

Abstract
Several attributes intuitively considered to be typical mammalian features, such as complex behavior, live birth, and malignant diseases like cancer, also appeared several times independently in so-called “lower” vertebrates. The genetic mechanisms underlying the evolution of these elaborate traits are poorly understood. The platyfish, Xiphophorus maculatus, offers a unique model to better understand the molecular biology of such traits. Herein we detail sequencing of the platyfish genome. Integrating genome assembly with extensive genetic maps uncovered that fish, in contrast to mammals, exhibit an unexpected evolutionary stability of chromosomes. Genes associated with viviparity show signatures of positive selection identifying new putative functional domains and rare cases of parallel evolution. We also discovered that genes implicated in cognition possess an unexpected high rate of duplicate gene retention after the teleost genome duplication suggesting a hypothesis for the evolution of the great behavioral complexity in fish, which exceeds that in amphibians and reptiles.
We sequenced the genome of a single platyfish female (XX, 2n=46 chromosomes, Jp163A strain Fig. 1) from generation 104 of continuous brother-sister matings. Total sequence coverage of 19.6-fold (Supplementary Note) produced an assembly with N50 contig and supercontig lengths of 22kb and 1.1Mb, respectively (Supplementary Table 1). Assembly errors, mostly single nucleotide indels, were corrected with Illumina paired-end reads. A total of 669Mb of the 750 – 950 Mb estimated genome length was assembled in contigs. Gene predictions revealed 20,366 coding genes, 348 non-coding genes and 28 pseudogenes (Supplementary Note).
Figure 1.

(A) Female (upper) and male (lower) platyfish, Xiphophorus maculatus of strain Jp163A with black pigment spots in the dorsal fin that develop when activity of a X-chromosomal oncogene is appropriately controlled. In hybrid genotypes this control is compromised and malignant melanoma develop from the spots. (B) Phylogenetic position of the platyfish relative to other species mentioned herein.
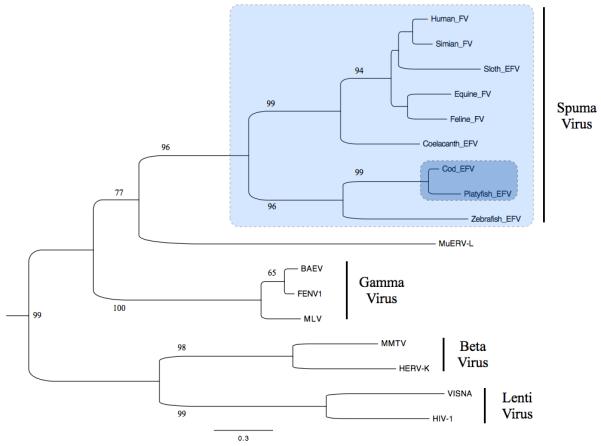
As in other teleosts, platyfish transposable elements (TEs) are highly diverse including many families absent from mammals1 and birds (Supplementary Note; Supplementary Fig. 1-3, Supplementary Tables 2-3). 4.8% of the transcriptome is derived from TE sequences representing about 40 different families, indicating that many of the platyfish TEs are most likely still active. The most active TEs are Tc1 DNA transposons (> 16,000 copies) followed by the RTE family (> 9,000 copies). Interestingly, we identified several almost intact envelope-encoding copies of a foamy retrovirus (Spumaviridae) integrated into the platyfish genome (Fig. 2). Foamy viruses (FV) are known as exogenous infectious agents in mammals2. Only recently, have endogenous FV sequences representing the “fossil record” of infections been described in the genomes of sloth 3 and aye-aye 4 in mammals, and in the coelacanth5. An FV-like sequence in zebrafish6, a sequence in cod discovered during this work, and the platyfish sequenced herein uncover an even broader spectrum of hosts. The molecular phylogeny of FV is consistent with their host phylogeny (Fig. 2). This result supports an ancient marine evolutionary origin of this type of virus, with possible host-virus coevolution5. The nearly intact copies of FV found in the genomes of some divergent fish species while absent from other sequenced fish genomes, might indicate independent germ-line introduction through infection. Exogenous FV had not been described in fish, however out results suggest that exogenous FV have been and might still be infectious in the fish lineage.
Figure 2.

Phylogenetic tree of endogenous retroviruses based on reverse transcriptase protein sequences. Note that the endogenous foamy virus sequences form two distinct tetrapod- and fish-specific phylogenetic groups. Alignment was made with clustalW (223 amino acids) and the phylogenetic tree was constructed with the PhyML package using maximum likelihood methods 38 with default bootstrap (shown at the beginning of branches) and optimized calculation options. FV, Foamy Virus; MuERV-L, Mus musculus Endogenous Retrovirus-L; BAEV, Baboon Endogenous Virus; FENV1, Feline Endogenous Virus; EFV, Endogenous Foamy Virus, MLV, Murine Leukemia Virus; HERV-K, Human Endogenous Retrovirus-K; MMTV, Mouse Mammary Tumour Virus; VISNA, Visna virus; HIV-1, Human Immunodeficiency Virus-1. Scale bar represents the number of substitution per sites.
