

Abstract
Background
Inflammatory bowel disease (IBD) is associated with defects in intestinal barriers that rely upon cellular tight junctions. Thus, identifying genes that could be targeted to enforce tight junctions and improve barrier function may lead to new treatment strategies for IBD.
Aims
This preclinical study aimed to evaluate an hypothesized role for the tumor suppressor gene Bin1 as a modifier of the severity of experimental colitis.
Methods
We ablated the Bin1 gene in a mosaic mouse model to evaluate its effects on experimental colitis and intestinal barrier function. Gross pathology, histology and inflammatory cytokine expression patterns were characterized and ex vivo physiology determinations were conducted to evaluate barrier function in intact colon tissue.
Results
Bin1 attenuation limited experimental colitis in a sexually dimorphic manner with stronger protection in female subjects. Colitis suppression was associated with an increase in basal transepithelial electrical resistance (TER) and a decrease in paracellular transepithelial flux, compared to control wild-type animals. In contrast, Bin1 attenuation did not affect short circuit current, nor did it alter the epithelial barrier response to non-inflammatory permeability enhancers in the absence of inflammatory stimuli.
Conclusions
Bin1 is a genetic modifier of experimental colitis that controls the paracellular pathway of transcellular ion transport regulated by cellular tight junctions. Our findings offer a preclinical validation of Bin1 as a novel therapeutic target for IBD treatment.
Keywords: IBD, colitis, inflammation, inflammatory cytokines, tight junctions, epithelial barrier
Introduction
Inflammatory bowel disease (IBD), encompassing ulcerative colitis (UC) and Crohn’s disease (CD), are chronic gastrointestinal tract inflammation produced by combinational interaction between genetic and environmental factors. Epidemiological evidence indicates that inappropriate activation of the immune system in IBD is likely responsible for promoting tumorigenesis within the epithelium [1]. Several experimental IBD mouse models have been developed to study this condition, including gene knockout (KO) models [2, 3], transgenic models [4], spontaneous colitis models [5], inducible colitis modes [6, 7], and adoptive transfer models [8, 9]. Among them, the inducible colitis model involving heparin-like polysaccharide dextran sodium sulfate (DSS) is used most commonly due to its simplicity and consistent colonic lesions [9]. In this model, low molecular weight DSS induces colitis by interfering with intestinal barrier function, causing epithelial damage and driving inflammatory responses that mimic the clinical and histological characteristics of human IBD.
There is considerable clinical evidence that epithelial barrier function is compromised in colitis patients [10–12]. However, TJ function may act as a disease modifier, insofar as targeted inactivation of TJs in the mouse is insufficient to cause intestinal disease but will instead drive mucosal immune responses and accelerate the onset and severity of immune-mediated colitis once induced [13]. In intestinal epithelia, fluid and solute transport occurs both transcellularly and paracellularly [14]. The transcellular pathway handles movement of solute across cells through plasma membrane transporters. Alternately, the paracellular pathway handles movement of solute across the intercellular space between adjacent epithelial cells through tight junctions (TJs) located at the most apical region of cells [15]. TJ complexes allow passive absorption of small hydrophilic molecules (nutrients and ions), but they restrict passage of large molecules which may be antigenic or toxic. Thus, TJ prevent antigens or infectious microbes in the intestinal lumen from travelling easily to the stroma, imposing a blockade to undesirable inflammation. Through their ability to selectively determine intestinal permeability [16], TJ strongly influence normal gut physiology and pathophysiology. Intestinal barrier function mediated by TJ can be quantitated by transepithelial electrical resistance (TER) [17, 18], which is closely correlated to permeability (leakiness) of the TJ in relatively low resistance tissues.
BAR adapter proteins such as those encoded by the Bin1 gene have been implicated in many cellular processes, including vesicle trafficking, transcription, stress signaling and tumor suppression [19, 20]. Bin1 is strongly expressed in colon and its attenuation in human and murine colon cancers is correlated with increased progression [21, 22], particularly in inflammatory models of colon carcinogenesis [23]. BAR adapter proteins encoded by Bin1 and its evolutionarily homologs functionally interact with Dlg-Scribble complex proteins that affect cell polarity, migration and tight junctional traffic [19, 24]. In this study, we made use of a unique genetic mosaic mouse knockout model developed in our laboratory to evaluate the effect of Bin1 attenuation on chemically induced colitis and colonic barrier function. Our findings suggest that attenuating Bin1 restricts the severity of induced colitis in a manner associated with an enhancement to epithelial barrier function.
