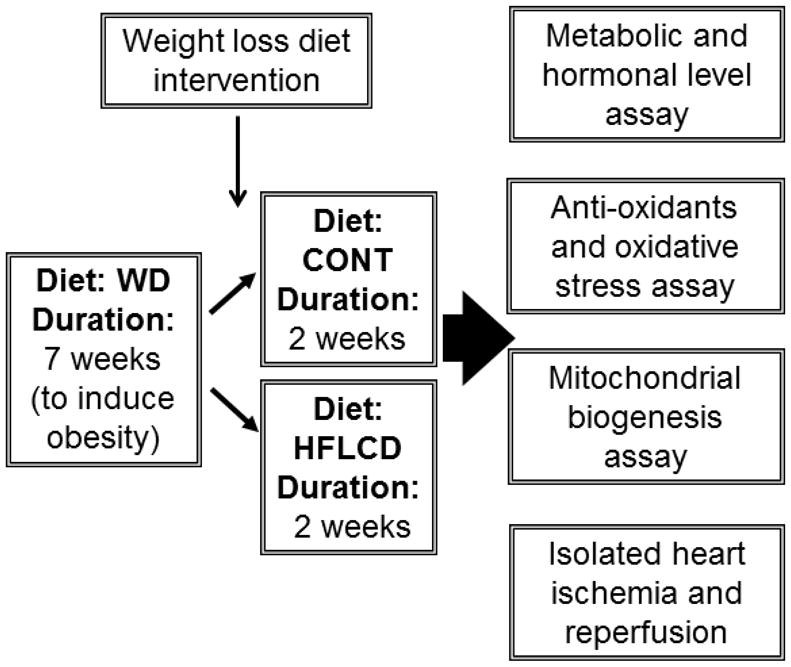
Simplified outline of cardiac effects of HFLCD
To provide an estimate of the number of animal experiments needed to demonstrate proof of the hypothesis, a power calculation was performed. As we did not have preliminary data to estimate the effect size and variance of many of the parameters tested in this work, we had to rely on available data regarding the cardiac effects of these diets from previous work [7]. This prior work demonstrated that, using these same diets in a non-obese rat model, HFLCD led to worsening of the left ventricular rate-pressure product (RPP) in the isolated heart during recovery following ischemia-reperfusion. In the prior work, we found that for HFLCD, RPP recovered to 18% of the initial pre-ischemic value, with standard deviation of 8.5%; for the CONT diet, RPP recovered to 43% of the pre-ischemic value with standard deviation of 14%. Using these values, and setting alpha value of 0.05 and power of 0.8, we find that a sample size of 4 animals in each group is sufficient to demonstrate a difference. Using a similar approach, we determined that a sample size of 5 to 6 animals was sufficient to demonstrate expected magnitudes of differences in ±dp/dt. Therefore, we used 5 to 6 animals per group for each of our assessed parameters.
2.3. Metabolic and hormonal level assay
Standard heparinized micro-hematocrit capillary tubes were used for blood collection. The blood was withdrawn through tail vein bleeding under fed condition, and the amount of blood withdrawn was no more than 1 % of the animal’s body weight (e.g., 1 ml from a 250g adult rat). Blood serum from tail vein bleeding was separated by centrifugation at 5000 rpm for 20 min and was stored at −80 °C. Whole left ventricle samples were taken immediately after the hearts were excised, under normoxic conditions, after two weeks of the study diet (HFLCD or CONT), then stored at −80 °C.
Rat serum insulin, leptin and adiponectin assays were done using Millipore radioimmunoassay kit (Billerica, MA). Myocardium glycogen content was measured by an enzymatic digestion method. Triglycerides LiquiColor® Test using glycerylphosphate oxidase (GPO) method (Stanbio Laboratory, Boerne, TX); Free fatty acids run on Stanbio Sirrus using WAKO NEFA-C reagent (Stanbio Laboratory, Boerne, TX). Glucose assay using sigma glucose assay kit.
2.4. Isolated heart perfusions
To determine the diets effect on LV function and ischemic injury, isolated heart experiments were performed according to the previous method. Animals were anesthetized with inhalational isoflurane (4%) and killed by decapitation between 2 and 4 hours after turning on lights. As rats are nocturnal feeders, this was considered the fed state. Hearts were quickly excised and were Langendorff-perfused with a modified Krebs-Henseleit buffer with perfusion pressure 75 mmHg. The buffer contained 3% bovine serum albumin (essentially fatty acid free; US Biological, Swampscott, MA) and the following (in mM). NaCl 118, KCl 4.8, MgSO4 1.2, CaCl2 1.4, KH2PO4 1.2, Na2HCO3 25, pyruvate 0.1, palmitate 0.3, glucose 5.0, lactate 1.0, 3-hydroxybutyrate 0.6, Insulin 30 μU/ml. The concentrations of free fatty acid (palmitate), insulin, and 3-hydroxybutyrate were chosen based on the previously measured in vivo, circulating concentration found in rats in the fed state, fed these same diets and all concentrations are in well accepted physiologic ranges; we used the same concentrations in both experimental groups to eliminate any effect of perfusate composition on the differences in cardiac function between groups. Baseline function under the standard flow, normal perfusion conditions was monitored and recorded (DASYLab, Measurement Computing Corp., Norton, MA). Heart rate (HR), systolic pressure (SP), end diastolic pressure (EDP), and peak positive and negative rates of change of pressure (+ dP/dt and − dP/dt) were used to compute indices of ventricular function. From these values, the left ventricular developed pressure (LVDP = SP − EDP), and the rate-pressure product (RPP = LVDP × HR) were used as the primary indices of cardiac systolic function or power. Perfusion was then decreased to 0.3 ml/min (approximately 97% reduction from baseline flow) for 60 min and then resumed at 75 mmHg perfusion pressure (“reperfusion”) for 60 minutes, with continuous recording of pressure-related parameters throughout the experiment. At the end of reperfusion, the percent recovery of the baseline values of RPP and ± dP/dt were computed as indices of the recovery of contractile and relaxation function.