Mammalian chromosome homology maps display a patchwork arrangement of about 35 large conserved synteny blocks (but about 80 in dog and 200 in mouse) and numerous small blocks assembled in different combinations among the varied species and spanning over 90 million years7. We constructed the most extensive meiotic genetic map for any vertebrate yet published, which allowed the ordering of X. maculatus scaffolds and the creation of precise conserved synteny comparing fish genomes (Supplementary Note). We used the innovative RAD-tag approach8 to construct a meiotic map consisting of 16,245 polymorphic markers that define 24 linkage groups equivalent to the haploid chromosome number of the platyfish9. Thus 90.17% of the total sequences in contigs could be assigned a chromosomal position. Long-range comparisons of gene orders across species10 revealed novel evolutionary relationships of platyfish and other teleost chromosomes. Medaka, the closest relative with a sequenced genome, also has 24 chromosomes and 19 of these show a strict one-to-one relationship (Fig. 3A, B). The remaining five platyfish chromosomes are also orthologous to a single medaka chromosome except for one or two short segments (≈1 Mb) that lie on another medaka chromosome (Fig. 3C, Supplementary Fig. 4). Thus, remarkably few short translocations have disrupted karyotypes since the divergence of medaka and platyfish 120 million years ago (mya)11,12. A similar picture emerges from comparisons of platyfish chromosomes to stickleback (divergence 180 mya11,12). These findings detail a previously unknown breadth to which the genetic content of chromosomes in these teleosts has been conserved over nearly 200 million years of evolution, a conservation much greater than that of mammals over about half that time7,11,12. This is somewhat surprising given the teleost genome duplication (TGD) because one might have thought that illegitimate pairing of paralogous chromosomes (paralogous chromosomes arising from the TGD) might have facilitated translocations. Mechanisms that might have mitigated such translocations remain unknown.
Figure 3.
Conserved syntenies between platyfish and medaka. (A). The medaka orthologs of genes on X. maculatus chromosome 9 (Xma9) tend to lie on O. latipes chromosome 4 (Ola4), showing that the genic content of these chromosomes has remained intact with no translocations in the 120 million years since the lineages of these species diverged. Each grey dot along the horizontal axis at the position labeled Xma9 represents the position of a platyfish gene whose medaka ortholog (as judged by reciprocal best blast hit analysis) lies directly vertical to the Xma9 gene plotted on the appropriate medaka chromosome 10. (B). Reciprocally, nearly all of the platyfish orthologs of genes on medaka chromosome Ola4 lie on Xma9. (C). Nearly all of the medaka orthologs of Xma19 lie on Ola22, except for a segment about 1 Mb long at position Ola22:20Mb that appears on Ola24.
The platyfish is a well-known model in cancer research13. Its genome contains a tumor control region (TCR). It includes the oncogene xmrk 14, which triggers melanoma development. The TCR also contains the tumor modifier mdl15,16. Mdl allelic variants control the body compartment, time onset and severity of tumors17. In addition, mdl manifest in platyfish as a high diversity of genetically defined pigment patterns. The mapped genome allows us to rule out many pigment genes as the responsible factors for these sex-associated pigment variants and melanoma modifiers. All known pigment genes18 are present in the XX female platyfish genome; thus, none is Y-specific. Only 6 of the 174 known pigment genes (asip2a, egfrb, muted, myca, rps20, tfap2a) are located on the X chromosome (Xma21). Of these six, only the proto-oncogene egfrb resides close enough to the melanoma oncogene xmrk (Supplementary Table 4) to be considered a candidate gene for mdl. Indeed, biochemical studies have shown that egfrb can cooperate with xmrk19, but expression levels of both genes are inversely regulated in melanoma20. Further studies are needed to evaluate egfrb function and to find other non-classical pigmentation gene candidates from this genomic region that may control both pigment pattern and melanoma phenotype.
Another so-far unidentified genetic component of the Xiphophorus melanoma model is the R/Diff gene. R/Diff suppresses melanoma formation in wild platyfish and, when eliminated by interspecific hybridization, allows tumor growth. R/Diff was mapped to a 10 cM interval on Xma5 near the cdkn2a/b locus21. Despite cdkn2a being a well-described tumor suppressor gene in certain human melanomas22, it was excluded from being R/Diff because it is not mutated but even overexpressed in Xiphophorus melanoma23. The Xma5 sequence now defines a number of R/Diff candidate genes for further exploration. For example, scaffold #182 (1,085,500 bp), which harbors cdkn2a/b, contains several genes that have a high potential to play a role as the R/Diff tumor suppressor (tet2, cxxc4, mtap, topo-rs, mdx4, pdcd4a, etc.). Alternatively, the region may represent a complex locus comprised of several genes that act in a synergistic or compensatory manner to regulate the xmrk oncogene consistent with previous reports on spontaneous and induced carcinogenesis among the many Xiphophorus interspecies hybrid tumor models24-26.
Viviparity is an elaborate reproductive mode involving diverse levels of maternal investment in offspring ranging from fully provisioning eggs prior to fertilization and retaining them through development to minimally provisioning eggs prior to fertilization, but doing so post fertilization via a placenta, as in mammals. The fish family Poeciliidae, a monophyletic clade of more than 260 species27, is unusual in including species that span the spectrum from negligible to extensive post-fertilization provisioning28,29. The platyfish genome is the first from a non-mammalian viviparous vertebrate. We analyzed in platyfish and for confirmation in a second livebearing fish, the swordtail X. hellerii, both having well provisioned eggs prior to fertilization30,31, three groups of viviparity genes (yolk-, placenta- and egg coat genes; n=34) for gene loss and positive selection compared to four species of egg-laying teleosts (medaka, tetraodon, stickleback, zebrafish).
In mammals, the rise of viviparity has been proposed to involve the progressive loss of vitellogenins (yolk precursors)32. In platyfish and swordtail all yolk-related genes (vitellogenins and their transporter/receptors, Supplementary Table 5) are present and evolved under purifying selection consistent with both species fully provision eggs prior to fertilization, except one gene evolving under positive selection, vitellogenin1 (Supplementary Fig. 5A).
