

Preface
Toll-like receptors (TLRs) are essential components of the innate immune system. Accessory proteins are required for TLR biosynthesis and activation. Here we summarize recent findings on TLR accessory proteins that are required for cell surface and endosomal TLR function, and classify these proteins based on their function as ligand recognition and delivery cofactors, chaperones and trafficking proteins. Because of their essential roles in TLR function, targeting of such accessory proteins may benefit strategies aimed at manipulating TLR activation for therapeutic applications.
Introduction
Pattern recognition receptors (PRRs) are germline-encoded innate immune receptors that were originally reported as sensors for pathogen-associated molecular patterns (PAMPs) 1. PRRs can also recognize endogenous molecules released in response to stress or tissue damage, thus behaving as sensors of alarmins. PRRs therefore sense PAMPs and alarmins, which together constitute damage-associated molecular patterns (DAMPs) 2. PRR engagement promotes the activation of innate and adaptive immune responses 1. Members of the Toll-like receptor (TLR) family are PRRs that recognize many pathogen-derived macromolecules, ranging from bacterial and yeast cell wall components to viral and bacterial nucleic acids. TLR ligation leads to activation of the transcription factors nuclear factor-κB (NF-κB) and interferon regulatory factors (IRFs), resulting in production of pro-inflammatory cytokines and type I interferons (IFNs), respectively.
Humans express ten functional TLRs (TLR1 to TLR10), whereas twelve TLRs (TLR1 to TLR9 and TLR11 to TLR13) have been identified in mice. Ligands have been identified for all TLRs except for human TLR10, mouse TLR12 and mouse TLR13. TLR1, TLR2, TLR4, TLR5, TLR6 and TLR11 reside at the plasma membrane, where they recognize molecular components located on the surface of pathogens. By contrast, TLR3, TLR7, TLR8 and TLR9 are found intracellularly, where they mediate recognition of nucleic acids. Thus, the subcellular distribution of TLRs correlates, to a significant degree, with the compartments in which their ligands are found (Table 1).
Table 1.
Localization and ligands of TLRs
| TLR | Subcelular Localization | Physiological ligands | Synthetic ligands |
|---|---|---|---|
| TLR1–TLR2 | Plasma membrane | Triacylated lipopeptides | Pam3CSK4 |
| TLR2 | Plasma membrane | Peptidoglycan, phospholipomannan, tGPI-mutin, hemagglutinin, porins, lipoarabinomannan, glucuronoxylomannan, HMGB1 | ND |
| TLR2–TLR6 | Plasma membrane | Diacylated lipopeptides, LTA, zymosan | FSL1, MALP2, Pam2CSK4 |
| TLR3 | Endosome | dsRNA | Poly(I:C) |
| TLR4 | Plasma membrane | LPS, VSV-G, RSV-F, MMTV-Env, mannan, glucuronoxylomannan, glycoinositolphospholipds, HSP60, HSP70, fibrinogen, nickel, HMGB1 | ND |
| TLR4–TLR6 | Plasma membrane | oxidized LDL, amyloid-β fibrils | ND |
| TLR5 | Plasma membrane | Flagellin | ND |
| TLR7 | Endosome | ssRNA | Imidazoquinoline compounds: imiquimod, resiquimod, loxoribine |
| TLR8 | Endosome | ssRNA | Resiquimod |
| TLR9 | Endosome | DNA, hemozoin | CpG-A, CpG-B, CpG-C |
| TLR11 (mouse) | Plasma membrane | Profilin | ND |
dsRNA, double stranded RNA; FSL1, S-(2,3-bispalmitoyloxypropyl)-CGDPKHSPKSF; LDL, low-density lipoprotein; LPS, lipopolysaccharide; LTA, lipoteichoic acid; HMGB1, high-mobility group box 1 protein; HSP, heat-shock protein; MALP2, macrophage-activating lipopeptide 2 kDa; MMTV-Env, envelope protein of mouse mammary tumour virus; ND, not determined; polyI:C, polyinosinic–polycytidylic acid; RSV-F, respiratory syncytial virus F protein; ssRNA, single stranded RNA; TLR, Toll-like receptor; VSV-G, vesicular stomatitis virus coat protein.
TLRs are type I transmembrane proteins composed of leucine-rich repeats (LRRs) in the ectodomain, a single transmembrane domain and a cytoplasmic Toll/IL-1 receptor (TIR) domain involved in recruitment of signaling adaptor molecules. TLRs form heterodimers or homodimers as a means of triggering a signal. Most TLRs form homodimers, with a few exceptions. For example, TLR2 forms heterodimers with TLR1 or TLR6, which enables differential recognition of lipopeptides: TLR1–TLR2 recognizes triacyl lipopeptides, whereas TLR2–TLR6 responds to diacyl lipopeptides (Table 1).
Both extracellular and endosomal TLRs are homologous in the sequence of their ectodomain, a feature that is in sharp contrast with the diversity of the ligands they recognize. One mode of ligand discrimination relies on the difference in residues present in the TLR ectodomain. The LRR modules located in the ectodomain of TLRs are composed of 20–30 amino acids containing the consensus sequence LxxLxLxxN. TLRs display different amino acid composition within these modules, leading to structural conformation variations that allow for ligand interaction 3. Amino acid variation and formation of heterodimers can only provide a limited platform for recognition of the varied set of TLR ligands. Thus, another mechanism that reflects the complexity and diversity of TLR ligand composition must exist to ensure proper PAMP detection and self/non-self discrimination. Specific accessory proteins or cofactors can fulfill that role. A given TLR dimer can bind to cofactors that deliver molecules of a particular composition while avoiding other ligands. These cofactors can also have roles in ensuring proper TLR folding in the endoplasmic reticulum (ER), localization to the appropriate subcellular compartment and protein processing, all of which ensure that TLRs reach their assigned subcellular compartments to bind ligand and initiate signaling. Thus, given the observed differences in TLR ligand binding and signaling outcomes, accessory proteins may modulate different aspects of TLR function.
For the purpose of this review, we use accessory proteins and cofactors synonymously, and we define various molecules as such when they fulfill the following roles: they are required for TLR function, they interact with a TLR or a TLR ligand and their ability to facilitate the interaction of a TLR with a ligand has been experimentally confirmed. This definition aims to refine the focus of the review on bonafide TLR cofactors, excluding scaffolding or adaptor proteins required for signaling (such as MYD88 and TRIF; reviewed in 4, 5), molecules involved in crosstalk of signaling pathways (reviewed in 6) and negative regulators (reviewed in 4, 7). We also discuss certain molecules that are essential for TLR functions but their role as cofactors is less well defined and they may also have roles in signaling crosstalk (Box 1). A separate category comprises receptors that can interact with and may passively modulate TLR functions (Box 2). Due to space constraints, we do not discuss the sequence and structure of TLRs (reviewed in ref 3), the possible crosstalk between TLRs and cytosolic innate immune receptors or TLR signaling (reviews in refs 4, 5, 8–11).
Box 1. Proteins with both TLR crosstalk and cofactor function.
Certain proteins have been suggested to have roles both as cofactors and molecules involved in crosstalk of TLR-dependent signaling pathways.
Vitronectin
Vitronectin is a glycoprotein present in the extracellular matrix that binds bacterial lipopeptides. Vitronectin enhances TLR2-mediated responses to lipopeptides and Staphylococcus aureus through interaction with its receptor integrin β3 86. Vitronectin also enhances responses to TLR4 ligands86 and integrins have been shown to facilitate TLR4 signaling by recruitment of the adaptor protein TIRAP to the plasma membrane 87.
Dectin-1
The signaling pathways triggered by dectin-1, a β-glucan receptor involved in the phagocytosis of yeast by macrophages, appear to crosstalk with TLR2 signaling induced by zymosan and β-glucan 88. Thus, dectin-1 and TLR2 collaborate in the response to fungal pathogens.
RP105
RP105 is a B cell specific LPS sensor 89. Its cell surface expression requires association with MD1. The role of RP105–MD1 in TLR4-mediated LPS responses seems to vary with cell type. While RP105 is required for full LPS responsiveness in B cells, expression of RP105–MD1 in dendritic cells and macrophages negatively regulates TLR4 responses to LPS 90. RP105 positively regulates a TLR2-dependent response to Mycobacterium tuberculosis lipoproteins in macrophages 91. Thus, RP105 is unique in its role in both enhancing and suppressing TLR responses in different cell types.
Box 2. Receptor-modulators of TLR responses.