2.5. Protein analysis
Cytoplasmic protein samples were extracted from heart tissue. Western blots were conducted using commercially available antibodies to the following proteins or chemical groups. NRF1, dilution 1.100; SOD2, dilution 1.200; and GAPDH, dilution 1.10000 (all from Santa Cruz Biotechnology); Catalase, dilution 1.1000 (Abcam); TFAM, dilution 1.100 (Acris); Nitrotyrosine, dilution 1.100 (Cayman); Secondary antibody, dilution 1.5000 (Santa Cruz Biotechnology). The Immunoblotting images were captured using KODAK image Station 4000R (Carestream Health Inc.) by developing the membranes in Supersignal West substrates (Thermo Scientific, 34080 or 34076), and analyzed with KODAK IM software (Ver 4.5.1).
2.6. Transcript analyses
Total RNA samples were extracted from 50 mg samples of left ventricles using a RNA extraction kit (Qiagen) and complementary DNA synthesized cDNA using cDNA synthesis kit (Clontech) according to the manufacturer’s instructions. Quantitative real-time RT-PCR analyses were carried out using the Roche SYBR green LightCycler 480 system (Roche) to determine transcript levels of target genes. Real time PCR results from each gene/primer pair were normalized to GAPDH, and the normalized value was compared across conditions. The primers used are listed in Table 2.
Table 2.
Primers used in the present study
| Gene | Forward | Reverse |
|---|---|---|
| GAPDH | 5′-ACCACAGTCCATGCCATCAC-3′ | 5′-TCCACCACCCTGTTGCTGTA-3′ |
| XDH | 5′-GACAAGCACTAACACCG-3′ | 5′-GAGCAAGCCACCCCATA-3′ |
| NOX4 | 5′-CTGTCTGCTTGTTTGG-3′ | 5′-AGGGACCTTCTGTGAT-3′ |
| SOD1 | 5′-CAGGGCGTCATTCACTTCG-3′ | 5′-CCTTTCCAGCAGCCACATT-3′ |
| SOD2 | 5′-GGAGCAAGGTCGCTTACAGAT-3′ | 5′-AACATTCTCCCAGTTGATTACATT-3′ |
| Catalase | 5′-CTATTGCCGTCCGATTCTC-3′ | 5′-GTCCCAGTTACCATCTTCAGTGT-3′ |
| Gpx1 | 5′-TCCACCGTGTATGCC-3′ | 5′-TGTCCGAACTGATTGC-3′ |
| NRF1 | 5′-CAGCAAGTTCAGCAGGTCCAT-3′ | 5′-GTCCGAGTCATCGTAAGAAGTGT-3′ |
| TFAM | 5′-GCAGAAACGCCTAAA-3′ | 5′-ATCACTTCGCCCAAC-3′ |
| COX4i1 | 5′-CCCATCCCTCATACCTTTG-3′ | 5′-CATTCATTCTTGTTGTAGTCCC-3′ |
| COX4i2 | 5′-AGGAGGACTCAGAACTCAAGG-3′ | 5′-AAGAAGACGCAGCCCATT-3′ |
| COX5b | 5′-GGCTTCTGGAGGTGGTG-3′ | 5′-TGCTAACGGATGGGACTA-3′ |
| COX6c | 5′-AAGCGTCTGCGGGTTCAT-3′ | 5′-ACACCAGCCTGCCTCATC-3′ |
| Cyto b | 5′-TTGCCTACGCTATTCTACGCTC-3′ | 5′-AATGGGTGTTCTACTGGTTGGC-3′ |
| Rcan1 | 5′-GCGAAAGTGAGACCAGGGC-3′ | 5′-CCAACAGGTGGAGAGGCAG-3′ |
| Cyto c | 5′-TACCCTGATGGAGTATTTGGA-3′ | 5′-GCTATTAGGTCTGCCCTTTCT-3′ |
| PPARα | 5′-ACTATGGAGTCCACGCATGTG-3′ | 5′-TGTCGTACGCCAGCTTTAGC-3′ |
| PGC1a | 5-AAGGCTCAAGAGGGACGAATA-3′ | 5′-CACAGGTGTAACGGTAGGTAAT-3′ |
| PGC1b | 5′-CGGTGAAGGTCGTGTGGT-3′ | 5′-GGCTCATTGCGTTTTCTCAG-3′ |
| Adiponectin | 5′-TCCTGGTCACAATGGGATACC-3′ | 5′-ATCTCCTGGGTCACCCTTAGG-3′ |
| AdipoR1 | 5′-GCTGGCCTTTATGCTGCTCG-3′, | 5′-TCTAGGCCGTAACGGAATTC-3′ |
| AdipoR2 | 5′-CCACAACCTTGCTTCATCTA-3′ | 5′-GATACTGAGGGGTGGCAAAC-3′ |
2.7. Analysis of mitochondrial DNA copies
The mitochondrial DNA copy number was evaluated by the methods described previously. Total genomic DNA was isolated from left ventricles and processed by standard procedures using a DNA extracting kit (Qiagen) and was digested with NcoI and subjected to real-time qPCR analysis. Cytochrome b (cyto b) was employed as a mitochondrial DNA (mtDNA) marker, and the regulator of calcineurin 1 (rcan1) as a nuclear DNA (nDNA) marker to quantify the amount of mtDNA.