Material and Methods
Transgenic mouse strains
The generation and maintenance of Bin1 mosaic −/− mice and the corresponding strain-matched wild-type mice has been described previously in detail [23]. Previous work has shown that in Bin1 mosaic −/− mice genetic deletions occur throughout the whole organ with cells harboring recombined (Bin1−/−) or non-recombined (Bin1+/+) alleles both present within the tissue [23]. Thus, this mosaic model mimics a partial loss of function not unlike that produced by an idealized drug-like inhibitor. All animal protocols employed were reviewed and approved by the Lankenau Animal Care and Use Committee.
Colitis induction
Mice of 7 weeks of age were administered 3% dextran sodium sulfate (DSS, MP Biomedicals, Cat# 160110, MW 36–50kDa) in drinking water. At 3 days and 7 days after DSS treatment, mice were euthanized and colons were inspected at necropsy for gross macroscopic lesions before histological processing and microscopic analysis. To evaluate the effects of Bin1 loss on survival, animals received 3% DSS continuously in drinking water until they expired or were euthanized due to signs of distress.
Inflammatory marker measurements
Serum collected from mice subjected to DSS treatment for 7 days was analyzed using a commercial cytometric bead array as described [25]. The inflammatory cytokines examined included IL-2, IL-4, IL-6, IL-12, TNF-α, and IFN-γ. Briefly, specimens were analyzed according to the vendor’s instructions on a FACSCanto II flow cytometer using FACSDIVA software, with cytokine concentrations calculated by comparison to standard curves using the cytometric bead array analysis software (BD Biosciences).
Tight junction physiology assays
Evaluation of transepithelial electrical resistance (TER) and transepithelial mannitol flux was done as described previously [26]. The mouse cecum was used for analysis in this study due to the restrictions in a Ussing chamber fit for the mouse colon. At necropsy, cecum was isolated by dissection, sliced longitudinally, and cleaned by ice-cold pH 7.35 bicarbonate-buffered saline (Krebs-Ringer bicarbonate, KRB). This tissue was then laid on its mucosal surface and the serosal membrane and muscularis propria were surgically removed. Remaining tissue was mounted in 4.2 cm2 Ussing chamber and the tissue bathed in 37°C KRB stirred by gas-lift (95% O2, 5% CO2) oxygenation. This chamber permits the separation and sampling of luminal versus anti-luminal fluid compartments, thereby allowing precise study of transport and barrier function by the epithelium. Transepithelial electrical resistance (TER) and transepithelial voltage were measured every 2 min using Ag/AgCl electrodes in series with 1 M NaCl agar bridges and a standard current/voltage clamp delivering 1 sec pulses of 40 μA current (McGrath Research and Technology, Phoenix, AZ). After 30–45 min of incubation, the mucosal tissue was physiologically stabilized and the maximal TER was measured (greater TER values indicate lower electrolyte permeability). Short circuit current and potential difference measurements were also performed using this apparatus. 14C-mannitol (Perkin-Elmer, Boston, MA) along with non-radiolabeled mannitol was then added to the mucosal hemichamber (final activity 0.1 μCi/ml, final concentration 0.1 mM). Aliquots from the serosal fluid hemichamber were taken every 15 min for 60 min for liquid scintillation counting. From linear regression of a graph of cpm per unit time, and using the measured specific activity of the non-metabolizable 14C-probe in the mucosal hemichamber, we calculated the transepithelial probe flux rate (pmol·min−1·cm−2 tissue). This value reflects the permeability of the TJs to that particular probe.
Results
Bin1 attenuation reduces colitis with a sexually dimorphic preference in females
In support of previous observations [21], histological examination of colon sections from Bin1 mosaic −/−mice of both sexes and a variety of ages up to one year revealed no differences in crypt or villus structures or any other evident abnormalities, compared to WT control mice (Fig. 1). Thus, on its own Bin1 attenuation did not increase susceptibility to colitis in the absence of an inflammatory stimulus. To determine whether Bin1 acted as a modifier in this setting, we compared the response of Bin1 mosaic −/− mice to treatment with 3% DSS in drinking water for 3 days or 7 days. At 3 days, we found that 40% of the WT mice but none of the Bin1 mosaic −/− mice displayed mild colitis (Table I). Histological examination revealed that by this time crypts in WT mice began to shorten and disappear and macrophages, lymphocytes and polymorphonuclear neutrophils were migrating into the colonic epithelial layer, evidencing the occurrence of an inflammatory reaction (Fig. 2). At 7 days of DSS administration, all WT mice displayed severe colitis marked by diarrhea, bloody stool and weight loss, whereas only 33% of the Bin1 mosaic −/− mice exhibited these effects (Table I). Confirming these effects, we documented a profound loss of crypts and colonic epithelium generally, an overall reduction in colon length, and an increase in inflammatory infiltrates as evidence of severe colitis (Fig. 2).