In contrast to the accessory proteins described in the main text, ‘passive’ receptor cofactors of TLR responses are membrane bound and do not necessarily interact with TLRs or their ligands. These receptors modulate TLR functions by passively delivering TLR agonists to their receptors as a result of their intracellular trafficking.
B cell receptor
Antigen recognition through the B cell receptor (BCR) triggers the B cell responses required for an adaptive immune response. Immune complexes comprised of IgG and chromatin extracts induce the proliferation of autoreactive B cells expressing BCR specific for IgG in a MYD88-dependent manner and B cell activation was blocked by TLR9 inhibitors 51. In addition, immune complexes composed of sequences containing CG-rich mammalian DNA activated autoreactive B cells in a TLR9-dependent manner 92, 93. Furthermore, RNA-containing immune complexes can trigger TLR7 activation through a similar mechanism 94. BCR ligation has been shown to affect trafficking of TLR9. BCR stimulation leads to TLR9 trafficking to an autophagosome-like compartment 95, and TLR9 activation by BCR-delivered ligands leads to a different cytokine profile compared with TLR9 stimulation by CpG in the absence of a BCR stimuli 96. Future studies should aim to clarify how BCR activation can crosstalk with trafficking and/or activation of TLRs in B cells.
RAGE
Receptor for advanced glycation end-products (RAGE) was the first reported receptor for high-mobility group box 1 protein (HMGB1) 97. RAGE was originally characterized as a receptor for non-enzymatical adducts of proteins, lipids and nucleic acids created in highly oxidative environments. RAGE ligation leads to nuclear factor-κB (NF-κB) activation and pro-inflammatory cytokine production, which is self-sustained and therefore dysregulated 98. RAGE was shown to modulate TLR9 functions in pDCs, where addition of HMGB1 potentiated CpG-dependent IFNα production, and this was dependent on RAGE 49. Whether HMGB1 loaded with RNA can trigger TLR3 or TLR7 responses through RAGE remains to be determined.
Cofactors for surface TLRs
LBP in ligand delivery to surface TLRs
LPS-binding protein (LBP) is a 481 amino acid acute-phase protein that binds with high affinity to lipopolysaccharide (LPS) derived from the outer membrane of gram-negative bacteria 12. This interaction facilitates the disaggregation and presentation of LPS to CD14, an accessory protein that, among other functions, mediates TLR4 responsiveness to LPS (Figure 1) 12, 13. LBP can also bind to lipoteichoic acid (LTA), peptidoglycan and lipopeptides and transfer them to CD14, suggesting that LBP may assist not only in the function of TLR4, but also in the function of TLR1, TLR2 and TLR6 14–16. Lbp−/− mice are highly susceptible to infection with the gram-negative bacterium Salmonella typhimurium compared with Lbp+/− mice 17. LBP also has a role in the in vivo immune response to gram-positive pneumococci, as leukocyte influx into the cerebrospinal fluid upon challenge with pneumococci, the hallmark of bacterial meningitis, was drastically reduced in Lbp −/− mice compared with Lbp+/− mice 18. LBP thus mediates the innate immune response to PAMPs derived from gram-negative and gram-positive bacteria.
Figure 1. Accessory molecules mediate ligand binding and delivery to surface and endosomal TLRs.

The TLR1-TLR2 heterodimer utilizes CD14 to respond to triacyl lipopetides. The TLR2-TLR6 heterodimer uses CD14 to respond to zymosan and both CD14 and CD36 to respond to lipoteichoic acid (LTA) and diacyl lipopeptides. LPS-binding protein (LBP) binds LPS and presents it to CD14, which is required for TRIF-dependent signaling in response to LPS and at low doses for MyD88-dependent signaling. TLR4 requires MD2 to bind LPS and homodimerize. CD36 is required by the TLR4-TLR6 heterodimer to respond to the altered self-components, amyloid-β and oxidized LDL (oxLDL). Endosomal TLRs utilize cofactors for nucleic acid delivery. CD14 and HMGBs bind to dsRNA, ssRNA, and DNA and mediate their delivery to TLR3, TLR7, and TLR9, respectively. LL37 binds both RNA and DNA and delivers it to TLR7, TLR8 (RNA) and TLR9 (DNA). Progranulin bind just to DNA, and mediate DNA delivery to TLR9. Signaling from TLRs culminates in the activation of the transcription factors AP1, NF-κB, and interferon-regulatory factors (IRFs) and the production of pro-inflammatory cytokines and type I interferons (not shown).
MD2 in ligand recognition by TLR4
MD2 is a 160 amino acid glycosylated soluble protein that associates with the extracellular domain of TLR4 and is required for TLR4 surface expression 19, 20. It is required for TLR4-dependent LPS responses in vivo 20 and Md2−/− B cells, macrophages and DCs are hyporesponsive to LPS. Md2−/− mice were shown to survive LPS-induced endotoxin shock and were more susceptible to Salmonella typhimurium than wild-type mice, demonstrating a phenotype identical to that of Tlr4−/− mice 20. The crystal structure of TLR4–MD2 complexed with E. coli LPS shows how MD2 facilitates TLR4 function 21: LPS buries five of its six lipid chains into the hydrophobic pocket of MD2. Two MD2–LPS complexes are essential for bridging two TLR4 molecules21. Of the TLRs whose structure has been determined in complex with a ligand (TLR1–TLR2, TLR2–TLR6, TLR3 and TLR4), TLR4 is unique in that it requires an accessory molecule for ligand binding 3. Since the two molecules of TLR4 in the TLR4–MD2 heterodimer have limited direct interaction, MD-2 is essential for both ligand binding and dimerization of TLR4 21 (Figure 1).
Ligand discrimination by CD36
CD36 is a 472 amino acid double-spanning membrane glycoprotein of the scavenger receptor type B family that is found in lipid rafts 22, 23. CD36 was first implicated in the function of TLR2–TLR6 heterodimers by a genetic screen that generated a mouse homozygous for a loss-of-function allele of Cd36 (Cd36obl/obl) 24. Cd36obl/obl macrophages showed impaired TNF production in response to the TLR2–TLR6 ligands LTA and the R-stereoisomer of the diacylated lipopeptide MALP2 (R-MALP2), but not to Pam2CSK4, Pam3CSK4, LPS, peptidoglycan, zymosan A, resiquimod, polyinosinic–polycytidylic acid (polyI:C) or CpG DNA 24. Thus, CD36 enhances immune responses to some TLR2–TLR6 ligands but not to others (Figure 1). In vivo, deficiency in CD36 results in increased susceptibility to infection by gram-positive Staphylococcus aureus 24, 25.
CD36 also mediates inflammatory responses to oxidized low-density lipoprotein (oxLDL) and amyloid-β fibrils through the assembly of a TLR4–TLR6 heterodimer 26. Tlr4−/−, Tlr6−/− and Cd36−/− macrophages and microglial cells failed to upregulate inflammatory mediators in response to oxLDL and fibrillar amyloid-β peptide (Aβ1–42), respectively 26. HEK293 cells expressing TLR4, TLR6, and CD36 induced higher expression of a NF-κB luciferase reporter gene in response to oxLDL or Aβ1–42 than HEK293 cells lacking CD36, suggesting that TLR4, TLR6 and CD36 function together to mediate responses to oxLDL and amyloid-β 26. Indeed, stimulation of THP1 enhanced the association of TLR4–TLR6 with CD36 26. monocytes with oxLDL or Aβ1–42 Whether the TLR4–TLR6–CD36 complex also recognizes and responds to PAMPs in addition to endogenous ligands remains to be determined.
How CD36 mediates the function of TLR2–TLR6 and TLR4–TLR6 is not completely understood, but the C-terminus of CD36 seems to have an important role. A point mutant in tyrosine 463 of CD36 (CD36Y463F) failed to induce activation of NF-κB in response to oxLDL and was unable to mediate TLR4–TLR6 dimerization in response to oxLDL 26. An interaction between the tyrosine kinase LYN with CD36 required residues 460–463 of CD36 and inhibition of LYN kinase activity impaired association of CD36 with TLR4–TLR6 and blocked NF-κB activation in response to oxLDL 26. Recruitment of LYN to the C-terminus of CD36 is thus important for the formation of a functional TLR4–TLR6–CD36 signaling complex26. The Y463F mutation in CD36 also abrogated CD36-mediated NF-κB activation in response to the ligand LTA25. Therefore, recruitment of LYN to CD36 may also be important for the formation of a signaling-competent TLR2–TLR6–CD36 complex. A C464S mutation in CD36 also abrogates NF-κB activation in response to LTA 25. Given that CD36 undergoes palmitoylation on residue 464, a signaling defect may be attributed to the inability of CD36C464S to be properly targeted to lipid rafts 27. Thus, fine-tuning of CD36-mediated TLR assembly and responses to ligand depend upon CD36 localization to plasma membrane microdomains for proper interaction with downstream components.