2.8. Xanthine oxidase (XO) assay
0.1g heart tissue was homogenized in 0.5ml of cold buffer (100mM Tris-HCl containing protease inhibitors, pH 7.5), and centrifuged at 10,000 g for 15 minutes at 4 degrees C. The xanthine oxidase activity of the supernatant was determined using the xanthine oxidase assay kit (Cayman Chemical, Item No. 10010895) according to the protocols provided. The fluorescence of the sample was measured using a spectrofluorescent multiwell plate reader (TECAN infinite M200 model) with 520–550 nm (excitation), 585–595 nm (emission). The XO activity (mU/g wet weight) was calculated using xanthine oxidase standard curve.
2.9. Statistical analyses
Data for comparison of two groups were analyzed using Student’s t-test using GraphPad Prism software (GraphPad Software Inc.). Values of quantitative results were expressed as means±SEM. Differences between groups and treatments were regarded as significant at the P<0.05 probability level.
3. Results
3.1. Whole body metabolism following seven-week obesity-promoting diet and two-week HFLCD feeding
A simplified outline of the experimental plan to determine the cardiac effects of HFLCD is summarized in Fig. 1. In order to create a group of obese animals, 250g male Sprague-Dawley rats were fed an obesity-promoting WD diet for 7 weeks. When compared to a group of rats of similar age and body size fed standard chow diet (N = 12), body weight increased by 15% in the WD group (N = 36) confirming the ability of the WD diet to cause obesity. Following the 7 weeks of WD, all WD rats were eventually distributed to the HFLCD and the CONT diets, HFLCD rats total energy intake was about 20% higher, and resulted in greater body weight gain than CONT (P < 0.05; Fig. 2A, 2B).
Figure 2. Body weight on the indicated diet.

A) Body weight of WD seven-week-fed (obesity-promoting) then changing to CONT and HFLCD for two weeks of feeding. B) Calorie intake of CONT and HFLCD group. Values are expressed as means±SEM, n=12–36 each group.
The whole body metabolic states following feeding with either CONT or HFLCD, are summarized in Table 3. There was no difference in heart weight between groups. HFLCD led to a 37.6% decrease in the fed-state insulin level compared with CONT. Change to HFLCD for 2 weeks also led to lower myocardial glycogen and a 28.5% decrease of plasma adiponectin with no difference in plasma triglycerides, free fatty acids, glucose, and leptin.
Table 3.
Metabolic characteristics
| Parameters | CONT | HFLCD |
|---|---|---|
| Heart weight (g) | 1.34±0.03 | 1.45±0.06 |
| Plasma Glucose (mM) | 7.3±0.2 | 7.2±0.2 |
| Myocardial Glycogen (mg/g tissue) | 2.22 ± 0.16 | 1.43 ± 0.2* |
| Plasma free fatty acids (mEq/l) | 0.52±0.03 | 0.53±0.06 |
| Plasma Triglyceride (mg/dl) | 183.9±17.3 | 207.9±21.6 |
| Plasma Leptin (ng/ml) | 29.2±2.2 | 30.2±3.4 |
| Plasma Adiponectin (μg/ml) | 3.68 ± 0.19 | 2.63 ± 0.21* |
| Plasma Insulin (μU/ml) | 66.5±4.5 | 41.5±9.0* |
Values are expressed as means ± SEM (n = 5–6).
P<0.05, vs HFLCD.
3.2. Hearts from obese rats fed HFLCD demonstrated impaired the I/R recovery of cardiac function
Baseline cardiac function was similar between groups (Table 4). HFLCD impaired the recovery of left ventricular function (RPP and ±dP/dt expressed as percent of baseline value recovered at the end of reperfusion; P < 0.05), decreasing RPP percent of baseline value by about 80%, ±dP/dt % baseline by about 60% each compared to CONT after ischemia-reperfusion (Fig. 3A,B). Total LDH release during reperfusion was an overall 3.5-fold greater compared to CONT (Fig. 3C), indicating greater ischemic injury in HFLCD.
Table 4.
Functional parameters of isolated rat hearts under baseline conditions
| Parameters | CONT | HFLCD |
|---|---|---|
| RPP (103 mmHg/min) | 31.7 ± 1.12 | 29.9 ± 1.1 |
| LVEDP (mmHg) | 6.6± 0.15 | 7.2±0.07 |
| +dP/dt (mmHg/s) | 3007±47.3 | 2953±28.5 |
| −dP/dt (mmHg/s) | −2204±40.2 | −2044±29.3 |
P not significant for all parameters, Results are expressed as means ± SEM (n = 6).
Figure 3. High fat low-carbohydrate diet increases susceptibility to myocardial ischemic injury.

Percent recovery of baseline A) RPP, B) ±dP/dt at the end of 60 min LFI and 60 min reperfusion. C) Total LDH release during the 60 min of reperfusion. Values are expressed as means±SEM, n=6, * P < 0.05 compared with CONT.
3.3. HFLCD diet leads to depressed endogenous anti-oxidants and increased oxidative stress in obese rats
We next examined whether HFLCD diet is involved in the regulation of important endogenous anti-oxidants and oxidative stress in the heart. The transcript expression of SOD2, catalase and GPX1 (antioxidant pathways) were decreased in HFLCD hearts relative to control hearts (CONT), while that of NADPH oxidase 4 (NOX4, ROS-generating) increased (Fig. 4A). Protein expression of both catalase and SOD2 were decreased in HFLCD hearts (Fig. 4B). HFLCD hearts exhibited augmented oxidative stress, illustrated by upregulated NOX4 gene transcription, increased nitrotyrosine protein modification by 37% (P < 0.05 compared to CONT), and stimulated xanthine oxidase activity (mU/g wet weight) (P = 0.01 compared to CONT) (Fig. 4A, C and D), along with decreased expression and activity of ROS-removal pathways (Fig. 4A, 4B). There was no change of SOD1 and xanthine dehydrogenase (xdh) transcript levels in myocardium from HFLCD hearts (Fig. 4A).