Figure 1. Bin1 loss does not affect normal colon histology.

Colons from dissected from untreated mice at necropsy, fixed in paraffin, processed for H&E staining and analyzed for microscopic histopathology. Representative colons from control Bin1+/+ and Bin1 mosaic −/− mice are shown. Original magnifications are 10x (left), 20x (right).
Table I. Genetic attenuation of Bin1 protects against experimental colitis.
Mice in WT and Bin1 mosaic −/− groups were administered 3% DSS in drinking water to induce colitis as described in the text. At the endpoints indicated, mice were euthanized and gross pathology of the colon was determined at necropsy before processing for histological evaluation. To score the severity of colitis, inflammation scoring was determined along the lines of methods that have been suggested for this model [27]. Minimal inflammation was determined by patchy shortening of the colonic crypts with only focal chronic inflammation. Mild inflammation was determined by more diffuse chronic inflammation of the lamina propria with shortening of the crypts. Severe inflammation was determined by dense diffuse chronic inflammation with acute cryptitis and marked distorsion and severe shortening of the colonic crypts.
| Genotype | DSS (days) | Normal colon | Minimal colitis | Mild colitis | Severe colitis |
|---|---|---|---|---|---|
| Bin1+/+ (WT) | 3 | 2/5 (40%) | 1/5 (20%) | 2/5 (40%) | 0/5 (0%) |
| Bin1 mosaic −/− | 3 | 6/6 (100%) | 0/6 (0%) | 0/6 (0%) | 0/6 (0%) |
| Bin1+/+ (WT) | 7 | 0/5 (0%) | 0/5 (0%) | 0/5 (0%) | 5/5 (100%) |
| Bin1 mosaic −/− | 7 | 1/6 (17%) | 1/6 (17%) | 2/6 (33%) | 2/6 (33%) |
Figure 2. Bin1 loss suppresses the development of DSS-induced experimental colitis.

(A) Three days DSS treatment. Colons processed as before from Bin1+/+ mice exhibited separation of the crypt base from the muscularis mucosa along with crypt loss, whereas colons from Bin1 mosaic −/− mice remained intact and histologically normal. (B) Seven days DSS treatment. Colons processed as before from Bin1+/+ mice showed prominent crypt loss and scar tissue that was not apparent in colons from Bin1 mosaic −/− mice which displayed a moderate phenotype. Original magnifications are 10x, 20x. N: normal colon crypt. I: inflammatory cell infiltration.
Survival experiments revealed a sexually dimorphic character in the benefits of Bin1 attenuation in protecting against colitis induction. In Bin1 mosaic −/− mice, we observed a significant increase in the survival of females but not males who received 3% DSS on a continuous basis in their drinking water (Fig. 3). While Bin1 mosaic −/− males were resistant to colitis on the basis of gross pathological and histological phenotypes noted above, the benefits of Bin1 attenuation in males were not associated with greater survival compared to WT males. In contrast, the phenotypic resistance of Bin1 mosaic −/− females was also associated with longer median survival when compared to WT females (Fig. 3).
Figure 3. Potency of Bin1 loss is sexually dimorphic with a preference in female mice.

Bin1+/+ and Bin1 mosaic −/− mice administered 3% DSS in drinking water and survival was monitored to expiry. Colons were obtained at necropsy and processed for analysis as before to confirm the presence experimental colitis at the levels of gross and microscopic histopathology.
The observed reduction in colitis due to Bin1 attenuation scored by histopathological examination was confirmed and mechanistically assessed by comparing levels for a panel of inflammatory cytokines in control versus Bin1 mosaic −/− females. Briefly, serum obtained from mice treated 7 days with DSS was analyzed using a commercial cytometric bead array as described previously [25]. Consistent with a reduced inflammatory response in Bin1 mosaic −/− animals, there was some reduction in the levels of TNF-α, interferon-γ and IL-4 and some elevation in the levels of IL-6 but these changes did not reach statistical significance (Supp. Fig. 1). The pattern of response argued that Bin1 did not exert a generalized effect on inflammation, but that it might act proximally to modify other functions that were coordinately regulated in colitis. Taken together, the results obtained established that Bin1 attenuation conferred a protective benefit against chemically induced colitis, with a sexually dimorphic preference to this benefit in female animals.