CD14, a cofactor for several TLRs
CD14 is a 375 amino acid leucine-rich repeat glycoprotein that is present in soluble form in the blood or as a glycosylphosphatidylinositol (GPI)-anchored membrane protein on myeloid cells. CD14 interacts with multiple TLR ligands and enhances their ability to activate TLRs (Figure 1). Direct binding studies with recombinant CD14 show that CD14 has the unusual ability to bind a variety of microbial products, including LPS, peptidoglycan, Pam3CSK4, polyI:C and CpG DNA 14, 28–31. The crystal structure shows CD14 to be a dimer and the two subunits together form a horseshoe-shaped structure reminiscent of the ectodomains of TLRs, each subunit equipped with a hydrophobic pocket located at its N-terminus 32. This pocket is the principal component of the LPS-binding site in CD14 32. The CD14 binding sites for different TLR ligands appear to overlap, as LPS can compete with DNA and peptidoglycan, LTA can compete with peptidoglycan, and dsDNA can (partially) compete with polyI:C for CD14 binding 14, 28, 30. How CD14 can bind ligands of such different molecular composition remains to be established, and crystal structures of CD14 complexed with ligands would help address this question.
CD14 was first implicated in TLR4-mediated immune responses. At doses of LPS or gram-negative bacteria that kill wild-type mice, Cd14−/− mice survive and produce negligible amounts of TNF and IL-6 33. In response to LPS, CD14 is required for TRIF-dependent signaling and at low doses for MyD88-dependent signaling 13, 34. While CD14 has been known to chaperone LPS from LBP to TLR4–MD2 at the cell surface, new evidence demonstrates that CD14 also mediates LPS-induced TLR4 endocytosis and delivery to a compartment from which TLR4 can engage TRIF-dependent signaling 34–37. Thus, for TLR4 activation, CD14 facilitates both ligand delivery and TLR4 endocytosis.
CD14 also enhances immune responses to the endosomal TLR ligands polyI:C, imiquimod and CpG DNA 28, 30. CD14 likely promotes the general internalization of nucleic acids, since addition of soluble CD14 enhanced internalization of polyI:C by CHO cells and Cd14−/− macrophages internalized less CpG DNA than wild-type macrophages 28, 30. However, responses to polyI:C, imiquimod and CpG DNA are not completely abrogated in the absence of CD14, suggesting the existence of additional factors that can mediate their delivery 28, 30. Although CD14 was found to associate with TLR3, TLR7 and TLR9 28, 30, it is unclear whether CD14 mediates trafficking of the endosomal TLRs as it does for TLR4 and may just mediate ligand trafficking.
Reflecting its ability to bind diverse ligands, CD14 also mediates TNF production in response to the TLR2–TLR6 ligands MALP2, LTA, zymosan A and Pam2CSK4 and participates in TLR-mediated immune responses to various viruses, including respiratory syncytial virus (RSV) through its fusion protein, vesicular stomatitis virus (VSV) through its glycoprotein G, human cytomegalovirus (CMV) and influenza A virus 13, 28, 38–40. How exactly CD14 is involved in these processes remains to be determined, but as shown for LPS and nucleic acids, CD14 may mediate interaction of ligands with several TLRs.
Delivery of TLR ligands by TRIL
TRIL (TLR4 interactor with leucine-rich repeats) is a type I transmembrane protein of 811 amino acids, containing 12 predicted leucine-rich repeats in its extracellular domain. TRIL is highly expressed in the brain and its expression is LPS- and polyI:C-inducible 41, 42. TRIL colocalizes with early endosomal markers in the human astrocytoma cell line U373, but was found on the surface of HEK293 cells, suggesting that TRIL may have cell type-specific localization patterns41, 42. Knockdown experiments demonstrated that TRIL mediates TLR3 and TLR4, but not TLR2 or TLR9 signaling41, 42. TRIL co-immunoprecipitates with LPS, TLR3 and TLR4, suggesting an involvement for TRIL in ligand delivery 41, 42. Future studies should clarify whether TRIL, similar to CD14, is involved in mediating ligand delivery to TLR3 and TLR4 and whether TRIL has a specialized function in the brain.
Ligand delivery to endosomal TLRs
Granulin delivers CpG to TLR9
Granulin is a cysteine-rich glycosylated multifunctional protein produced as a result of proteolytic processing of the 593 amino acid long precursor progranulin by the serine proteases elastase and proteinase 3 43, 44. Multiple cell types constitutively secrete progranulin, and it is present at high levels in serum 43–45. Granulin fragments were identified as full-length TLR9 interactors by immunoprecipitation of RAW264.7 macrophages treated with the broad cysteine protease inhibitor z-FA-FMK. Addition of exogenous progranulin enhanced TNF secretion by RAW macrophages in response to the synthetic oligodeoxynucleotides (ODNs) CpG-B and CpG-C, and mice deficient in progranulin (Grn−/−) had a defect in TNF production 45. Grn−/− bone marrow-derived macrophages (BMDMs) were less able to bind CpG than their wild-type counterparts, and this defect could be corrected by addition of exogenous progranulin. Progranulin binds not only CpG ODNs, but also ODNs with inhibitory activity, suggesting that granulin binds ODNs independently of sequence. Binding of CpG ODN to C-terminal TLR9 was impaired in Grn−/− mice, thus granulin may facilitate delivery of CpG to lysosomal compartments (Figure 1). Inhibition of elastase activity reduced TNF responses to CpG, suggesting that processing of progranulin into its fragments is required for its contribution for TLR9 signaling 45. Taken together these results demonstrate that granulin helps deliver CpG to the appropriate compartment to promote TLR9 responses. It is still unclear whether granulins interact with a surface receptor, whether they can bind double-stranded DNA, and what determines the enhancement of TLR9 responses to CpG-B and CpG-C but not CpG-A. Future studies should aim to clarify these issues and also determine which forms of granulin are required for TLR9 activation.
HMGB1 delivery of RNA and DNA
Members of the high mobility group box (HMGB) family are nuclear proteins associated with chromatin, involved in making DNA available for regulation of transcription 46. HMGB1 is the most studied member of this family, and mediates its pro-inflammatory functions through interactions with its receptors TLR2, TLR4 and receptor for advanced glycation end-products (RAGE). HMGB1 has a role in sterile inflammation (injury) and infection 47. HMGB1 is a 215 amino acid soluble protein composed of two DNA-binding domains of basic amino acid composition (the A and B boxes) and an acidic tail, and binds DNA in a sequence-independent manner. HMGB1 displays pro-inflammatory functions once secreted by the cell and this activity led to the exploration of whether HMGB1 could deliver ligands to TLR9 and other endosomal TLRs.
HMGB1 was described as a TLR9 cofactor based on its ability to bind CpG DNA, to interact with TLR9 and to enhance delivery of TLR9 to endosomal compartments in response to CpG. Exogenous addition of HMGB1 enhances IFNα and pro-inflammatory cytokine production in response to CpG in DCs and macrophages 48, 49. IFNα secretion was dependent on HMGB1 interaction with RAGE 49, which is a “passive” receptor cofactor of TLR9 (Box 2). The absence of HMGB1 decreases the ability of CpG DNA to upregulate type I IFN expression and secrete pro-inflammatory cytokines, such as IL-6, TNF and IL-12p40 in DCs 48. In patients with lupus, HMGB1 forms complexes with nucleosomes that circulate in the blood as a result of increased apopotosis. Such HMGB1–nucleosome complexes induced pro-inflammatory cytokine production by peripheral blood mononuclear cells 50. Immune complexes containing mammalian DNA, HMGB1 and IgG can activate autoreactive B cells in a TLR9-dependent manner 49, 51. HMGB1 binds both CG-rich and CG-poor DNA, but only delivery of CG-rich mammalian DNA — in the form of HMGB1–DNA–IgG immune complexes through the B cell receptor (Box 2) — promotes TLR9-dependent B cell activation 52. Therefore, HMGB1 is required for TLR9 responses to CpG DNA and may exacerbate autoimmune disease due to its ability to bind DNA.