Figure 4. Endogenous anti-oxidants and oxidative stress.

A) Real-time PCR analysis of transcript expression of SOD1, SOD2, catalase, GPX, XDH and NOX4 in samples from CONT and HFLCD hearts. B) and C) Western blot analysis of protein levels of SOD2, catalase and nitrotyrosine in samples from CONT and HFLCD hearts. D) Xanthine oxidase activity of samples from CONT and HFLCD hearts. Values are expressed as means±SEM, n=5–6 each, * P < 0.05 compared with CONT.
3.4. HFLCD diet leads to depressed mitochondrial biogenesis in obese rats
Oxidative stress plays an essential role in regulating the transcriptional expression of key determinants of mitochondrial biogenesis and function, and high fat diets are known to augment oxidative stress. Therefore we hypothesized that HFLCD would result in low mitochondrial copy number. Realtime PCR revealed that TFAM, NRF1, and NRF1 target genes COX4i2, 5b and 6c were all downregulated in HFLCD hearts, although COX4i1 was upregulated significantly. There was no change of cytochrome c and cytochrome b transcript levels in myocardium from HFLCD hearts (Fig. 5A). PPARα, PGC1α and PGC1b transcript level in myocardium did not differ between these diet groups (data not shown). Protein expression of key mitochondrial determinants, such as NRF1 and TFAM were decreased by about 31% and 21%, respectively (Fig. 5B). Consequently, associated with these changes in mitochondrial-biogenetic proteins, HFLCD hearts exhibited an attenuation of the relative mitochondrial DNA copy number (Fig. 5C).
Figure 5. Expression of key determinants of mitochondrial biogenesis and mitochondrial proteins.

A) Real-time PCR measurement of transcript levels of mitochondrial protein NRF-1, TFAM, and COX in samples from CONT and HFLCD hearts. B) Western blot analyses of levels of NRF1 and TFAM in samples of CONT and HFLCD hearts. C) Relative Mitochondrial DNA copy number in samples from CONT and HFLCD hearts. Values are expressed as means±SEM, n=5–6 each, * P < 0.05 compared with CONT.
3.5. HFLCD diet leads to depressed local cardiac-specific adiponectin system in obese rat heart
Adiponectin is a protein hormone involved in maintaining energy homeostasis in metabolically active tissues. Adiponectin and its receptors are expressed in adult ventricular cardiomyocytes. Real-time PCR revealed that adiponectin and adiponectin receptor1,2 were all downregulated in the HFLCD heart (Fig. 6).
Figure 6. Detection of adiponectin and its receptors expressed in the heart.
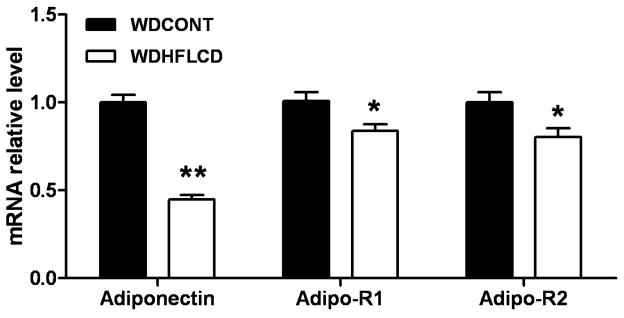
Real-time PCR measurement of transcript levels of adiponectin and adiponectin receptor1,2 in samples from CONT and HFLCD hearts. Values are expressed as means±SEM, n=5–6 each, * P < 0.05 compared with CONT.
4. Discussion
High fat intake, in the range of 45–60% of total kcals, is known to promote increases in ROS and mitochondrial damage and obesity is associated with oxidative stress and mitochondrial and myocardial dysfunction; therefore we hypothesized that HFLCD may lead to enhanced oxidative injury in the heart, providing a potential mechanism of our previous observation of increased ischemic injury in HFLCD. To add relevance to studies of human obesity and dietary intervention, we chose to work with obese rats in the current study. Therefore, the goal of this study was to demonstrate the effect of HFLCD on oxidative stress, and determine the effect on myocardial ischemia-reperfusion injury in a non-genetic, diet-induced model of obesity in the rat. Our findings demonstrate that HFLCD suppresses cardiac expression of essential endogenous antioxidants and transcriptional regulators of mitochondrial biogenesis, resulting in mitochondrial depletion, along with cardiac dysfunction following myocardial ischemia-reperfusion. Thus, the hypothesis is accepted.
We found that HFLCD did not affect the baseline heart function under non-stress conditions. An important finding in the present study is that in the isolated heart following ischemia- reperfusion, HFLCD resulted in lower recovery of cardiac function and greater total LDH release, indicating greater ischemic injury. These differences reflect an adverse response to physiologic stress, which was not apparent under non-stress conditions. Similar work by others has shown left ventricular failure was exacerbated after myocardial infarction in high-fat diet (HFD)-induced obese type 2 diabetic mice, and was associated with increased oxidative stress. A high-fat diet increases risk of ventricular arrhythmia in female rats during ischemia reperfusion injury. Following myocardial infarction, tertiary prevention to maintain left ventricular systolic function and avoiding progression to heart failure is of great importance. Thus, our preliminary work here using animal models may have important implications for diet selection in obese patients with ischemic heart disease.