Three of 13 platyfish genes, whose mammalian orthologs are related to placenta development, evolved under positive selection (Supplementary Table 5, Fig. 4A, Supplementary Fig. 5B-D). Igf2, which in mouse regulates placenta permeability33, evolved under strong positive selection in platyfish (Fig. 4a) particularly in the region distal to the proteolysis site. The igf2 sequence33 was also available from another poeciliid, the desert topminnow Poeciliopsis lucida, which shares a livebearing ancestor with Xiphophorus but differs in having evolved placentation recently. In the desert topminnow the same region as in platyfish was evolving under positive selection but even stronger (Supplementary Fig. 5B) suggesting on-going molecular adaptive evolution since the two genera diverged several mya. The two other placental genes pparg and ncoa6 have multiple regions with signals for positive selection outside known functional domains, suggesting novel regions important for viviparity. The same genes under selection in livebearing fish, however, do not show positive selection signatures when orthologous genes from the egg-laying platypus, from marsupials and placental mammals are analyzed (Supplementary Table 6). This result is in line with the fact that placentas of mammals and fish are convergent but not homologous structures.
Figure 4.

Posterior probabilities for sites classes under alternative models along the gene for each amino acid site calculated by Bayes Empirical Bayes analysis. Class 1 (yellow) is the probability of this site being under purifying selection (ka/ks ratio about 0), class 2 (grey) the probability of this site of being under neutral selection (ka/ks ratio about 1), class 3 (red) the probability of this site being under positive selection in Xiphophorus species. (A) Insulin-like growth factor 2 (igf2). The colored bars show known functional domains, from left to right: signal peptide (1 to 52), insulin motif (54 to 110) and IGF2 C-terminal domain (147 to 202). The arrowhead shows the position of the proteolysis site (between 118 and 119) (B) choriogeninH minor, upper: comparison of egg-laying vs. livebearing fish, lower: comparison placental vs. non-placental mammals, showing the same regions under positive selection in fishes and mammals.
Zona pellucida (Zpc) genes, which produce a glycoprotein rich coat surrounding the oocyte plasma membrane, show the most dramatic changes. Alveolin was lost from the platyfish genome. Conversely, choriogeninH minor, choriolysinL, choriolysinH and zvep, evolved under positive selection (Fig. 4B, Supplementary Fig. 5E-G, Supplementary Table 5). In Xenopus, zpc genes control species-specific sperm binding and help ensure that only conspecific sperm released into the aqueous environment fertilizes eggs34. Viviparous fish, however, have internal fertilization, where species-specific sperm recognition would not be as crucial. Compared to egg-laying fish the eggshell is expected to have adapted to development inside the mother because it is no longer essential for protection but must facilitate gas and material exchange. Hatching enzyme genes, zvep and choriolysinH exhibit fast evolving sites generally located adjacent to the catalytic domains (Supplementary Fig. 4F-G) indicating that during evolution of viviparity these enzymes might have altered interactions with target or regulatory proteins. Interestingly, in choriogeninH minor the same regions in particular in the zona pellucida domain evolved under positive selection in both mammals and fishes (Fig. 4B). This is a striking example of how convergent evolution at the molecular level manifests on the physiological and ultimately morphology, levels.
Our analyses of the consequences of the TGD uncovered a functional class of genes that raised our interest because Xiphophorus fish in particular, and teleosts in general, show a remarkably high level of behavioral complexity35 that other groups of ”coldblooded“ vertebrates like amphibians and reptiles do not achieve. Utilizing the platyfish genome and gene annotations from six other sequenced teleosts, we asked whether duplicate gene retention from the TGD could produce through neofunctionalization and/or sub/neofunctionalization36, the acquisition of more complex behaviors. We compared 190 cognition-related genes (Supplementary Note, Supplementary Table 7) to pigmentation (133 genes, for which increased gene repertoires have been connected to the high complexity and diversity of teleost coloration) and liver functions (187 genes)18 as a control. Analysis of cognition-related genes revealed an outstanding high duplicate retention rate of 45% in platyfish and similar values in other teleosts (Fig. 5, Supplementary Fig. 6) compared to pigmentation (30%) and liver (15%) genes. The average duplicate retention rate over all genes in teleost genomes is estimated at 12-24%37. We found no bias for genes from all three functional categories (cognition/pigmentation/liver) that were retained after the TGD to be dosage sensitive or members of protein complexes (Supplementary Note, Supplementary Table 8, 9), but a bias in the cognition genes (but not for liver and pigmentation) for particularly large proteins (>1000 amino acids) (Supplementary Note, Supplementary Table 10, Supplementary Fig. 7). Plotting gene losses on the phylogenetic tree revealed that cognition gene retention was already fixed shortly after the TGD and before teleosts diversification. This finding supports the hypothesis that paralog retention from the TGD may have supported the high level of behavioral complexity in Xiphophorus and other teleosts.
Figure 5.

Differential retention of gene duplicates in cognition, pigmentation and liver function classes in teleosts after the teleost genome duplication (TGD). (A) Retention rates for TGD duplicates of cognition, pigmentation, and liver genes in seven teleost genomes. Time points during teleost evolution that involve the lineage leading to Xiphophorus are connected by lines. (B) Phylogenetic mapping of gene losses for 190 pairs of cognition gene duplicates after the TGD. Losses are indicated with negative values. The number of retained TGD paralog pairs for each individual teleost genome is given in brackets. TGD paralog losses were mapped onto the teleost phylogeny provided by Setiamarga et al. 39 following the parsimony principle. The TGD event was set to 350 million years ago. The retention rate of TGD paralogs is defined by the pairs of TGD duplicates present in a specific lineage divided by the number of pairs of TGD duplicates present at the time of the TGD 18
The platyfish genome reveals new perspectives for several prominent features of this fish model including its livebearing reproductive mode, variation in pigmentation patterns, sex chromosome evolution in action, complex behavior and both spontaneous and induced carcinogenesis17. Teleosts dominate the extant fish fauna, and within teleosts (Fig. 1B), the family Poeciliidae, including platyfish, swordtails, guppies and mollies, is a paradigmatic example of this wide spectrum of adaptations. Our study of this first genome of a poeciliid fish illuminates some teleost evolutionary adaptations and provides a critical resource to advance the study of melanoma and other segregating phenotypes.