HMGB proteins may be universal mediators of innate immune responses to nucleic acids. HMGB1 binds both DNA and RNA, and the closely related proteins HMGB2 and HMGB3 bind DNA and RNA, respectively. HMGB proteins were required for type I IFN and pro-inflammatory cytokine production in response to RNA through TLR3 and TLR7, and DNA through TLR9. Although direct PRR binding was not demonstrated, absence of HMGB function decreased responses to DNA and RNA. HMGB proteins are thus required for normal inflammatory immune responses to nucleic acids 53. However, it is not clear how HMGB proteins distinguish between DNA and RNA. How can they resist degradation once outside cells? Is their binding to nucleic acids regulated? If HMGB proteins are implicated in “promiscuous sensing” of nucleic acids, what then prevents immune responses to self-DNA and self-RNA? Future studies should address these questions to further clarify the role of HMGB proteins in responses to nucleic acids.
LL37 ligand delivery to endosomal TLRs
LL37 has been reported to be a TLR9 cofactor, and was implicated in the delivery of self-DNA to TLR9 in pDCs 54. LL37 is a 37 amino acid-long amphipathic peptide, activated upon cleavage of its precursor by a serine protease 55, 56. LL37–DNA complexes are resistant to degradation by DNAses and are internalized by plasmacytoid DCs (pDCs) and localize to early endosomes, from where they mediate TLR9-dependent IFNα production 54. Patients with psoriasis, a skin autoimmune disease characterized by local activation of DCs and T cells, have an infiltration of pDCs in the skin and high expression of LL37 in keratinocytes 55. LL37 drives pDC activation and IFNα production in psoriasis through binding to DNA released by injured cells. There is no evidence for a direct interaction between LL37 and TLR9, suggesting that LL37 may serve mostly as a DNA-delivery molecule in situations of cell injury. LL37 has also been shown to form complexes with self-RNA, and delivery of these complexes to pDCs for TLR7 and TLR8-dependent IFN production. LL37-self RNA complexes are also found in psoriatic skin lesions 57. Thus, LL37 — similar to HMGB proteins — can bind self nucleic acids to mediate delivery to endosomal TLRs. Whether LL37 is important for host defense in the context of TLR-driven responses remains to be established.
Chaperones, trafficking and processing factors for TLRs
TLR folding by Gp96 and PRAT4A
Gp96 is the 803 amino acid endoplasmic reticulum (ER) paralogue of the HSP90 family of chaperones that mediate protein folding. Gp96 is ubiquitously expressed and exists as an obligate soluble homodimer, with each unit composed of a N-terminal ATP-binding domain, a highly charged middle domain and a C-terminal dimerization domain 58. The viability of B cells, macrophages and embryonic stem cells does not require gp96, thus gp96 is not essential for global protein quality control in the ER 59–62.
So far, a limited number of targets have been identified for gp96, including integrins, platelet glycoprotein complexes and TLRs 58–60, 62, 63. The function of TLR1, TLR2, TLR4, TLR5, TLR7 and TLR9, but not that of TLR3, requires gp96 59, 60, 62. Gp96 co-immunoprecipitates with TLR1, TLR2, TLR4 and TLR9; it is required for surface expression of TLR1, TLR2 and TLR4 and for maturation and cleavage of TLR9 60, 62, 64. Thus, gp96 mediates the folding and maturation of TLRs to allow exit from the ER. Exactly at what stage gp96 intervenes in TLR folding, and how, is not known. Until recently, it was unclear whether the function of gp96 — similar to HSP90 — required co-chaperones. Recent evidence suggests that protein associated with TLR4 A (PRAT4A) may fulfill that role.
PRAT4A is a ubiquitous and highly conserved soluble 276 amino acid ER luminal protein that was identified as a protein that co-immunoprecipitates with TLR4 65. Bone marrow-derived DCs (BMDCs), macrophages and B cells isolated from Prat4a−/− mice displayed reduced cytokine production in response to ligands for TLR1, TLR2, TLR4, TLR7 and TLR9, but not for TLR3, a phenotype similar to that of gp96-conditional knockout mice 66. Short hairpin RNA (shRNA)-mediated knockdown of Prat4a in B cell lines impeded passage of TLR1 and TLR4 through the Golgi and prevented ligand-induced trafficking of TLR9 from the ER to endolysosomes 66. Thus, PRAT4A, similar to gp96, is important for the maturation of multiple TLRs in the ER, but it is not a chaperone for general membrane glycoprotein synthesis: PRAT4A-deficient BMDCs showed normal surface expression of CD14, MHC class I molecules and CD11c 66.
PRAT4A and gp96 work together to ensure proper folding of TLRs (Figure 2). PRAT4A and gp96 can interact directly in vitro 64. Mutations of gp96 (E103A) and PRAT4A (M145K) that prevent exit of TLRs from the ER also prevent gp96 and PRAT4A from associating in vivo 60, 64, 67. Knockdown of PRAT4A or gp96 impaired the association of the other protein with TLR9 64, indicating that PRAT4A and gp96 are dependent on each other for TLR9 binding. For other TLRs, a similar folding mechanism may operate.
Figure 2. ER chaperones and trafficking and processing factors of TLRs.

The ER luminal chaperones gp96 and PRAT4A are responsible for proper folding and function of TLR1, TLR2, TLR4, TLR7 and TLR9, but not TLR3. The ER membrane protein UNC93B1 is required for translocation of endosomal TLR7 and TLR9 to endolysosomes, where these TLRs are cleaved by cathepsins and asparagine endopeptidase (AEP). The cleaved TLRs bind ligand (DNA or RNA) triggering recruitment of signaling components leading to NF-κB-dependent proinflammatory cytokine production. The adaptor protein 3 (AP3) mediates translocation of TLR9 to LAMP2+ lysosome or lysosome-related organelles (LRO), where the IRF7 signaling pathway is initiated leading to the expression of type I interferon genes.
As an ER luminal protein, gp96 interacts with the ectodomain of TLRs, confirmed by co-immunoprecipitation of gp96 with fusion proteins that contain the ectodomains of TLR4, TLR9, or TLR11 fused to the transmembrane domain of the platelet-derived growth factor receptor 64. Such fusion proteins of TLR4, TLR9 and TLR11 were expressed on the cell surface in wild-type pre-B cells but not gp96-deficient pre-B cells 64. Similar results were found when measuring surface expression of TLR fusion proteins on RAW264.7 cells transduced with Prat4a-targeting shRNA 64. The ectodomains of TLRs thus require both gp96 and PRAT4A to mediate proper folding, such that they may exit the ER.
Both gp96 and PRAT4A are essential for folding of several TLRs, but many questions remain. What features of folding, dimerization and/or stability of TLRs (except TLR3) dictate a need for gp96 and PRAT4A? Notwithstanding its structural similarity to the other TLRs, does TLR3 require a different set of specialized chaperones in place of gp96–PRAT4A, or is it inherently less chaperone-dependent? The structural motif in TLRs recognized by gp96 and PRATA remains to be molecularly defined and leaves open the possibility of identification of additional client proteins through a search for proteins that contain the relevant motif(s). TLRs appear to have unique folding requirements when compared to other glycoproteins (Box 3).
Box 3. Unique folding of TLRs in the ER.
Several proteins associate with TLRs early in the course of their biosynthesis. These include chaperones such as gp96 and PRAT4A, which are soluble proteins that reside in the ER lumen, as well as ER membrane proteins that possess multiple membrane spanning segments such as UNC93B1. Some of these proteins, such as gp96, clearly assist in the biogenesis of glycoproteins other than TLRs. Others, such as UNC93B1, appear to be far more selective for TLRs. Little is known about the oligomeric state of these TLR-associated proteins themselves, but the current incomplete picture that has emerged is that of a highly complex launching pad that prepares TLRs for their release from the ER in a properly assembled form. Other accessory molecules may not associate with TLRs until they have reached their final destination.
The proper assembly of multi-protein complexes such as the T cell receptor is a prerequisite for their exit from the ER, a concept referred to as architectural editing: the absence of a single subunit compromises assembly, egress from the ER and hence surface display. A similar concept may apply to the formation of signaling-competent TLR assemblies. However, we have little mechanistic information on how each of the proteins discussed in this article participates in the generation of active TLRs. This, clearly, is an area in need of further exploration.
Endosomal TLR trafficking by UNC93B1
Uncoordinated 93B1 (UNC93B1) is a 598 amino acid ER-resident glycoprotein that is predicted to span the membrane twelve times 68. Mice homozygous for a missense mutation (H412R) located in the ninth predicted transmembrane domain of UNC93B1 (known as 3d mice) have impaired signaling via TLR3, TLR7 and TLR9 and show increased susceptibility to a variety of viral and bacterial pathogens 68. Similarly, cells from human patients with mutations that result in truncated UNC93B1 transcripts have defective TLR3, TLR7, TLR8 and TLR9 signaling 69. Thus, UNC93B1 is required for endosomal TLR responses (Figure 2).