Bin1 loss improves basal intestinal barrier function in cecum
We used cecum tissue to evaluate the effects of Bin1 attenuation on intestinal epithelial cell barrier function and its involvement in colitis. Cecal segments from mice were mounted in Ussing chambers for measurement of mucosal electrophysiology and 14C-mannitol flux. Cecum mucosal permeability was determined by measuring diffusion of 14C-mannitol from the luminal to the antiluminal fluid compartment. Because mannitol has no affinity for membrane transporters, its movement across epithelia is mainly through the paracellular pathway. We found that Bin1 mosaic −/− mice displayed a higher basal transepithelial electrical resistance in cecum compared to WT mice (Fig. 4A). Consistent with this observation, there was also a lower transepithelial flux of 14C-mannitol across the cecum in Bin1 mosaic −/− mice compared to WT mice (Fig. 4B). By establishing a relative decrease in paracellular leak in Bin1 mosaic −/− mice, these results also directly implied an increased epithelial barrier function.
Figure 4. Bin1 loss enhances epithelial barrier function in cecum.
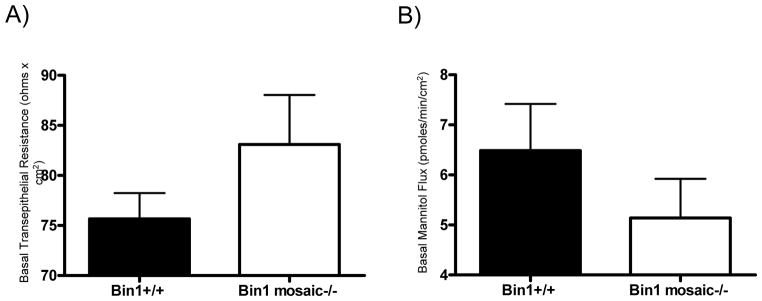
(A) Transepithelial electrical resistance (Rt, ohms x cm2) was determined across cecum obtained from Bin1+/+ (75.7±2.6, n=6) and Bin1 mosaic −/− mice (83.1±5.0, n=6). (B) Transepithelial 14C-mannitol influx (pmoles/min/cm2) was determined across cecum obtained from Bin1+/+ (6.5±0.9, n=6) and Bin1 mosaic −/− mice (5.1±0.8, n=6).
Since Bin1 acted as a modifier of barrier function in the setting of DSS-induced inflammation, we sought to evaluate the specificity of this effect, that is, whether Bin1 attenuation might also affect non-inflammatory mucosal electrophysiology or 14C-mannitol permeability in response to chemical enhancers of colon permeability. Stimulating cecum with PDBU produced an immediate sharp increase followed by a decrease in the short circuit current, but no obvious differences in the response patterns in cecum from WT or Bin1 mosaic −/− mice (Fig. 5A). PDBU also affected TER and transepithelial mannitrol flux, as expected, but again with similar kinetics in each group (Figs. 5B,C). Similarly, cecum stimulation with sodium caprate induced changes in these parameters, but again with the same kinetics in both groups (Figs. 6A–C). Taken together, we interpreted these results to mean that Bin1 acted to modify intestinal barrier function by a specific mechanism that was associated with inflammatory stimuli and distinct from established pathways of permeability control influenced by PDBU or sodium caprate. In summary, we concluded that Bin1 attenuation reduced the severity of experimental inflammatory colitis in a manner associated with an TJ-dependent increase in intestinal barrier function.
Figure 5. Bin1 does not affect epithelial barrier function perturbation by PDBU.

After tissue electrical parameters were stabilized, cecum was stimulated by 5x10−6 M phorbol 12,13-dibutyrate (PDBU) and the following physiological parameters were monitored. (A) Short circuit current (μamps/cm2). (B) Transepithelial electrical resistance (Rt, ohms x cm2). (C) Transepithelial 14C-mannitol flux (CPMS).
Figure 6. Bin1 does not affect epithelial barrier function perturbation by sodium caprate.

After tissue electrical parameters were stabilized, cecum was stimulated by 5 mM sodium caprate and the following physiological parameters were monitored. (A) Short circuit current (μamps/cm2). (B) Transepithelial electrical resistance (Rt, ohms x cm2). (C) Transepithelial 14C-mannitol flux (CPMS).