It is well known that metabolic and hormonal factors play an important role in determining the severity and extent of ischemic myocardial injury. We previously demonstrated that short-term HFLCD feeding in the young, normal weight rat resulted in a number of metabolic and hormonal abnormalities, including lower circulating insulin levels and higher ketone bodies. Our present study, using an older, obese rat model, shows that HFLCD lowered plasma and cardiac adiponectin levels, and reduced adiponectin receptor-1 and receptor-2 transcription in the heart, accompanied by increasing body weight and impairing the recovery of cardiac contractile function. This may suggest that lower plasma adiponectin and local cardiac-specific adiponectin system depression play a role in the myocardial ischemia-reperfusion injury. Though adiponectin is mainly produced by adipocytes, its serum levels are significantly lower in obese patients compared with nonobese controls and are inversely correlated with body mass index; this appears to be due to the fact that visceral fat secretes lower amounts of adiponectin. Clinical work shows that ROS is produced in greater amounts and adiponectin in lower amounts in human obese subjects with visceral fat accumulation. The heart expresses adiponectin and adiponectin receptors suggesting there is a direct effect of adiponectin in the heart. Adiponectin also plays a role in protection from ischemia/reperfusion injury, involving the reduction of oxidative/nitrative stress. Lower circulating levels of adiponectin results in insulin resistance and greater ROS production. Using an obese mouse model, Furukawa et al. demonstrated that production of ROS increased through augmented expression of NADPH oxidase and decreased expression of antioxidative enzymes SOD2 and catalase in adipose tissue which was associated with dysregulation of various adipocytokines, including decreased production of adiponectin. However, any consideration of the role of adiponectin in as an explanation of our findings remains speculative, since the isolated heart perfusion experiments that were performed did not directly address this possibility.
It is well-known that ROS exert deleterious effects on myocardium. We found evidence of a potential role for increased oxidative stress in HFLCD as a cause of the increased ischemic injury. In the present study, we demonstrate that HFLCD upregulated myocardial NADPH oxidase-4 (NOX4) gene transcription and stimulated xanthine oxidase activity, suggesting increased activity of pathways leading to ROS formation. The potential defense mechanisms against ROS-mediated cardiac injury including several antioxidant enzymes such as SOD, catalase, and glutathione peroxidase were significantly downregulated in the heart of obese rats fed HFLCD, further suggesting that augmented oxidative stress may be playing a detrimental role in cardiac physiology. Furthermore, as a direct measure of ROS-mediated damage to the cardiomyocytes, we found that nitrate-modified protein content (tyrosine residue nitration) was increased. Because such protein nitration occurs through reaction with peroxynitrite, produced by reaction of O with nitric oxide, our data indicate that HFLCD might induce an increase in superoxide anion production.
Mitochondrial biogenesis (the growth and division of existing mitochondria) is essential for the cell to cope with changes in energy demand. The link between mitochondrial biogenesis and cardiac pathologies such as the hypertrophied or failing heart is a rapidly emerging field. In heart, a prior study showed that high fat diet reduced levels of nuclear respiratory factors 1 and 2, mitochondrial transcription factor A, and mitochondrial DNA copy number in FVB mice. In the present study, we found that HFLCD led to a decreased transcript and protein expression of both nuclear respiratory factor-1 (NRF1) and transcription factor A-mitochondrial (TFAM), well-documented transcriptional determinants of mitochondrial biogenesis. Furthermore, it is plausible that the reduced NRF1 and TFAM expression is directly related to decreased expression of cytochrome c oxidase subunit 4i2 (COX4i2), 5b and 6c in the heart of obese rats fed HFLCD. With increased oxidative stress attributable to diminished expression of key antioxidant pathway proteins and increased superoxide production, the data suggest that depressed mitochondrial biogenesis in the heart may be a secondary response to oxidative stress. Mitochondria are highly susceptible to ROS, since they affect mitochondrial function by damaging mtDNA and impairing the electron transport chain. Consistent with this, we found that the mitochondrial DNA copy number was reduced. This lower mitochondrial number may play a role in our observed impaired recovery of function following ischemia-reperfusion. The additional effects of impaired mitochondrial DNA copy number and deranged mitochondrial antioxidant defense system in the heart of rats fed HFLCD similar to the carbohydrate-restricted diets favored by humans likely further contribute to our finding of impaired recovery following ischemia and reperfusion.
A limitation of our work is that contrary to the effect of HFLCD in humans, in whom this diet has been shown to lead to weight loss, our rats fed HFLCD actually gained more weight than rats fed the CONT diet. One of the key concepts underpinning the theoretical adoption of HFLCD as a weight loss mechanism is that restriction of CHO with more allowance for protein and fat results in greater satiety, leading to less overall food intake, and is therefore effective for weight loss. However, we observed THAT obese rats on HFLCD actually gained more weight, with higher total energy consumption compared to the CONT diet. A similar observation was made by Focardi et al. using a form of HFLCD in the Obese Zucker rat model; their rats fed HFLCD gained more weight than counterparts fed a control diet. It is possible that regulation of feeding behavior differs between humans and rats, potentially explaining this relative hyperphagia and weight gain. Others have shown that HFLCD increases rat total fat mass; the response of mice to a carbohydrate-free diet also was greater weight gain. Though the precise mechanisms leading to this greater observed weight gain are not clear, our study clearly demonstrates that a final result of HFLCD in the rat is that it leads to metabolic and hormonal dysregulation and worsened response to myocardial ischemiareperfusion. Therefore, these rodent models to study HFLCD effect on the heart may be more useful to examine direct physiologic diet-induced changes in the response to myocardial ischemia, rather than to study the effect of these diets on weight and eating behavior. Another limitation is that we only relied on measurements of enzymes involved in the production or removal of ROS or measurements of the direct downstream effect of ROS, including nitrated proteins used frozen tissue. We did not directly measure the mitochondrial electron transport chain function or endogenous reactive oxygen species content in this study. We used frozen tissue and hence were unable to directly measure the short-lived, unstable ROS. This will be the subject of future work. Furthermore, future experiments will examine the effect of systemic antioxidants and anti-inflammatory mediators, which would provide additional mechanistic data to support the hypothesis.