Co-immunoprecipitation experiments show that UNC93B1 interacts with TLR3, TLR7, TLR8, TLR9 and TLR13, but not with TLR4, and its interaction with TLRs is eliminated by the H412R mutation 70. Exchange of the transmembrane domains of TLR3 or TLR9 with that of TLR4 resulted in a TLR chimera that is unable to interact with UNC93B1, whereas exchange of the transmembrane domain of TLR4 with that of TLR3 or TLR9 resulted in chimeric proteins that could interact with UNC93B1. The transmembrane domain of endosomal TLRs thus controls association with UNC93B1 70. Ligand-induced trafficking of UNC93B1, TLR7 and TLR9 to CpG-containing endolysosomal compartments is defective in BMDCs from 3d mice 71. Nucleic acid-sensing TLRs must thus interact with UNC93B1 via their transmembrane domains so that UNC93B1 can mediate their delivery to endolysosomes, where they can bind and respond to their respective ligands.
UNC93B1 discriminates between various nucleic acid-sensing TLRs. A D34A missense mutation in UNC93B1 renders TLR7 hyperresponsive and TLR9 hyporesponsive, whereas TLR3 is unaffected. This is due to a stronger association of this mutant with TLR7 72. Mice homozygous for Unc93b1D34A/D34A die prematurely due to systemic inflammation73. TLR7 is responsible for the pathologies of the Unc93b1D34A/D34A mice, because Unc93b1D34A/D34A Tlr7−/− mice showed normal survival and splenic cell numbers. Increased trafficking of TLR7 to endolysosomes in the absence of ligand was observed in Unc93b1D34A/D34A stem cell-derived DCs compared with wild-type DCs 73. The D34A mutation in UNC93B1 thus leads to aberrant trafficking and activation of TLR7.
The role of UNC93B1 in TLR biology is intriguing. How the cell perceives and processes the signals initially required for the trafficking of UNC93B1–TLR complexes from the ER to their endolysosomal destination remains unsolved. Are there small numbers of functional nucleic acid sensing TLRs at the cell surface that could transmit this signal? Are additional nucleic acid sensors involved? It is also becoming apparent that the intracellular distribution of nucleic acid-sensing TLRs mediated by UNC93B1 may differ between cell types: in B cells from transgenic mice expressing green florescent protein-linked TLR9, TLR9 was shown to preferentially localize to an endolysosomal compartment even in the absence of any obvious stimulation (A.M.A., M.M.B and H.L.P., unpublished observations.) Also, how TLR–UNC93B1 oligomeric structures assemble in the ER remains a black box.
Divergence of TLR9 responses by AP3
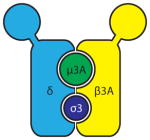
Adaptor protein 3 (AP3) is a required component of the trafficking machinery of TLR9. Members of the AP family are tetrameric proteins that mediate the sorting of membrane proteins in the secretory and endocytic pathways 74. AP3, which consists of four subunits: δ, μ3A, β3A and σ3, recruits cargo into endosomes for delivery to lysosomes and lysosome-related organelles (LROs) 74. BMDMs lacking the β3A subunit of AP3 (Ap3b1−/−) had reduced IFN expression in response to CpG-A complexed with the cationic lipid DOTAP (DOTAP–CpG-A), PolyI:C and LPS 75. pDCs from mice with mutations in Ap3b1 show a similar defect in IFN production in response to CpG-A, VSV and influenza virus 75, 76. Upon DOTAP–CpG-A treatment of AP3-deficient BMDMs, TLR9 failed to enter LAMP2+ compartments at 6 hours post-stimulation, suggesting a role for AP3 in recruiting TLR9 to lysosomes or LROs 75. Furthermore, IRF7 recruitment to CpG-A-containing lysosomes was impaired in AP3-deficient BMDMs 75. Thus, failure of TLR9 to reach LAMP2+ compartments and lack of IRF7 recruitment are responsible for the defective IFN response in AP3-deficient BMDMs.
These findings contrast with observations made at earlier time points (90 minutes) when CpG-A is observed in endosomes of pDCs 77, 78. DOTAP-mediated retention of CpG-A in endosomes of macrophages, which do not normally produce IFNs, promoted recruitment of IRF7 and MyD88 to CpG-A containing endosomes, and expression of IFN 78. Thus, the endosomal compartment was thought to be crucial for IFN production. It is necessary to reconcile these findings with the observation that AP3-mediated trafficking of TLR9 to LAMP2+ compartments is necessary for IFN production by BMDMs stimulated with DOTAP–CpG-A. Further trafficking studies thus need to be conducted to clarify this point and to determine whether AP3 is also involved in TLR3, TLR4 or TLR7 trafficking.
Proteases involved in TLR cleavage
TLR9 undergoes proteolytic processing upon arrival to endolysosomal compartments, and possibly also in early endosomes endowed with low pH and proteases 79, 80. Proteases that process TLRs must interact with TLR9, at least transiently, and are thus considered cofactors. The endosomal/lysosomal complement of proteases is composed of mostly cathepsins. Cathepsins were first implicated in TLR9 function when it was found that inhibition of cathepsin K ameliorated disease in an adjuvant-induced mouse model of arthritis, and cathepsin K deficiency resulted in a decreased BMDC cytokine response to CpG but not to TLR3, TLR7, TLR8 or TLR2–TLR6 ligands 81. By functional cDNA cloning, cathepsin B, cathepsin L, cathepsin S and cathepsin F were identified as factors associated with TLR9 function in a B cell line. Furthermore, the inhibition of these cathepsins by small molecules blocked TLR3-, TLR7- and TLR9-mediated responses in primary B cells 82. The combined action of cathepsin L and cathepsin S results in cleavage of TLR9 80, an event required for signaling 79, 80. Inhibitors for individual cathepsins failed to fully inhibit cleavage and TLR-driven responses 79, 80. The activity of multiple cathepsins is therefore required for full TLR9 activity. Asparagine endopeptidase (AEP) is a lysosomal protease that cleaves C-terminal to asparagine residues. AEP can cleave TLR9 and thus lead to its activation in DCs 83. Therefore, AEP and cathepsins appear to have redundant and/or sequential roles in the cleavage of TLR9 in different types of antigen-presenting cells 84.
Proteolytic processing has also been reported for TLR3 and TLR7 79, 84, and cleavage may therefore be a general occurrence for endosomal TLR activation. Partial inhibition of cleavage and activation by cathepsin and AEP inhibitors suggests that proteolysis, although importantly, may not be essential for TLR3 and TLR7 activation 84. A differential requirement for either AEP or cathepsins in the proteolytic conversion of TLR9 and its activation most likely reflects variation in protease expression in different tissues and cell types. A better understanding of the regulation of TLR3, TLR7 and TLR9 cleavage and function demands a cell-specific exploration of the proteases required for activation. However, it remains to be established whether these results can be generalized to other species, including human TLRs.
Cofactors and advances in TLR biology
The identification of novel TLR accessory molecules and the elucidation of their mechanism of action have led to a greater understanding of TLR biology. From a biological perspective, ligand discrimination by different TLRs can be accomplished through the use of different cofactors that aid in specificity of ligand recognition. Yet, many accessory proteins (exemplified by UNC93B and CD14) appear to overlap in their function for different TLRs, suggesting an additional layer of complexity in the mechanisms by which TLRs distinguish one ligand from another (Table 2). Many questions still remain regarding the role of cofactors in different aspects of TLR biosynthesis, trafficking, ligand recognition and activation.
Table 2.