Discussion
The intestinal epithelium forms a physiological barrier that protects the body from many antigens and microorganisms [10]. Dysfunction in the TJ complex leads to an increase in intestinal permeability [28, 29], allowing for diffusion of toxic luminal bacterial products like endotoxin into the intestinal submucosa. These unwanted substances may cause an abnormal immune response (loss of tolerance) resulting in the development of IBD [30–32]. A relationship between inflammation and cancer has long been suspected and IBD is associated with increased risk of developing colorectal cancer [33]. Based on our earlier findings that Bin1 attenuations are common in human colon cancers and that genetic deletions are sufficient to drive progression of colon carcinogenesis [23], we hypothesized that Bin1 attenuation might heighten colitis, which resembles human IBD in its symptoms of diarrhea, bloody stool and weight loss. In fact, we found that Bin1 attenuation inhibited colitis with a sexually dimorphic preference in females. Thus, a complex relationship with cancer was indicated given that Bin1 attenuation inhibits DSS-induced colitis whereas it promotes the development of colon carcinogenesis [23], in this case in the absence of any sexual dimorphism. The pattern of susceptibilities differs with the human setting, insofar as gender differences are negligible in IBD [34] but are more highly linked to males in colon cancer [35]. Acknowledging the complex relationship between inflammation, barrier function and cancer, the present findings establish that Bin1 attenuation restricts colitis development in a manner associated with enhanced colonic barrier function.
The beneficial effects of Bin1 loss in restricting development of colitis associated with enhanced intestinal barrier function were determined by electrophysiological and radiolabeled tracer methods. Bin1 mosaic −/− mice displayed an elevation in basal transepithelial electrical resistance (TER) as well as a corresponding decrease in mannitol permeability, both of which directly implicated improvements in TJ function responsible for mediating barrier function. Cecum epithelia does not generate a series circuit but rather a parallel circuit, where the total resistance equals the addition of the reciprocals of the individual resistors in the circuit such that a resistor with low resistance controls the total resistance. Considering a mosaic epithelium is a parallel patchwork array of two different epithelial populations, moderately increased basal TER in Bin1 mosaic −/− mice indicates that recombined (Bin1−/−) cells likely have a dramatically higher TER than nonrecombined (Bin1+/+) cells. If recombined (Bin1−/−) cells had created a lower resistance (more leaky TJ), then total resistance would have been decreased and bigger difference of total resistance between the groups would have been expected. Thus, the moderate difference in TER value can be explained by interpreting how the mosaic model affects barrier resistance. This improved barrier function in Bin1 mosaic −/− mice was confirmed by reduced basal transepithelial paracellular flux of a radioactive tracer (14C-mannitol) across the cecum mucosa. These results were consistent in strongly supporting an enhanced tight junction function in Bin1 mosaic −/− mice as an explanation for the improved barrier function. The lack of difference in the short circuit current argues that Bin1 does not affect Na+ transcellular transport, underscoring the conclusion that a change in TJ permeability rather than the transcellular permeability pathway is responsible for increased barrier function. The lack of any effect of Bin1 status on permeability enhancement by PBDU or sodium caprate further strengthens this conclusion.
How might enhanced TJ function translate to reduced inflammation? While a full mechanistic understanding has yet to be developed, two explanations might be forwarded that could reduce overall pro-inflammatory signaling. First, enhanced TJ function may more strongly block diffusion of pro-inflammatory stimuli across the intestinal barrier, thereby directly reducing inflammatory responses. There is some evidence that changes in TJ function are accompanied by changes in expression of TJ component proteins, although specific causal relationships remain obscure. For example, enhancement or loosening of barrier function in the colon is associated with increased expression of TJ structural components claudin-3 or claudin-2, respectively [36, 37]. However, in surveying the expression of TJ component proteins claudin-1,2,3,7 and occludin in Bin1 mosaic −/− colons we found no significant differences in expression (M.Y.C. and J.M.M., unpublished observations). Another potential link between Bin1-mediated effects on TJ function and inflammation might consider the immune modulatory enzyme indoleamine 2,3-dioxygenase (IDO), which is a critical target of Bin1 in manifesting its tumor suppressor activity [38]. IDO promotes immune tolerance and formation of a chronic inflammatory state that is associated with altered T cell function [39, 40]. In addition to enhancing TJ function, Bin1 loss also leads to upregulation of IDO that may alter inflammatory responses [38]. Indeed, evidence exists that IDO induction in the colon is sufficient to limit experimental colitis [41]. While further work is needed to explore mechanistic questions, the findings of this study suggest that Bin1-targeting strategies might offer general utility in principle as a novel approach to manage IBD and related chronic inflammatory conditions in the lower bowel.
Supplementary Material
Serum was collected from mice administered 3% DSS in drinking water for 7 days and the level of the immune cytokines indicated was determined by a cytometric bead array. The data represent the determination of at least three data points per sample.