In conclusion, the results of the present study confirm our hypothesis that feeding obese rats with HFLCD results in derangements of mitochondrial biogenesis, enhanced myocardial ROS and ROS-induced damage, and impaired recovery of cardiac contractile function following myocardial ischemia-reperfusion. These findings may have important consequences for humans in choosing appropriate lifestyle modification for weight loss. Further experiments will be aimed at determining mechanisms of myocardial damage associated with these diets in the presence of myocardial ischemia and reperfusion, including apoptosis and inflammation.
Acknowledgments
The present study was supported by Scientist Development Grant 0735212N from the American Heart Association and in part by NIH grant P30DK056336. The authors would like to thank Qinglin Yang and C. Roger White for assistance with the experiment.
Abbreviations
- Gpx1
Glutathione peroxidase 1
- SOD1
Cu/Zn-Superoxide dismutase
- SOD2
Manganese superoxide dismutases
- NOX4
NADPH oxidase 4
- XDH
Xanthine dehydrogenase
- TFAM
Transcription factor A, mitochondrial
- NRF
Nuclear respiratory factor
- Cox4i1
Cytochrome c oxidase subunit IV isoform 1
- Cox4i2
Cytochrome c oxidase subunit IV isoform 2
- Cox5b
Cytochrome c oxidase subunit Vb
- Cox6c
Cytochrome c oxidase subunit VIc
- Cyto c
Cytochrome c, mitochondrion, complete genome
- Cyto b
Cytochrome b, mitochondrion, complete genome
- Rcan1
Regulator of calcineurin 1
- PPAR
Peroxisome proliferator-activated receptor
- PGC1
PPARgamma coactivator-1
- AdipoR
Adiponectin receptor
- XO
Xanthine oxidase
- HFLCD
High fat, low carbohydrate diet
- ROS
Reactive oxygen species
- I/R
Ischemia reperfusion
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.The Lancet. Curbing the obesity epidemic. Lancet. 2006;367:1549–1549. doi: 10.1016/S0140-6736(06)68664-9. [DOI] [PubMed] [Google Scholar]
- 2.Atkins RC. Dr Atkins’ New Diet Revolution. New York: Avon; 2001. Revised ed. [Google Scholar]
- 3.Bravata DM, Sanders L, Huang J, Krumholz HM, Olkin I, Gardner CD, Bravata DM. Efficacy and Safety of Low-Carbohydrate Diets. A Systematic Review. JAMA. 2003;289:1837–1850. doi: 10.1001/jama.289.14.1837. [DOI] [PubMed] [Google Scholar]
- 4.Lagiou P, Sandin S, Lof M, Trichopoulos D, Adami H-O, Weiderpass E. Low carbohydrate-high protein diet and incidence of cardiovascular diseases in Swedish women: prospective cohort study. BMJ. 2012;344:e4026. doi: 10.1136/bmj.e4026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Trichopoulou A, Psaltopoulou T, Orfanos P, Hsieh CC, Trichopoulos D. Low-carbohydrate-high-protein diet and long-term survival in a general population cohort. Eur J Clin Nutr. 2007;61:575–581. doi: 10.1038/sj.ejcn.1602557. [DOI] [PubMed] [Google Scholar]
- 6.Halton TL, Willett WC, Liu S, Manson JE, Albert CM, Rexrode K, Hu FB. Low-Carbohydrate-Diet Score and the Risk of Coronary Heart Disease in Women. N Engl J Med. 2006;355:1991–2002. doi: 10.1056/NEJMoa055317. [DOI] [PubMed] [Google Scholar]
- 7.Wang P, Tate JM, Lloyd SG. Low carbohydrate diet decreases myocardial insulin signaling and increases susceptibility to myocardial ischemia. Life Sci. 2008;83:836–844. doi: 10.1016/j.lfs.2008.09.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Marsh SA, Dell’italia LJ, Chatham JC. Interaction of diet and diabetes on cardiovascular function in rats. Am J Physiol Heart Circ Physiol. 2009;296:H282–292. doi: 10.1152/ajpheart.00421.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Maulik SK, Kumar S. Oxidative stress and cardiac hypertrophy a review. Toxicol Mech Methods. 2012;22:359–366. doi: 10.3109/15376516.2012.666650. [DOI] [PubMed] [Google Scholar]
- 10.James AM, Murphy MP. How mitochondrial damage affects cell function. J Biomed Sci. 2002;9:475–487. doi: 10.1159/000064721. [DOI] [PubMed] [Google Scholar]
- 11.Sreekumar R, Unnikrishnan J, Fu A, Nygren J, Short KR, Schimke J, Barazzoni R, Nair KS. Impact of high-fat diet and antioxidant supplement on mitochondrial functions and gene transcripts in rat muscle. Am J Physiol Endocrinol Metab. 2002;282:E1055–1061. doi: 10.1152/ajpendo.00554.2001. [DOI] [PubMed] [Google Scholar]