Accessory molecules for TLR function
| Name | Protein Domain Structure | Localization | Interacting TLR | Interacting ligand | References | |
|---|---|---|---|---|---|---|
| Mediators of ligand delivery and/or recognition | LBP |
|
Secreted | None demonstrated | LPS | 12 |
| MD2 |

|
Plasma membrane associated | TLR4 | LPS | 19–21 | |
| CD36 |
|
Plasma membrane, Golgi | TLR2, TLR4, TLR6 | FSL1, LTA, oxLDL, amyloid-β fibrils | 23, 26, 99–101 | |
| CD14 |
|
Secreted, Plasma membrane (GPI-linked), Endolysosomes | TLR2, TLR3, TLR4, TLR7, TLR8, TLR9 | LPS, PGN, Pam3CSK4, Poly(I:C), CpG DNA | 14, 23, 29–31, 102, 103 | |
| TRIL |
|
Plasma Membrane, Early Endosomes | TLR3, TLR4 | LPS | 41, 42 | |
| Progranulin |
|
Secreted, Endolysosomes | TLR9 | CpG-A, CpG-B, CpG-C, and inhibitory ODNs | 45 | |
| HMGB1 |
|
Nucleus, cytoplasm, can be secreted upon TLR ligation | TLR9, possibly TLR3 and TLR7 | CpG-A, CpG- B, DNA and RNA | 48, 49, 53 | |
| LL37 |
|
Early endosomes | TLR9 | Mammalian DNA | 54 | |
| Chaperones | Gp96 |
|
ER | TLR1, TLR2, TLR4, TLR9 | None demonstrated | 58, 60, 62, 64 |
| PRAT4A |

|
ER | TLR1, TLR2, TLR4, TLR9 | None demonstrated | 64–67 | |
| Molecules that facilitate trafficking of endosomal TLRs | UNC93B1 |
|
ER, Endolysosomes | TLR3, TLR7, TLR8, TLR9, TLR13 | None demonstrated | 68, 70, 71 |
| AP3 |
|
TGN, Endolysosomes, LROs | TLR9 | None demonstrated | 75 | |
| TLR processing enzymes | Cathepsins |

|
Endosomes, lysosomes | TLR9, possibly TLR3 and TLR7 | None demonstrated | 79, 80, 84 |
| AEP |

|
Endosomes, lysosomes | TLR9, possibly TLR3 and TLR7 | None demonstrated | 83, 84 |
AEP, asparagine endopeptidase; AP3, adaptor protein 3; ER, endoplasmic reticulum; FSL1, S-(2,3-bispalmitoyloxypropyl)-CGDPKHSPKSF; GPI, glycosylphosphatidylinositol; HMGB1, high-mobility group box 1 protein; LBP, LPS binding protein; LPS, lipopolysaccharide; LRO, lysosomes-related organelles; LTA, lipoteichoic acid; ODN, oligodeoxynucleotide; oxLDL, oxidized low-density lipid; PGN, peptidoglycan; polyI:C, polyinosinic–polycytidylic acid; PRAT4A, protein associated with TLR4; TGN, trans-golgi network; TLR, Toll-like receptor; UNC93B1, uncoordinated 93B1.
The involvement of UNC93B1 in the trafficking of signaling competent TLR3, TLR7 and TLR9 and how this polytopic protein regulates interaction with its client TLRs is incompletely understood. The emerging picture is that UNC93B1 function is more complex than that of merely serving as a delivery platform for endosomal TLRs. Much remains to be learnt about the assembly of UNC93B1–TLR complexes in the ER, their exit sites and targeting to organelles for proper TLR function. Trafficking factors such as AP3 are important for compartment-specific TLR signaling regulation. But are all cells subject to this differential TLR distribution? With the discovery of AP3 as a cell- and compartment-specific cofactor, together with the recent identification of viperin as a possible TLR7 and TLR9 adaptor signaling molecule specific for production of IFNα 85, this area of TLR biology is coming into focus.
The ability of surface TLRs to sense a wide variety of diverse ligands contrasts with the restricted specificity of endosomal TLRs (Table 1). Is this due to the presence of more surface TLR cofactors for ligand discrimination or to the variety of extracellular PAMPs found in nature? The more restricted pattern of endosomal TLR ligand recognition likely evolved as an adaptation to prevent recognition of self-nucleic acids that mimic those of microbial origin. The use of cofactors for ligand recognition makes a special case for TLR7 and TLR8: their activation by small molecules such as the imidazoquinolines is not easily reconciled with the mode of natural ligand binding to other TLRs, and the existance of a TLR7- or TLR8-associated cofactor would be an obvious solution to this conundrum. Importantly, how ubiquitous cofactors, such as granulin and HMGB1, which can potentially carry pro-inflammatory endogenous nucleic acids, enter cells and reach TLR-containing compartments remains to be determined.
Conclusions and future perspectives
The TLR field has rapidly evolved from the initial discovery of receptors that recognize widely different PAMPs, yet display structurally conserved ectodomains. Many molecules that contribute to ligand discrimination and receptor signaling have been identified, and such molecules display different functions: cofactors, signaling adaptors and molecules, and regulators of TLR function. The final result of TLR specificity and activation must stem from a combination of such mediators, resulting in complex signaling platforms.
Because of their contributions to TLR function, the study of cofactors that help activate TLRs yields the obvious dividend of a better understanding of TLR pathways that control innate and adaptive immunity. Whether such knowledge can be applied to devise new therapies is impossible to gauge, but additional means of manipulating TLRs remains a highly desirable goal. We may thus anticipate important advances in our understanding of the roles of TLR accessory proteins.
Glossary terms
- Alarmins
Endogenous mediators that are released by necrotic cells in response to infection or injury and that interact with PRRs to activate innate immune cells
- Acute-phase proteins
A group of proteins, including C-reactive protein, serum amyloid A, fibrinogen and α1-acid glycoprotein, that are secreted into the blood in increased or decreased quantities by hepatocytes in response to trauma, inflammation or disease. These proteins can be inhibitors or mediators of inflammatory processes
- Lipid raft
Structures that are proposed to arise from phase separation of different plasma membrane lipids as a result of the selective coalescence of certain lipids on the basis of their physical properties. This results in the formation of distinct and stable lipid domains in membranes that might provide a platform for membrane-associated protein organization
- Sterile inflammation
Inflammatory response triggered by tissue damage in the absence of infection
- Amphipathic peptides
Peptides containing hydrophilic and hydrophobic domains that allows them to interact both with charged residues and lipophilic structures
- Endosomes
Vesicles of the endocytic pathway that transport proteins from the plasma membrane and the Golgi compartment and contain mild acidic pH
- Paralogue
A homologous gene that resulted from a gene duplication event
- Short hairpin RNA
One of the two most common forms of short (usually 21-base-pairs long) double-stranded RNAs used for gene silencing. The other form is known as small interfering RNA (siRNA)
- Lysosomes
Organelles involved in protein degradation that are low in pH and correspond to the last step of the endocytic pathway
- Lysosome-related organelles (LROs)
Cell-specific compartments that share properties with lysosomes but have specialized functions. LROs include melanosomes, lytic granules, MHC class II compartments, platelet-dense granules, basophil granules and azurophil granules
References
- 1.Janeway CA, Jr, Medzhitov R. Introduction: the role of innate immunity in the adaptive immune response. Semin Immunol. 1998;10:349–350. doi: 10.1006/smim.1998.0142. [DOI] [PubMed] [Google Scholar]
- 2.Bianchi ME. DAMPs, PAMPs and alarmins: all we need to know about danger. J Leukoc Biol. 2007;81:1–5. doi: 10.1189/jlb.0306164. [DOI] [PubMed] [Google Scholar]
- 3.Kang JY, Lee JO. Structural biology of the toll-like receptor family. Annu Rev Biochem. 2011;80:917–941. doi: 10.1146/annurev-biochem-052909-141507. [DOI] [PubMed] [Google Scholar]
- 4.Kawai T, Akira S. The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol. 2010;11:373–384. doi: 10.1038/ni.1863. [DOI] [PubMed] [Google Scholar]
- 5.O’Neill LA, Bowie AG. The family of five: TIR-domain-containing adaptors in Toll-like receptor signalling. Nat Rev Immunol. 2007;7:353–364. doi: 10.1038/nri2079. [DOI] [PubMed] [Google Scholar]
- 6.Trinchieri G, Sher A. Cooperation of Toll-like receptor signals in innate immune defence. Nat Rev Immunol. 2007;7:179–190. doi: 10.1038/nri2038. [DOI] [PubMed] [Google Scholar]