Acknowledgments
We thank Gwen Guillard for tissue sectioning and histology. This work was supported in part by NCI R01 grants CA100123 and CA10954 with additional support from the Charlotte Geyer Foundation and the Lankenau Medical Center Foundation (G.C.P.). A.J.M. is the recipient of grants from the Lance Armstrong Foundation, the DoD Breast Cancer Research Program, and the State of Pennsylvania Department of Health (CURE/Tobacco Settlement Award). J.M.M is a recipient of a grant from the Sharpe-Strumia Foundation and the Prevent Cancer Foundation.
References
- 1.Clevers H. At the crossroads of inflammation and cancer. Cell. 2004;118:671–674. doi: 10.1016/j.cell.2004.09.005. [DOI] [PubMed] [Google Scholar]
- 2.Sadlack B, Merz H, Schorle H, Schimpl A, Feller AC, Horak I. Ulcerative colitis-like disease in mice with a disrupted interleukin-2 gene. Cell. 1993;75:253–261. doi: 10.1016/0092-8674(93)80067-o. [DOI] [PubMed] [Google Scholar]
- 3.Kuhn R, Lohler J, Rennick D, Rajewsky K, Muller W. Interleukin-10-deficient mice develop chronic enterocolitis. Cell. 1993;75:263–274. doi: 10.1016/0092-8674(93)80068-p. [DOI] [PubMed] [Google Scholar]
- 4.Hibi T, Ogata H, Sakuraba A. Animal models of inflammatory bowel disease. J Gastroenterol. 2002;37:409–417. doi: 10.1007/s005350200060. [DOI] [PubMed] [Google Scholar]
- 5.Cong Y, Brandwein SL, McCabe RP, Lazenby A, Birkenmeier EH, Sundberg JP, Elson CO. Cd4+ t cells reactive to enteric bacterial antigens in spontaneously colitic c3h/hejbir mice: Increased t helper cell type 1 response and ability to transfer disease. J Exp Med. 1998;187:855–864. doi: 10.1084/jem.187.6.855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Morris GP, Beck PL, Herridge MS, Depew WT, Szewczuk MR, Wallace JL. Hapten-induced model of chronic inflammation and ulceration in the rat colon. Gastroenterology. 1989;96:795–803. [PubMed] [Google Scholar]
- 7.Okayasu I, Hatakeyama S, Yamada M, Ohkusa T, Inagaki Y, Nakaya R. A novel method in the induction of reliable experimental acute and chronic ulcerative colitis in mice. Gastroenterology. 1990;98:694–702. doi: 10.1016/0016-5085(90)90290-h. [DOI] [PubMed] [Google Scholar]
- 8.Steinhoff U, Brinkmann V, Klemm U, Aichele P, Seiler P, Brandt U, Bland PW, Prinz I, Zugel U, Kaufmann SH. Autoimmune intestinal pathology induced by hsp60-specific cd8 t cells. Immunity. 1999;11:349–358. doi: 10.1016/s1074-7613(00)80110-7. [DOI] [PubMed] [Google Scholar]
- 9.Elson CO, Sartor RB, Tennyson GS, Riddell RH. Experimental models of inflammatory bowel disease. Gastroenterology. 1995;109:1344–1367. doi: 10.1016/0016-5085(95)90599-5. [DOI] [PubMed] [Google Scholar]
- 10.Hollander D. The intestinal permeability barrier. A hypothesis as to its regulation and involvement in crohn’s disease. Scand J Gastroenterol. 1992;27:721–726. doi: 10.3109/00365529209011172. [DOI] [PubMed] [Google Scholar]
- 11.Soderholm JD, Peterson KH, Olaison G, Franzen LE, Westrom B, Magnusson KE, Sjodahl R. Epithelial permeability to proteins in the noninflamed ileum of crohn’s disease? Gastroenterology. 1999;117:65–72. doi: 10.1016/s0016-5085(99)70551-2. [DOI] [PubMed] [Google Scholar]
- 12.Schmitz H, Barmeyer C, Fromm M, Runkel N, Foss HD, Bentzel CJ, Riecken EO, Schulzke JD. Altered tight junction structure contributes to the impaired epithelial barrier function in ulcerative colitis. Gastroenterology. 1999;116:301–309. doi: 10.1016/s0016-5085(99)70126-5. [DOI] [PubMed] [Google Scholar]
- 13.Su L, Shen L, Clayburgh DR, Nalle SC, Sullivan EA, Meddings JB, Abraham C, Turner JR. Targeted epithelial tight junction dysfunction causes immune activation and contributes to development of experimental colitis. Gastroenterology. 2009;136:551–563. doi: 10.1053/j.gastro.2008.10.081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schneeberger EE, Lynch RD. Structure, function, and regulation of cellular tight junctions. Am J Physiol. 1992;262:L647–661. doi: 10.1152/ajplung.1992.262.6.L647. [DOI] [PubMed] [Google Scholar]