Methods
Methods and any associated references are available in the online version of the paper.
Online Methods
Source Material
DNA for genome sequencing was derived from a single female of Xiphophorus maculatus, strain Jp 163A (sample id: XMAC-090115_JP163A) from the Xiphophorus Genetic Stock Center, Texas State University, San Marcos, Texas, USA (XGSC; http://www.xiphophorus.txstate.edu/). The Jp163A line is maintained exclusively by brother-sister matings. The sequenced fish came from generation 104. A female fish was chosen because of its XX sex chromosome constitution. RNA that was sequenced to assemble the Jp163A reference transcriptome was isolated from two stages of pooled embryos (stages 15 and 25), a single individual 5 day old and a 1 month old fry, a single male and female at 2 months of age, one 9 month old female, one 15 month old male fish, and the testes and ovaries from single 10 month old fish.
A Jp163A BAC library (average insert size 160 kb; 10× genome coverage with a total of 43,192 clones available at http://bacpac.chori.org/library.php?id=353)40 was produced from subline WLC#1247 maintained at the Biocenter Fish Facility (BFF), University of Würzburg, Germany. WLC#1247 was separated from the XGSC Jp163A line after about generations 50 and then maintained by inbreeding at the BFF.
For RAD-tag mapping one X. maculatus Jp 163A male (WLC#1325, BFF) was crossed with a female of X. hellerii (strain Rio Lancetilla, Db-, WLC#1337, BFF). Two F1 hybrid females from this cross were then backcrossed to X. hellerii males and DNAs from 267 backcross individuals were used for analysis.
Genome Sequencing
All genomic sequences for de novo assembly were generated on Roche 454 Titanium and Illumina GAIIx instruments with the exception of the BAC-end sequences, which were generated on an ABI3730.
Physical map
A physical map indicating tiling paths of Xiphophorus maculatus contigs was constructed by generating fingerprints from the WLC-1247 BAC library (http://bacpac.chori.org, 40).
Genome assembly
Two independent assemblies were built with all sequence data, using the Newbler (Roche) and PCAP 41 algorithms from ~19.6× total sequence coverage in whole-genome shotgun reads, a combination of 12× fragments, 9× 3kb, 0.38× 20kb and 0.02× BAC-end read pairs. A merged assembly was achieved by assigning the Newbler assembly as the reference and aligning the PCAP assembly via BLAT followed by assimilation of all aligned scaffolds using an established graph accordance method 42. Assembly consensus base error correction was accomplished by aligning Illumina reads (75 base paired-end reads, insert size 200bp), the same DNA source used for the reference, to the reference assembly using the Genomics Workbench v.4.03 software (CLC Bio). A consensus sequence was then created that factored the quality scores of both the reference assembly and the individual Illumina reads.
Transcriptome sequencing and annotation
Total RNA was isolated from platyfish tissues using the RiboPure Total RNA Isolation kit (Ambion). mRNA was isolated from total RNA using the Micro-PolyA Purist kit (Ambion). mRNA was reverse transcribed with SuperScript III Reverse Transcriptase (Invitrogen) using random hexamer primers (Invitrogen). Second-strand cDNA was synthesized using random primers and 15 units of Klenow DNA polymerase exo-minus (Epicentre). Double stranded cDNA was sheared in a Bioruptor (Diagenode) for 30 cycles (30 sec on, 60 sec off). Sheared DNA was end repaired with the End-It DNA repair kit (Epicentre) and dA overhangs were added with Klenow DNA polymerase exo-minus. Adapters were ligated to cDNA overnight and 100 ng was PCR amplified for 12 cycles with Phusion DNA polymerase (New England Biolabs). Each mRNA sample was sequenced on an Illumina GAII sequencer (60 bp). The X. maculatus transcriptome was assembled combining sequences from several tissues including heart, liver, brain, ovaries, and testes, as well as from embryonic stages 15 and 25. For the X. hellerii transcriptome, RNA from 1 month old whole fish, and from brain, liver, ovaries and testes of mature fishes was sequenced and assembled. The transcriptome sequences were aligned to the genome assembly contigs using Bowtie43, then assembled using the Velvet44/Oases package (http://www.ebi.ac.uk/~zerbino/oases/), reporting putative transcripts and splice variants using a coverage cutoff of 4, an insert length estimate of 120, and other parameters at default values.
Gene models and annotation
Gene annotation using Ensembl genebuild was done on assembly Xipmac4.4.2 (GenBank Assembly ID GCA_000241075.1; http://www.ncbi.nlm.nih.gov/genome/assembly/?term=GCA_000241075.1 The annotated platyfish genome can be found at http://www.ensembl.org/Xiphophorus_maculatus/Info/Index.
Another gene identification analysis was performed by a combination of gene prediction and transcriptome integration. We used ab initio modeling with Augustus 45 that had been trained on the medaka gene set and on the alignment of full-length gene models of medaka and zebrafish (both from Ensembl) using BLATX 46. Transcriptome sequences were aligned to the assembly scaffolds using Bowtie 43, then this alignment was adjusted for the most likely exon-intron boundaries using TopHat 47, and then gene models created using Cufflinks 48. Only those transcripts containing a complete ORF and a transcript read coverage of at least 3× were retained, and then these were reconciled into a single set of 33,756 unique potential protein-encoding genes. These gene models were further culled to a subset of 17,783 that are amenable to phylogenetic analysis for entry into a whole genome evolutionary interpretation using the PHRINGE (Phylogenetic Resources for the Interpretation of Genomes) system (http://genomeprojectsolutions.com/PHRINGE_pipeline.html) by eliminating any transcripts shorter than 300 nucleotides and retaining only the longest version of any splice variant at each locus (Supplementary Note).