- 7.Liew FY, Xu D, Brint EK, O’Neill LA. Negative regulation of toll-like receptor-mediated immune responses. Nat Rev Immunol. 2005;5:446–458. doi: 10.1038/nri1630. [DOI] [PubMed] [Google Scholar]
- 8.Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140:821–832. doi: 10.1016/j.cell.2010.01.040. [DOI] [PubMed] [Google Scholar]
- 9.Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell. 2010;140:805–820. doi: 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 10.Ting JP, Duncan JA, Lei Y. How the noninflammasome NLRs function in the innate immune system. Science. 2010;327:286–290. doi: 10.1126/science.1184004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tschopp J, Schroder K. NLRP3 inflammasome activation: The convergence of multiple signalling pathways on ROS production? Nat Rev Immunol. 2010;10:210–215. doi: 10.1038/nri2725. [DOI] [PubMed] [Google Scholar]
- 12.Ulevitch RJ, Tobias PS. Receptor-dependent mechanisms of cell stimulation by bacterial endotoxin. Annu Rev Immunol. 1995;13:437–457. doi: 10.1146/annurev.iy.13.040195.002253. [DOI] [PubMed] [Google Scholar]
- 13.Jiang Z, et al. CD14 is required for MyD88-independent LPS signaling. Nat Immunol. 2005;6:565–570. doi: 10.1038/ni1207. [DOI] [PubMed] [Google Scholar]
- 14.Dziarski R, Tapping RI, Tobias PS. Binding of bacterial peptidoglycan to CD14. J Biol Chem. 1998;273:8680–8690. doi: 10.1074/jbc.273.15.8680. [DOI] [PubMed] [Google Scholar]
- 15.Schroder NW, et al. Lipopolysaccharide binding protein binds to triacylated and diacylated lipopeptides and mediates innate immune responses. J Immunol. 2004;173:2683–2691. doi: 10.4049/jimmunol.173.4.2683. [DOI] [PubMed] [Google Scholar]
- 16.Schroder NW, et al. Lipoteichoic acid (LTA) of Streptococcus pneumoniae and Staphylococcus aureus activates immune cells via Toll-like receptor (TLR)-2, lipopolysaccharide-binding protein (LBP), and CD14, whereas TLR-4 and MD-2 are not involved. J Biol Chem. 2003;278:15587–15594. doi: 10.1074/jbc.M212829200. [DOI] [PubMed] [Google Scholar]
- 17.Jack RS, et al. Lipopolysaccharide-binding protein is required to combat a murine gram-negative bacterial infection. Nature. 1997;389:742–745. doi: 10.1038/39622. [DOI] [PubMed] [Google Scholar]
- 18.Weber JR, et al. Recognition of pneumococcal peptidoglycan: an expanded, pivotal role for LPS binding protein. Immunity. 2003;19:269–279. doi: 10.1016/s1074-7613(03)00205-x. [DOI] [PubMed] [Google Scholar]
- 19.Shimazu R, et al. MD-2, a molecule that confers lipopolysaccharide responsiveness on Toll-like receptor 4. J Exp Med. 1999;189:1777–1782. doi: 10.1084/jem.189.11.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nagai Y, et al. Essential role of MD-2 in LPS responsiveness and TLR4 distribution. Nat Immunol. 2002;3:667–672. doi: 10.1038/ni809. [DOI] [PubMed] [Google Scholar]
- 21.Park BS, et al. The structural basis of lipopolysaccharide recognition by the TLR4-MD-2 complex. Nature. 2009;458:1191–1195. doi: 10.1038/nature07830. This paper is the first description of the crystal structure of a TLR in complex with its ligand and an accessory molecule. [DOI] [PubMed] [Google Scholar]
- 22.Calvo D, Dopazo J, Vega MA. The CD36, CLA-1 (CD36L1), and LIMPII (CD36L2) gene family: cellular distribution, chromosomal location, and genetic evolution. Genomics. 1995;25:100–106. doi: 10.1016/0888-7543(95)80114-2. [DOI] [PubMed] [Google Scholar]
- 23.Triantafilou M, et al. Membrane sorting of toll-like receptor (TLR)-2/6 and TLR2/1 heterodimers at the cell surface determines heterotypic associations with CD36 and intracellular targeting. J Biol Chem. 2006;281:31002–31011. doi: 10.1074/jbc.M602794200. [DOI] [PubMed] [Google Scholar]
- 24.Hoebe K, et al. CD36 is a sensor of diacylglycerides. Nature. 2005;433:523–527. doi: 10.1038/nature03253. [DOI] [PubMed] [Google Scholar]
- 25.Stuart LM, et al. Response to Staphylococcus aureus requires CD36-mediated phagocytosis triggered by the COOH-terminal cytoplasmic domain. J Cell Biol. 2005;170:477–485. doi: 10.1083/jcb.200501113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Stewart CR, et al. CD36 ligands promote sterile inflammation through assembly of a Toll-like receptor 4 and 6 heterodimer. Nat Immunol. 2010;11:155–161. doi: 10.1038/ni.1836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tao N, Wagner SJ, Lublin DM. CD36 is palmitoylated on both N- and C-terminal cytoplasmic tails. J Biol Chem. 1996;271:22315–22320. doi: 10.1074/jbc.271.37.22315. [DOI] [PubMed] [Google Scholar]
- 28.Baumann CL, et al. CD14 is a coreceptor of Toll-like receptors 7 and 9. J Exp Med. 2010;207:2689–2701. doi: 10.1084/jem.20101111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hailman E, et al. Lipopolysaccharide (LPS)-binding protein accelerates the binding of LPS to CD14. J Exp Med. 1994;179:269–277. doi: 10.1084/jem.179.1.269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee HK, Dunzendorfer S, Soldau K, Tobias PS. Double-stranded RNA-mediated TLR3 activation is enhanced by CD14. Immunity. 2006;24:153–163. doi: 10.1016/j.immuni.2005.12.012. This paper provided the first concrete evidence that CD14 is important for endosomal TLRs in addition to surface TLRs. [DOI] [PubMed] [Google Scholar]
- 31.Nakata T, et al. CD14 directly binds to triacylated lipopeptides and facilitates recognition of the lipopeptides by the receptor complex of Toll-like receptors 2 and 1 without binding to the complex. Cell Microbiol. 2006;8:1899–1909. doi: 10.1111/j.1462-5822.2006.00756.x. [DOI] [PubMed] [Google Scholar]
- 32.Kim JI, et al. Crystal structure of CD14 and its implications for lipopolysaccharide signaling. J Biol Chem. 2005;280:11347–11351. doi: 10.1074/jbc.M414607200. [DOI] [PubMed] [Google Scholar]
- 33.Haziot A, et al. Resistance to endotoxin shock and reduced dissemination of gram-negative bacteria in CD14-deficient mice. Immunity. 1996;4:407–414. doi: 10.1016/s1074-7613(00)80254-x. [DOI] [PubMed] [Google Scholar]
- 34.Zanoni I, et al. CD14 Controls the LPS-Induced Endocytosis of Toll-like Receptor 4. Cell. 2011;147:868–880. doi: 10.1016/j.cell.2011.09.051. First description of LPS-induced TLR4 trafficking by CD14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Akashi-Takamura S, Miyake K. TLR accessory molecules. Curr Opin Immunol. 2008;20:420–425. doi: 10.1016/j.coi.2008.07.001. [DOI] [PubMed] [Google Scholar]
- 36.da Silva Correia J, Soldau K, Christen U, Tobias PS, Ulevitch RJ. Lipopolysaccharide is in close proximity to each of the proteins in its membrane receptor complex. transfer from CD14 to TLR4 and MD-2. J Biol Chem. 2001;276:21129–21135. doi: 10.1074/jbc.M009164200. [DOI] [PubMed] [Google Scholar]
- 37.Gioannini TL, et al. Isolation of an endotoxin-MD-2 complex that produces Toll-like receptor 4-dependent cell activation at picomolar concentrations. Proc Natl Acad Sci U S A. 2004;101:4186–4191. doi: 10.1073/pnas.0306906101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Compton T, et al. Human cytomegalovirus activates inflammatory cytokine responses via CD14 and Toll-like receptor 2. J Virol. 2003;77:4588–4596. doi: 10.1128/JVI.77.8.4588-4596.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Georgel P, et al. Vesicular stomatitis virus glycoprotein G activates a specific antiviral Toll-like receptor 4-dependent pathway. Virology. 2007;362:304–313. doi: 10.1016/j.virol.2006.12.032. [DOI] [PubMed] [Google Scholar]
- 40.Kurt-Jones EA, et al. Pattern recognition receptors TLR4 and CD14 mediate response to respiratory syncytial virus. Nat Immunol. 2000;1:398–401. doi: 10.1038/80833. [DOI] [PubMed] [Google Scholar]
- 41.Carpenter S, et al. TRIL, a functional component of the TLR4 signaling complex, highly expressed in brain. J Immunol. 2009;183:3989–3995. doi: 10.4049/jimmunol.0901518. [DOI] [PubMed] [Google Scholar]