- 15.Cano-Cebrian MJ, Zornoza T, Granero L, Polache A. Intestinal absorption enhancement via the paracellular route by fatty acids, chitosans and others: A target for drug delivery. Curr Drug Deliv. 2005;2:9–22. doi: 10.2174/1567201052772834. [DOI] [PubMed] [Google Scholar]
- 16.Madara JL. Regulation of the movement of solutes across tight junctions. Annu Rev Physiol. 1998;60:143–159. doi: 10.1146/annurev.physiol.60.1.143. [DOI] [PubMed] [Google Scholar]
- 17.Kitajima S, Takuma S, Morimoto M. Changes in colonic mucosal permeability in mouse colitis induced with dextran sulfate sodium. Exp Anim. 1999;48:137–143. doi: 10.1538/expanim.48.137. [DOI] [PubMed] [Google Scholar]
- 18.Soderholm JD, Olaison G, Peterson KH, Franzen LE, Lindmark T, Wiren M, Tagesson C, Sjodahl R. Augmented increase in tight junction permeability by luminal stimuli in the non-inflamed ileum of crohn’s disease. Gut. 2002;50:307–313. doi: 10.1136/gut.50.3.307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Prendergast GC, Muller AJ, Ramalingam A, Chang MY. Bar the door: Cancer suppression by amphiphysin-like genes. Biochim Biophys Acta. 2009;1795:25–36. doi: 10.1016/j.bbcan.2008.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ren G, Vajjhala P, Lee JS, Winsor B, Munn AL. The bar domain proteins: Molding membranes in fission, fusion, and phagy. Microbiol Mol Biol Rev. 2006;70:37–120. doi: 10.1128/MMBR.70.1.37-120.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chang M, Boulden J, Katz JB, Wang L, Meyer TJ, Soler AP, Muller AJ, Prendergast GC. Bin1 ablation increases susceptibility to cancer during aging, particularly lung cancer. Cancer Res. 2007;67:7605–7612. doi: 10.1158/0008-5472.CAN-07-1100. [DOI] [PubMed] [Google Scholar]
- 22.DuHadaway JB, Lynch FJ, Brisbay S, Bueso-Ramos C, Troncoso P, McDonnell T, Prendergast GC. Immunohistochemical analysis of bin1/amphiphysin ii in human tissues: Diverse sites of nuclear expression and losses in prostate cancer. J Cell Biochem. 2003;88:635–642. doi: 10.1002/jcb.10380. [DOI] [PubMed] [Google Scholar]
- 23.Chang MY, Boulden J, Katz JB, Wang L, Meyer TJ, Soler AP, Muller AJ, Prendergast GC. Bin1 ablation increases susceptibility to cancer during aging, particularly lung cancer. Cancer Res. 2007;67:7605–7612. doi: 10.1158/0008-5472.CAN-07-1100. [DOI] [PubMed] [Google Scholar]
- 24.Grelle G, Kostka S, Otto A, Kersten B, Genser KF, Muller EC, Walter S, Boddrich A, Stelzl U, Hanig C, Volkmer-Engert R, Landgraf C, Alberti S, Hohfeld J, Strodicke M, Wanker EE. Identification of vcp/p97, carboxyl terminus of hsp70-interacting protein (chip), and amphiphysin ii interaction partners using membrane-based human proteome arrays. Mol Cell Proteomics. 2006;5:234–244. doi: 10.1074/mcp.M500198-MCP200. [DOI] [PubMed] [Google Scholar]
- 25.Scott GN, DuHadaway J, Pigott E, Ridge N, Prendergast GC, Muller AJ, Mandik-Nayak L. The immunoregulatory enzyme ido paradoxically drives b cell-mediated autoimmunity. J Immunol. 2009;182:7509–7517. doi: 10.4049/jimmunol.0804328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mullin JM, Soler AP, Laughlin KV, Kampherstein JA, Russo LM, Saladik DT, George K, Shurina RD, O’Brien TG. Chronic exposure of llc-pk1 epithelia to the phorbol ester tpa produces polyp-like foci with leaky tight junctions and altered protein kinase c-alpha expression and localization. Exp Cell Res. 1996;227:12–22. doi: 10.1006/excr.1996.0244. [DOI] [PubMed] [Google Scholar]
- 27.Cooper HS, Murthy SNS, Shah RS, Sedergran DJ. Clinicopathologic study of dextran sulfate sodium experimental murine colitis. Lab Invest. 1993;69:238–248. [PubMed] [Google Scholar]