Estimation of gene number by transcriptome similarity
We identified known genes by reciprocal BLASTX 49 searches of the de novo transcriptome assembly against medaka, stickleback, fugu, tetraodon, zebrafish, and human Ensembl gene libraries. In order to control for the inclusion of alternate transcript forms, we grouped these by the ‘locus’ number as reported by Oases 50.
Estimation of novel genes
In order to identify novel genes, we first reduced the redundancy of the platyfish transcriptome by clustering similar (>95% identity) sequences. Sequences from clusters with no identifiable members were filtered to remove sequences that mapped (by GMAP 51) with less than 99% identity to the genome or had predicted coding sequences shorter than 300bp. Finally, identities for the remaining sequences were sought in the non-redundant database (NCBI). A separate clustering by genomic distance (1kb) produced a very similar gene number estimate. (Supplementary note).
Annotation of non-coding RNAs
To detect snoRNA, snRNA, miRNA and rRNA a homology-based prediction was done using the multispecies RNA database (http://www.ensembl.org/info/data/ftp/index.html) and combined with zebrafish, stickleback, medaka and Takifugu ncRNA libraries. tRNAs were annotated using tRNAscan-SE .21 software locally on UNIX 52. rRNA, miRNA, snRNA and snoRNA were predicted by BLASTN using other fish ncRNA database as query and duplicates were removed from the output files (Supplementary Tables 11-12). Fish databases were downloaded from Ensembl on the following genome versions: zv9 (Danio rerio), BROADS1 (Gasterosteus aculeatus), HdrR (Oryzias latipes) and FUGU4.0 (Takifugu rubripes). miRNA sequences were identified with the Vienna RNA package of MiRscan(http://genes.mit.edu/mirscan/).
Annotation of transposable elements
Both manual and automatic classification of transposable elements (TE), based on Wicker’s nomenclature 53, were performed and combined into a single library. Two TE elements were considered as different if their sequence diverged by more than 20% at the nucleotide level. Manual classification was done by searching TE sequence homology using CENSOR 54 software, by homology searching specific TE proteins using TBLATN and BLASTP, by identifying terminal repeat features (TIRs, LTRs and TSDs) using BLASTN2 and LTR_FINDER software 55, and by reconstructing phylogeny using ClustalW alignment and maximum likelihood calculation (default aLRT) using the PhyML package 38. Phylogenetic reconstructions for the DNA, LINE and LTR classes (Supplementary Fig. 1-3) were based either on transposase or reverse transcriptase proteins. An automatic repeat library was built with RepeatScout software using default parameters on the supercontig assembly corrected for the homopolymer errors. The percentage of transposable elements in the genome was determined from unassembled reads by running locally RepeatMasker software (A.F.A. Smit, R. Hubley & P. Green RepeatMasker at http://repeatmasker.org) on the UNIX system.
Construction of a meiotic map using RAD-tags
Genomic DNA from map cross parents and progeny was digested with the restriction enzyme SbfI (New England Biolabs) and adapters with five nt barcodes each differing by at least two nucleotides were ligated onto fragments. RAD-tag libraries were made as described 8. A 50 ng aliquot of size-selected DNA was PCR amplified for 12 cycles and fragments 200 to 500-bp long were gel purified and sequenced using 80 nt single-end reads on an Illumina HiSeq2000 sequencer. Equal quantities of bar-coded DNA from 16 progeny were loaded onto each lane. Low quality reads and ambiguous barcodes were discarded. We used Stacks software 56 to sort retained reads into loci and to genotype individuals by implementing the likelihood-based SNP calling algorithm 57 to distinguish SNPs from sequencing errors. Stacks exported data into JoinMap 4.0 (Wageningen, The Netherlands) for linkage analysis using markers that were present in at least 200 of 267 individuals
Assigning scaffolds to map positions
To finalize assembly scaffold order and orientation we utilized the high density meiotic map for assigning genome contigs to the genetic map. Using 14,391 marker sequences, we could reliably align 1,950 scaffolds to all linkage groups. Of these, 231 scaffolds mapped to multiple linkage groups, suggesting a misassembly event and were manually split (Supplementary Note).
Genome synteny
For the analysis of conserved syntenies, the Synteny Database 10 was employed using parameters as described. To construct the dot plots, for each gene along a specific platyfish chromosome, the Synteny Database identifies orthologs and paralogs by reciprocal best BLAST analysis and plots positive results on the chromosomes of the same or other species directly above the index gene on the index chromosome.
Analyses of viviparity genes
Thirty-four protein-coding genes known to function in yolk production, placenta-related characteristics, and zona pellucida structures were selected as candidate genes (Supplementary Notes) for the evolution of viviparity among Xiphophorus fishes. Eighteen randomly selected genes were used for control. Orthologous sequences for these genes from four fish species (O. latipes, G. aculeatus, T. nigroviridis, D. rerio) were retrieved from the Ensembl database and then aligned using the MAFFT translation alignment. PAML (version 4.4, linux 64bit) was implemented to test if genes are under positive selection using branch-site specific model 58. Genes with p-value less than 0.05 from likelihood ratio tests were designated as positively selected in Xiphophorus and the Bayes empirical Bayes method 59 was further used to calculate the selection pressure at each site.