- 12.Park J, Lee J, Choi C. Mitochondrial network determines intracellular ROS dynamics and sensitivity to oxidative stress through switching inter-mitochondrial messengers. PLoS One. 2011;6:e23211. doi: 10.1371/journal.pone.0023211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dai D-F, Rabinovitch PS, Ungvari Z. Mitochondria and Cardiovascular Aging. Circulation Research. 2012;110:1109–1124. doi: 10.1161/CIRCRESAHA.111.246140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kurdi M, Booz GW. Focus on mitochondria dysfunction and dysregulation in heart failure towards new therapeutic strategies to improve heart function. Congest Heart Fail. 2011;17:255–256. doi: 10.1111/j.1751-7133.2011.00269.x. [DOI] [PubMed] [Google Scholar]
- 15.Feillet-Coudray C, Sutra T, Fouret G, Ramos J, Wrutniak-Cabello C, Cabello G, Cristol JP, Coudray C. Oxidative stress in rats fed a high-fat high-sucrose diet and preventive effect of polyphenols. Involvement of mitochondrial and NAD(P)H oxidase systems. Free Radi Biol & Med. 2009;46:624–632. doi: 10.1016/j.freeradbiomed.2008.11.020. [DOI] [PubMed] [Google Scholar]
- 16.Tsutsui H, Kinugawa S, Matsushima S. Mitochondrial oxidative stress and dysfunction in myocardial remodelling. Cardiovas Res. 2009;81:449–456. doi: 10.1093/cvr/cvn280. [DOI] [PubMed] [Google Scholar]
- 17.Naya FJ, Black BL, Wu H, Bassel-Duby R, Richardson JA, Hill JA, Olson EN. Mitochondrial deficiency and cardiac sudden death in mice lacking the MEF2A transcription factor. Nat Med. 2002;8:1303–1309. doi: 10.1038/nm789. [DOI] [PubMed] [Google Scholar]
- 18.Liu J, Wang P, He L, Li Y, Luo J, Cheng L, Qin Q, Brako LA, Lo WK, Lewis W, Yang Q. Cardiomyocyte-Restricted Deletion of PPARbeta/delta in PPARalpha-Null Mice Causes Impaired Mitochondrial Biogenesis and Defense, but No Further Depression of Myocardial Fatty Acid Oxidation. PPAR Res. 2011;2011:372854. doi: 10.1155/2011/372854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roberts CK, Barnard RJ, Sindhu RK, Jurczak M, Ehdaie A, Vaziri ND. Oxidative stress and dysregulation of NAD(P)H oxidase and antioxidant enzymes in diet-induced metabolic syndrome. Metabolism. 2006;55:928–934. doi: 10.1016/j.metabol.2006.02.022. [DOI] [PubMed] [Google Scholar]
- 20.Ding G, Qin Q, He N, Francis-David SC, Hou J, Liu J, Ricks E, Yang Q. Adiponectin and its receptors are expressed in adult ventricular cardiomyocytes and upregulated by activation of peroxisome proliferator-activated receptor gamma. J Mol Cell Cardiol. 2007;43:73–84. doi: 10.1016/j.yjmcc.2007.04.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Leonardi DS, Feres MB, Portari GV, Zanuto ME, Zucoloto S, Jordao AA. Low-carbohydrate and high-fat diets on the promotion of hepatic steatosis in rats. Exp Clin Endocrinol Diabetes. 2010;118:724–729. doi: 10.1055/s-0030-1255021. [DOI] [PubMed] [Google Scholar]
- 22.Dong F, Li Q, Sreejayan N, Nunn JM, Ren J. Metallothionein prevents high-fat diet induced cardiac contractile dysfunction. role of peroxisome proliferator activated receptor gamma coactivator 1alpha and mitochondrial biogenesis. Diabetes. 2007;56:2201–2212. doi: 10.2337/db06-1596. [DOI] [PubMed] [Google Scholar]
- 23.Keaney JF, Jr, Larson MG, Vasan RS, Wilson PW, Lipinska I, Corey D, Massaro JM, Sutherland P, Vita JA, Benjamin EJ, Framingham S. Obesity and systemic oxidative stress. clinical correlates of oxidative stress in the Framingham Study. Arterioscler Thromb Vasc Biol. 2003;23:434–439. doi: 10.1161/01.ATV.0000058402.34138.11. [DOI] [PubMed] [Google Scholar]
- 24.Matsushima S, Kinugawa S, Yokota T, Inoue N, Ohta Y, Hamaguchi S, Tsutsui H. Increased myocardial NAD(P)H oxidase-derived superoxide causes the exacerbation of postinfarct heart failure in type 2 diabetes. Am J Physiol Heart Circ Physiol. 2009;297:H409–416. doi: 10.1152/ajpheart.01332.2008. [DOI] [PubMed] [Google Scholar]
- 25.Aubin MC, Cardin S, Comtois P, Clément R, Gosselin H, Gillis MA, Le Quang K, Nattel S, Perrault LP, Calderone A. A high-fat diet increases risk of ventricular arrhythmia in female rats: enhanced arrhythmic risk in the absence of obesity or hyperlipidemia. J Appl Physiol. 2010;108:933–40. doi: 10.1152/japplphysiol.01281.2009. [DOI] [PubMed] [Google Scholar]
- 26.Wang P, Lloyd SG, Chatham JC. Impact of High Glucose/High Insulin and Dichloroacetate Treatment on Carbohydrate Oxidation and Functional Recovery After Low-Flow Ischemia and Reperfusion in the Isolated Perfused Rat Heart. Circulation. 2005;111:2066–2072. doi: 10.1161/01.CIR.0000162466.06150.D4. [DOI] [PubMed] [Google Scholar]