- 28.Madsen KL, Malfair D, Gray D, Doyle JS, Jewell LD, Fedorak RN. Interleukin-10 gene-deficient mice develop a primary intestinal permeability defect in response to enteric microflora. Inflamm Bowel Dis. 1999;5:262–270. doi: 10.1097/00054725-199911000-00004. [DOI] [PubMed] [Google Scholar]
- 29.Mullin JM, Agostino N, Rendon-Huerta E, Thornton JJ. Keynote review: Epithelial and endothelial barriers in human disease. Drug Discov Today. 2005;10:395–408. doi: 10.1016/S1359-6446(05)03379-9. [DOI] [PubMed] [Google Scholar]
- 30.Hawker PC, McKay JS, Turnberg LA. Electrolyte transport across colonic mucosa from patients with inflammatory bowel disease. Gastroenterology. 1980;79:508–511. [PubMed] [Google Scholar]
- 31.Hollander D. Crohn’s disease--a permeability disorder of the tight junction? Gut. 1988;29:1621–1624. doi: 10.1136/gut.29.12.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marin ML, Greenstein AJ, Geller SA, Gordon RE, Aufses AH., Jr A freeze fracture study of crohn’s disease of the terminal ileum: Changes in epithelial tight junction organization. Am J Gastroenterol. 1983;78:537–547. [PubMed] [Google Scholar]
- 33.Pohl C, Hombach A, Kruis W. Chronic inflammatory bowel disease and cancer. Hepatogastroenterology. 2000;47:57–70. [PubMed] [Google Scholar]
- 34.Brant SR, Nguyen GC. Is there a gender difference in the prevalence of crohn’s disease or ulcerative colitis? Inflamm Bowel Dis. 2008;14 (Suppl 2):S2–3. doi: 10.1002/ibd.20540. [DOI] [PubMed] [Google Scholar]
- 35.Nelson RL, Dollear T, Freels S, Persky V. The relation of age, race, and gender to the subsite location of colorectal carcinoma. Cancer. 1997;80:193–197. doi: 10.1002/(sici)1097-0142(19970715)80:2<193::aid-cncr4>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 36.Ramalingam A, Wang X, Gabello M, Valenzano MC, Soler AP, Ko A, Morin PJ, Mullin JM. Dietary methionine restriction improves colon tight junction barrier function and alters claudin expression pattern. Am J Physiol Cell Physiol. 299:C1028–1035. doi: 10.1152/ajpcell.00482.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Roxas JL, Koutsouris A, Bellmeyer A, Tesfay S, Royan S, Falzari K, Harris A, Cheng H, Rhee KJ, Hecht G. Enterohemorrhagic e. Coli alters murine intestinal epithelial tight junction protein expression and barrier function in a shiga toxin independent manner. Lab Invest. 90:1152–1168. doi: 10.1038/labinvest.2010.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Muller AJ, DuHadaway JB, Sutanto-Ward E, Donover PS, Prendergast GC. Inhibition of indoleamine 2,3-dioxygenase, an immunomodulatory target of the tumor suppressor gene bin1, potentiates cancer chemotherapy. Nature Med. 2005;11:312–319. doi: 10.1038/nm1196. [DOI] [PubMed] [Google Scholar]
- 39.Katz JB, Muller AJ, Metz R, Prendergast GC. Indoleamine 2,3-dioxygenase in t-cell tolerance and tumoral immune escape. Immunol Rev. 2008;222:206–221. doi: 10.1111/j.1600-065X.2008.00610.x. [DOI] [PubMed] [Google Scholar]
- 40.Prendergast GC, Metz R, Muller AJ. Towards a genetic definition of cancer-associated inflammation: Role of the ido pathway. Am J Pathol. 2010;176:2082–2087. doi: 10.2353/ajpath.2010.091173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ciorba MA, Bettonville EE, McDonald KG, Metz R, Prendergast GC, Newberry RD, Stenson WF. Induction of ido-1 by immunostimulatory DNA limits severity of experimental colitis. J Immunol. 2010;184:3907–3916. doi: 10.4049/jimmunol.0900291. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Serum was collected from mice administered 3% DSS in drinking water for 7 days and the level of the immune cytokines indicated was determined by a cytometric bead array. The data represent the determination of at least three data points per sample.