Analysis of post-TGD gene retention
Cognition, pigmentation, and liver gene orthologs of human, mouse, and teleosts were obtained from Ensembl65 and missing gene annotations identified with TBLASTN (Supplementary Notes, Supplementary Table 7). EnsemblCompara GeneTrees were checked for teleost duplications and TGD-based duplications were confirmed using the Synteny Database 10. Xiphophorus orthologs were identified from transcriptome v4 and the genome using BLAST searches and assignment confirmed with the Synteny Database. Potential bias in TGD duplicated retention for dosage sensitivity, protein complex membership, and gene length was tested (Supplementary Notes).
Supplementary Material
Acknowledgements
The authors would like to thank the staff of the Xiphophorus Genetic Stock Center and the Biocenter Fish Facility for maintaining the pedigreed fish lines used in this study. We gratefully acknowledge sequencing efforts of Catrina Fronick, Kim Delehaunty, and the production sequencing group at the Genome Institute. This work was supported by National Institutes of Health, NCRR and ORIP, Division of Comparative Medicine grants R24 RR024790 and R24 OD011120 (RBW) including an American Recovery and Reinvestment Act supplement to this award, and R24OD011199 (RBW), R24 RR032658-01 (WCW), R01 RR020833 and R01 OD011116 (JHP), by the Deutsche Forschungsgemeinschaft, TR 17 (MS), and the Agence Nationale de Recherche (JNV).
Footnotes
URLs. Xiphophorus Genetic Stock Center http://www.xiphophorus.txstate.edu/; BAC library http://bacpac.chori.org/library.php?id=353; Oases software package http://www.ebi.ac.uk/~zerbino/oases/; PHRINGE resource http://genomeprojectsolutions.com/PHRINGE_pipeline.html; MiRscan tool http://genes.mit.edu/mirscan/; RepeatMasker http://repeatmasker.org; Genious software package www.geneious.com/; Transcriptome http://avogadro.tr.txstate.edu/cgibin/gb2/gbrowse/XM_ncbi442/ http://avogadro.tr.txstate.edu/Xiph_data_link/stable/Xm_transcriptome_v4.0/,. Gene models http://xiphophorus.genomeprojectsolutions-databases.com/ http://avogadro.tr.txstate.edu/Xiph_data_link/stable/Xm_JB_gene_models/,; Platyfish Genome at Ensembl http://www.ensembl.org/Xiphophorus_maculatus/Info/Index; GenBank Assembly ID GCA_000241075.1; http://www.ncbi.nlm.nih.gov/genome/assembly/?term=GCA_000241075.1; Genomic variants database http://dgvbeta.tcag.ca/dgv/app/home; Human Protein Reference Database www.hprd.org
Accession codes
All sequence data have been deposited in the NCBI database with accession numbers AGAJ00000000.
Note: Supplementary information is available in the online version of the paper
Author contributions
M.S., R.B.W., J.H.P., W.C.W. principal investigators, conceived the project, analyzed data, wrote the manuscript; R.B.W., M.S. provided the inbred Jp163A fish and RNA samples; W.C.W. did BAC and whole genome sequencing, produced assembly, testing and submission; P.M. built the assembly; R.K.W. coordinated genome sequencing and assembly; T.G., R.B.W., Y.S., provided RNA sequencing and assembled a JP163A reference transcriptome, J.B., S.F. improved the genome assembly consensus by incorporating Illumina sequencing reads, created gene models, and performed whole-genome evolutionary analysis using PHRINGE; J.C. wrote software for RAD-tag mapping and aligned the transcriptome and genomic contigs to the genetic map; J.H.P led the construction of the genetic map, performed the conserved synteny analyses; M.S. did the mapping cross; A.A. constructed the genetic map; D.C., T.G., J.N.V. performed repeat analysis, ncRNA and TE annotation, Y.S. analyzed the viviparity genes; A.B. developed the TGD/cognition hypothesis; I.B. designed Fig. 1b, analyzed TGD paralog retention and pigmentation gene locations, contributed to manuscript writing; I.B., K.P.L., M.S. analyzed cognition genes.
Competing financial interests
The authors declare no competing financial interests.