- 27.Wang P, Lloyd SG, Zeng H, Bonen A, Chatham JC. Impact of altered substrate utilization on cardiac function in isolated hearts from Zucker diabetic fatty rats. Am J Physiol Heart Circ Physiol. 2005;288:H2102–2110. doi: 10.1152/ajpheart.00935.2004. [DOI] [PubMed] [Google Scholar]
- 28.Yamauchi T, Kamon J, Ito Y, Tsuchida A, Yokomizo T, Kita S, Sugiyama T, Miyagishi M, Hara K, Tsunoda M, et al. Cloning of adiponectin receptors that mediate antidiabetic metabolic effects. Nature. 2003;423:762–769. doi: 10.1038/nature01705. [DOI] [PubMed] [Google Scholar]
- 29.Arita Y, Kihara S, Ouchi N, Takahashi M, Maeda K, Miyagawa J, Hotta K, Shimomura I, Nakamura T, Miyaoka K, et al. Paradoxical decrease of an adipose-specific protein, adiponectin, in obesity. Biochem Biophys Res Commun. 1999;257:79–83. doi: 10.1006/bbrc.1999.0255. [DOI] [PubMed] [Google Scholar]
- 30.Lopaschuk GD, Folmes CD, Stanley WC. Cardiac energy metabolism in obesity. Circ Res. 2007;101:335–347. doi: 10.1161/CIRCRESAHA.107.150417. [DOI] [PubMed] [Google Scholar]
- 31.Lihn AS, Bruun JM, He G, Pedersen SB, Jensen PF, Richelsen B. Lower expression of adiponectin mRNA in visceral adipose tissue in lean and obese subjects. Mol Cell Endocrinol. 2004;219:9–15. doi: 10.1016/j.mce.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 32.Fujita K, Nishizawa H, Funahashi T, Shimomura I, Shimabukuro M. Systemic oxidative stress is associated with visceral fat accumulation and the metabolic syndrome. Circ J. 2006;70:1437–1442. doi: 10.1253/circj.70.1437. [DOI] [PubMed] [Google Scholar]
- 33.Tao L, Gao E, Jiao X, Yuan Y, Li S, Christopher TA, Lopez BL, Koch W, Chan L, Goldstein BJ, Ma XL. Adiponectin cardioprotection after myocardial ischemia/reperfusion involves the reduction of oxidative/nitrative stress. Circulation. 2007;115:1408–1416. doi: 10.1161/CIRCULATIONAHA.106.666941. [DOI] [PubMed] [Google Scholar]
- 34.Shibata R, Sato K, Pimentel DR, Takemura Y, Kihara S, Ohashi K, Funahashi T, Ouchi N, Walsh K. Adiponectin protects against myocardial ischemia-reperfusion injury through AMPK- and COX-2-dependent mechanisms. Nat Med. 2005;11:1096–1103. doi: 10.1038/nm1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Medina-Bravo P, Meza-Santibanez R, Rosas-Fernandez P, Galvan-Duarte R, Saucedo-Garcia R, Velazquez-Lopez L, Torres-Tamayo M. Decrease in serum adiponectin levels associated with visceral fat accumulation independent of pubertal stage in children and adolescents. Arch Med Res. 2011;42:115–121. doi: 10.1016/j.arcmed.2011.03.002. [DOI] [PubMed] [Google Scholar]
- 36.Furukawa S, Fujita T, Shimabukuro M, Iwaki M, Yamada Y, Nakajima Y, Nakayama O, Makishima M, Matsuda M, Shimomura I. Increased oxidative stress in obesity and its impact on metabolic syndrome. J Clin Invest. 2004;114:1752–1761. doi: 10.1172/JCI21625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hock MB, Kralli A. Transcriptional control of mitochondrial biogenesis and function. Annu Rev Physiol. 2009;71:177–203. doi: 10.1146/annurev.physiol.010908.163119. [DOI] [PubMed] [Google Scholar]
- 38.Ide T, Tsutsui H, Hayashidani S, Kang D, Suematsu N, Nakamura K, Utsumi H, Hamasaki N, Takeshita A. Mitochondrial DNA damage and dysfunction associated with oxidative stress in failing hearts after myocardial infarction. Circ Res. 2001;88:529–535. doi: 10.1161/01.res.88.5.529. [DOI] [PubMed] [Google Scholar]
- 39.Van Houten B, Woshner V, Santos JH. Role of mitochondrial DNA in toxic responses to oxidative stress. DNA Repair (Amst) 2006;5:145–152. doi: 10.1016/j.dnarep.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 40.Focardi M, Dick GM, Picchi A, Zhang C, Chilian WM. Restoration of coronary endothelial function in obese Zucker rats by a low-carbohydrate diet. Am J Physiol Heart Circ Physiol. 2007;292:H2093–2099. doi: 10.1152/ajpheart.01202.2006. [DOI] [PubMed] [Google Scholar]
- 41.Caton SJ, Bielohuby M, Bai Y, Spangler LJ, Burget L, Pfluger P, Reinel C, Czisch M, Reincke M, Obici S, et al. Low-carbohydrate high-fat diets in combination with daily exercise in rats. effects on body weight regulation, body composition and exercise capacity. Physiol Behav. 2012;106:185–192. doi: 10.1016/j.physbeh.2012.02.003. [DOI] [PubMed] [Google Scholar]
- 42.Borghjid SPD, Feinman RDPD. Response of C57Bl/6 mice to a carbohydrate-free diet. Nutr Metab (Lond) 2012;9:69. doi: 10.1186/1743-7075-9-69. [DOI] [PMC free article] [PubMed] [Google Scholar]