References
- 1.Volff JN, Bouneau L, Ozouf-Costaz C, Fischer C. Diversity of retrotransposable elements in compact pufferfish genomes. Trends in genetics : TIG. 2003;19:674–8. doi: 10.1016/j.tig.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 2.Gifford R, Tristem M. The evolution, distribution and diversity of endogenous retroviruses. Virus genes. 2003;26:291–315. doi: 10.1023/a:1024455415443. [DOI] [PubMed] [Google Scholar]
- 3.Katzourakis A, Gifford RJ, Tristem M, Gilbert MT, Pybus OG. Macroevolution of complex retroviruses. Science. 2009;325:1512. doi: 10.1126/science.1174149. [DOI] [PubMed] [Google Scholar]
- 4.Han GZ, Worobey M. An endogenous foamy virus in the Aye-Aye (Daubentonia madagascariensis) Journal of Virology. 2012;86:7696–8. doi: 10.1128/JVI.00650-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Han GZ, Worobey M. An endogenous foamy-like viral element in the coelacanth genome. PLoS Pathogens. 2012;8:e1002790. doi: 10.1371/journal.ppat.1002790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Llorens C, Munoz-Pomer A, Bernad L, Botella H, Moya A. Network dynamics of eukaryotic LTR retroelements beyond phylogenetic trees. Biology direct. 2009;4:41. doi: 10.1186/1745-6150-4-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ferguson-Smith MA, Trifonov V. Mammalian karyotype evolution. Nature Reviews Genetics. 2007;8:950–62. doi: 10.1038/nrg2199. [DOI] [PubMed] [Google Scholar]
- 8.Amores A, Catchen J, Ferrara A, Fontenot Q, Postlethwait JH. Genome evolution and meiotic maps by massively parallel DNA sequencing: spotted gar, an outgroup for the teleost genome duplication. Genetics. 2011;188:799–808. doi: 10.1534/genetics.111.127324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Walter RB, et al. A microsatellite genetic linkage map for Xiphophorus. Genetics. 2004;168:363–72. doi: 10.1534/genetics.103.019349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Catchen JM, Conery JS, Postlethwait JH. Automated identification of conserved synteny after whole-genome duplication. Genome Research. 2009;19:1497–505. doi: 10.1101/gr.090480.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miya M, et al. Major patterns of higher teleostean phylogenies: a new perspective based on 100 complete mitochondrial DNA sequences. Molecular phylogenetics and evolution. 2003;26:121–38. doi: 10.1016/s1055-7903(02)00332-9. [DOI] [PubMed] [Google Scholar]
- 12.Steinke D, Salzburger W, Meyer A. Novel relationships among ten fish model species revealed based on a phylogenomic analysis using ESTs. Journal of molecular evolution. 2006;62:772–84. doi: 10.1007/s00239-005-0170-8. [DOI] [PubMed] [Google Scholar]
- 13.Patton EE, Mitchell DL, Nairn RS. Genetic and environmental melanoma models in fish. Pigment Cell and Melanoma Research. 2010;23:314–37. doi: 10.1111/j.1755-148X.2010.00693.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wittbrodt J, et al. Novel putative receptor tyrosine kinase encoded by the melanoma-inducing Tu locus in Xiphophorus. Nature. 1989;341:415–421. doi: 10.1038/341415a0. [DOI] [PubMed] [Google Scholar]
- 15.Kallman KD. The platyfish Xiphophorus maculatus. In: King RC, editor. Handbook of Genetics. Vol. 4. Plenum Press; New York: 1975. pp. 81–132. [Google Scholar]
- 16.Gutbrod H, Schartl M. Intragenic sex-chromosomal crossovers of Xmrk oncogene alleles affect pigment pattern formation and the severity of melanoma in Xiphophorus. Genetics. 1999;151:773–83. doi: 10.1093/genetics/151.2.773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Meierjohann S, Schartl M. From Mendelian to molecular genetics: the Xiphophorus melanoma model. Trends in genetics : TIG. 2006;22:654–61. doi: 10.1016/j.tig.2006.09.013. [DOI] [PubMed] [Google Scholar]
- 18.Braasch I, Brunet F, Volff JN, Schartl M. Pigmentation pathway evolution after whole-genome duplication in fish. Genome Biology and Evolution. 2009:479–493. doi: 10.1093/gbe/evp050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Laisney JA, Mueller TD, Schartl M, Meierjohann S. Hyperactivation of constitutively dimerized oncogenic EGF receptors by autocrine loops. Oncogene. 2012 doi: 10.1038/onc.2012.267. [DOI] [PubMed] [Google Scholar]
- 20.Regneri J, Schartl M. Expression regulation triggers oncogenicity of xmrk alleles in the Xiphophorus melanoma system. Comparative biochemistry and physiology. Toxicology & pharmacology : CBP. 2012;155:71–80. doi: 10.1016/j.cbpc.2011.04.003. [DOI] [PubMed] [Google Scholar]
- 21.Kazianis S, et al. Localization of a CDKN2 gene in linkage group V of Xiphophorus fishes defines it as a candidate for the DIFF tumor suppressor. Genes, chromosomes & cancer. 1998;22:210–20. [PubMed] [Google Scholar]
- 22.Chatzinasiou F, et al. Comprehensive field synopsis and systematic metaanalyses of genetic association studies in cutaneous melanoma. Journal of the National Cancer Institute. 2011;103:1227–35. doi: 10.1093/jnci/djr219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Butler AP, et al. Regulation of CDKN2A/B and Retinoblastoma genes in Xiphophorus melanoma. Comparative biochemistry and physiology. Toxicology & pharmacology : CBP. 2007;145:145–55. doi: 10.1016/j.cbpc.2006.07.013. [DOI] [PubMed] [Google Scholar]
- 24.Walter RB, Kazianis S. Xiphophorus interspecies hybrids as genetic models of induced neoplasia. ILAR Journal of the Institute of Laboratory Animal Resources. 2001;42:299–321. doi: 10.1093/ilar.42.4.299. [DOI] [PubMed] [Google Scholar]
- 25.Nairn RS, et al. Genetic analysis of susceptibility to spontaneous and UV-induced carcinogenesis in Xiphophorus hybrid fish. Marine Biotechnology. 2001;3:S24–36. doi: 10.1007/s1012601-0004-7. [DOI] [PubMed] [Google Scholar]
- 26.Kazianis S, et al. Genetic analysis of neoplasia induced by N-nitroso-Nmethylurea in Xiphophorus hybrid fish. Marine Biotechnology. 2001;3:S37–43. doi: 10.1007/s10126001-0025-2. [DOI] [PubMed] [Google Scholar]
- 27.Hrbek T, Seckinger J, Meyer A. A phylogenetic and biogeographic perspective on the evolution of poeciliid fishes. Molecular Phylogenetics and Evolution. 2007;43:986–98. doi: 10.1016/j.ympev.2006.06.009. [DOI] [PubMed] [Google Scholar]
- 28.Pollux BJA, Pires MN, Banet AI, Reznick DN. Evolution of Placentas in the Fish Family Poeciliidae: An Empirical Study of Macroevolution. Annu. Rev. Ecol. Evol. Syst. 2009;40:271–289. [Google Scholar]
- 29.Turner CL. Pseudoamnion, pseudochorion, and follicular pseudoplacenta in poeciliid fishes. Journal of Morphology. 1940;67:59–87. [Google Scholar]
- 30.Tavolga WN, Rugh R. Development of the platyfish, Platypoecilius maculatus. Zoologica. 1947;32:1–15. [PubMed] [Google Scholar]
- 31.Scrimshaw NS. Embryonic development in poeciliid fishes. Biological Bulletin. 1945;88:233–246. [Google Scholar]
- 32.Brawand D, Wahli W, Kaessmann H. Loss of egg yolk genes in mammals and the origin of lactation and placentation. PLoS Biology. 2008;6:e63. doi: 10.1371/journal.pbio.0060063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sibley CP, et al. Placental-specific insulin-like growth factor 2 (Igf2) regulates the diffusional exchange characteristics of the mouse placenta. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:8204–8. doi: 10.1073/pnas.0402508101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Vo LH, Hedrick JL. Independent and hetero-oligomeric-dependent sperm binding to egg envelope glycoprotein ZPC in Xenopus laevis. Biol Reprod. 2000;62:766–74. doi: 10.1095/biolreprod62.3.766. [DOI] [PubMed] [Google Scholar]
- 35.Bshary R, Wickler W, Fricke H. Fish cognition: a primate’s eye view. Animal cognition. 2002;5:1–13. doi: 10.1007/s10071-001-0116-5. [DOI] [PubMed] [Google Scholar]
- 36.Force A, et al. Preservation of duplicate genes by complementary, degenerative mutations. Genetics. 1999;151:1531–45. doi: 10.1093/genetics/151.4.1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Braasch I, Postlethwait J.H.I.e. Polyploidy in fish and the teleost genome duplication. In: Soltis PS, Soltis DE, editors. Polyploidy and Genome Evolution. Springer; 2012. in press. [Google Scholar]
- 38.Galtier N, Gouy M, Gautier C. SEAVIEW and PHYLO_WIN: two graphic tools for sequence alignment and molecular phylogeny. Comput Appl Biosci. 1996;12:543–548. doi: 10.1093/bioinformatics/12.6.543. [DOI] [PubMed] [Google Scholar]
- 39.Setiamarga DH, et al. Divergence time of the two regional medaka populations in Japan as a new time scale for comparative genomics of vertebrates. Biology letters. 2009;5:812–6. doi: 10.1098/rsbl.2009.0419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Froschauer A, et al. Construction and initial analysis of bacterial artificial chromosome (BAC) contigs from the sex-determining region of the platyfish Xiphophorus maculatus. Gene. 2002;295:247–54. doi: 10.1016/s0378-1119(02)00684-4. [DOI] [PubMed] [Google Scholar]
- 41.Huang X, Wang J, Aluru S, Yang SP, Hillier L. PCAP: a whole-genome assembly program. Genome research. 2003;13:2164–70. doi: 10.1101/gr.1390403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yao G, et al. Graph accordance of next-generation sequence assemblies. Bioinformatics. 2012;28:13–6. doi: 10.1093/bioinformatics/btr588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biology. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zerbino DR, Birney E. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008;18:821–9. doi: 10.1101/gr.074492.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stanke M, Waack S. Gene prediction with a hidden Markov model and a new intron submodel. Bioinformatics. 2003;19(Suppl 2):ii215–25. doi: 10.1093/bioinformatics/btg1080. [DOI] [PubMed] [Google Scholar]
- 46.Kent WJ. BLAT - the BLAST-like alignment tool. Genome Research. 2002;12:656–64. doi: 10.1101/gr.229202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25:1105–11. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Trapnell C, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nature Biotechnology. 2010;28:511–5. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Camacho C, et al. BLAST+: architecture and applications. BMC Bioinformatics. 2009;10:421. doi: 10.1186/1471-2105-10-421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schulz MH, Zerbino DR, Vingron M, Birney E. Oases: robust de novo RNAseq assembly across the dynamic range of expression levels. Bioinformatics. 2012;28:1086–92. doi: 10.1093/bioinformatics/bts094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wu TD, Watanabe CK. GMAP: a genomic mapping and alignment program for mRNA and EST sequences. Bioinformatics. 2005;21:1859–75. doi: 10.1093/bioinformatics/bti310. [DOI] [PubMed] [Google Scholar]
- 52.Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Research. 1997;25:955–64. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wicker T, et al. A unified classification system for eukaryotic transposable elements. Nature Reviews Genetics. 2007;8:973–82. doi: 10.1038/nrg2165. [DOI] [PubMed] [Google Scholar]
- 54.Kohani O, Gentles AJ, Hankus L, Jurka J. Annotation, submission and screening of repetitive elements in Repbase: RepbaseSubmitter and Censor. BMC Bioinformatics. 2006;7:474. doi: 10.1186/1471-2105-7-474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Xu Z, Wang H. LTR_FINDER: an efficient tool for the prediction of full-length LTR retrotransposons. Nucleic Acids Research. 2007;35:W265–8. doi: 10.1093/nar/gkm286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Catchen JM, Amores A, Hohenlohe P, Cresko W, Postlethwait JH. Stacks: building and genotyping Loci de novo from short-read sequences. G3. 2011;1:171–82. doi: 10.1534/g3.111.000240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hohenlohe PA, et al. Population genomics of parallel adaptation in threespine stickleback using sequenced RAD tags. PLoS genetics. 2010;6:e1000862. doi: 10.1371/journal.pgen.1000862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Drummond AJ, et al. Geneious v5.5. 2010 Available from http://www.geneious.com.
- 59.Yang Z, Wong WS, Nielsen R. Bayes empirical bayes inference of amino acid sites under positive selection. Molecular Biology and Evolution. 2005;22:1107–18. doi: 10.1093/molbev/msi097. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.