- 42.Carpenter S, Wochal P, Dunne A, O’Neill LA. Toll-like Receptor 3 (TLR3) Signaling Requires TLR4 Interactor with Leucine-rich Repeats (TRIL) J Biol Chem. 2011;286:38795–38804. doi: 10.1074/jbc.M111.255893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kessenbrock K, et al. Proteinase 3 and neutrophil elastase enhance inflammation in mice by inactivating antiinflammatory progranulin. J Clin Invest. 2008;118:2438–2447. doi: 10.1172/JCI34694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhu J, et al. Conversion of proepithelin to epithelins: roles of SLPI and elastase in host defense and wound repair. Cell. 2002;111:867–878. doi: 10.1016/s0092-8674(02)01141-8. [DOI] [PubMed] [Google Scholar]
- 45.Park B, et al. Granulin is a soluble cofactor for toll-like receptor 9 signaling. Immunity. 2011;34:505–513. doi: 10.1016/j.immuni.2011.01.018. Here granulin was shown to have an essential role in ligand delivery for TLR9 activation. [DOI] [PubMed] [Google Scholar]
- 46.Lotze MT, Tracey KJ. High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nat Rev Immunol. 2005;5:331–342. doi: 10.1038/nri1594. [DOI] [PubMed] [Google Scholar]
- 47.Andersson U, Tracey KJ. HMGB1 is a therapeutic target for sterile inflammation and infection. Annu Rev Immunol. 2011;29:139–162. doi: 10.1146/annurev-immunol-030409-101323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ivanov S, et al. A novel role for HMGB1 in TLR9-mediated inflammatory responses to CpG-DNA. Blood. 2007;110:1970–1981. doi: 10.1182/blood-2006-09-044776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tian J, et al. Toll-like receptor 9-dependent activation by DNA-containing immune complexes is mediated by HMGB1 and RAGE. Nat Immunol. 2007;8:487–496. doi: 10.1038/ni1457. This work first reported a role for HMGB1 in delivering DNA to TLR9. [DOI] [PubMed] [Google Scholar]
- 50.Urbonaviciute V, et al. Induction of inflammatory and immune responses by HMGB1-nucleosome complexes: implications for the pathogenesis of SLE. J Exp Med. 2008;205:3007–3018. doi: 10.1084/jem.20081165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Leadbetter EA, et al. Chromatin-IgG complexes activate B cells by dual engagement of IgM and Toll-like receptors. Nature. 2002;416:603–607. doi: 10.1038/416603a. [DOI] [PubMed] [Google Scholar]
- 52.Avalos AM, et al. RAGE-independent autoreactive B cell activation in response to chromatin and HMGB1/DNA immune complexes. Autoimmunity. 2010;43:103–110. doi: 10.3109/08916930903384591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yanai H, et al. HMGB proteins function as universal sentinels for nucleic-acid-mediated innate immune responses. Nature. 2009;462:99–103. doi: 10.1038/nature08512. [DOI] [PubMed] [Google Scholar]
- 54.Lande R, et al. Plasmacytoid dendritic cells sense self-DNA coupled with antimicrobial peptide. Nature. 2007;449:564–569. doi: 10.1038/nature06116. This paper demonstrated the role of LL37 in DNA delivery to TLR9 in pDCs. [DOI] [PubMed] [Google Scholar]
- 55.Gilliet M, Lande R. Antimicrobial peptides and self-DNA in autoimmune skin inflammation. Curr Opin Immunol. 2008;20:401–407. doi: 10.1016/j.coi.2008.06.008. [DOI] [PubMed] [Google Scholar]
- 56.Zanetti M, Gennaro R, Romeo D. The cathelicidin family of antimicrobial peptide precursors: a component of the oxygen-independent defense mechanisms of neutrophils. Ann N Y Acad Sci. 1997;832:147–162. doi: 10.1111/j.1749-6632.1997.tb46244.x. [DOI] [PubMed] [Google Scholar]
- 57.Ganguly D, et al. Self-RNA-antimicrobial peptide complexes activate human dendritic cells through TLR7 and TLR8. J Exp Med. 2009;206:1983–1994. doi: 10.1084/jem.20090480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yang Y, Li Z. Roles of heat shock protein gp96 in the ER quality control: redundant or unique function? Mol Cells. 2005;20:173–182. [PubMed] [Google Scholar]
- 59.Liu B, Li Z. Endoplasmic reticulum HSP90b1 (gp96, grp94) optimizes B-cell function via chaperoning integrin and TLR but not immunoglobulin. Blood. 2008;112:1223–1230. doi: 10.1182/blood-2008-03-143107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Randow F, Seed B. Endoplasmic reticulum chaperone gp96 is required for innate immunity but not cell viability. Nat Cell Biol. 2001;3:891–896. doi: 10.1038/ncb1001-891. [DOI] [PubMed] [Google Scholar]
- 61.Wanderling S, et al. GRP94 is essential for mesoderm induction and muscle development because it regulates insulin-like growth factor secretion. Mol Biol Cell. 2007;18:3764–3775. doi: 10.1091/mbc.E07-03-0275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yang Y, et al. Heat shock protein gp96 is a master chaperone for toll-like receptors and is important in the innate function of macrophages. Immunity. 2007;26:215–226. doi: 10.1016/j.immuni.2006.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Staron M, et al. Heat-shock protein gp96/grp94 is an essential chaperone for the platelet glycoprotein Ib-IX-V complex. Blood. 2011;117:7136–7144. doi: 10.1182/blood-2011-01-330464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Liu B, et al. Folding of Toll-like receptors by the HSP90 paralogue gp96 requires a substrate-specific cochaperone. Nat Commun. 2010;1:79. doi: 10.1038/ncomms1070. This paper shows that gp96 and PRAT4A work together to chaperone TLRs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wakabayashi Y, et al. A protein associated with toll-like receptor 4 (PRAT4A) regulates cell surface expression of TLR4. J Immunol. 2006;177:1772–1779. doi: 10.4049/jimmunol.177.3.1772. [DOI] [PubMed] [Google Scholar]
- 66.Takahashi K, et al. A protein associated with Toll-like receptor (TLR) 4 (PRAT4A) is required for TLR-dependent immune responses. J Exp Med. 2007;204:2963–2976. doi: 10.1084/jem.20071132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kiyokawa T, et al. A single base mutation in the PRAT4A gene reveals differential interaction of PRAT4A with Toll-like receptors. Int Immunol. 2008;20:1407–1415. doi: 10.1093/intimm/dxn098. [DOI] [PubMed] [Google Scholar]
- 68.Tabeta K, et al. The Unc93b1 mutation 3d disrupts exogenous antigen presentation and signaling via Toll-like receptors 3, 7 and 9. Nat Immunol. 2006;7:156–164. doi: 10.1038/ni1297. [DOI] [PubMed] [Google Scholar]
- 69.Casrouge A, et al. Herpes simplex virus encephalitis in human UNC-93B deficiency. Science. 2006;314:308–312. doi: 10.1126/science.1128346. [DOI] [PubMed] [Google Scholar]
- 70.Brinkmann MM, et al. The interaction between the ER membrane protein UNC93B and TLR3, 7, and 9 is crucial for TLR signaling. J Cell Biol. 2007;177:265–275. doi: 10.1083/jcb.200612056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kim YM, Brinkmann MM, Paquet ME, Ploegh HL. UNC93B1 delivers nucleotide-sensing toll-like receptors to endolysosomes. Nature. 2008;452:234–238. doi: 10.1038/nature06726. This paper provides evidence for the role of UNC93B1 in the trafficking of endosomal TLRs. [DOI] [PubMed] [Google Scholar]
- 72.Fukui R, et al. Unc93B1 biases Toll-like receptor responses to nucleic acid in dendritic cells toward DNA- but against RNA-sensing. J Exp Med. 2009;206:1339–1350. doi: 10.1084/jem.20082316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fukui R, et al. Unc93B1 restricts systemic lethal inflammation by orchestrating Toll-like receptor 7 and 9 trafficking. Immunity. 2011;35:69–81. doi: 10.1016/j.immuni.2011.05.010. [DOI] [PubMed] [Google Scholar]
- 74.Nakatsu F, Ohno H. Adaptor protein complexes as the key regulators of protein sorting in the post-Golgi network. Cell Struct Funct. 2003;28:419–429. doi: 10.1247/csf.28.419. [DOI] [PubMed] [Google Scholar]
- 75.Sasai M, Linehan MM, Iwasaki A. Bifurcation of Toll-like receptor 9 signaling by adaptor protein 3. Science. 2010;329:1530–1534. doi: 10.1126/science.1187029. This paper first demonstrated that AP3 has a role in TLR9-dependent IFN responses. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Blasius AL, et al. Slc15a4, AP-3, and Hermansky-Pudlak syndrome proteins are required for Toll-like receptor signaling in plasmacytoid dendritic cells. Proc Natl Acad Sci U S A. 2010;107:19973–19978. doi: 10.1073/pnas.1014051107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Guiducci C, et al. Properties regulating the nature of the plasmacytoid dendritic cell response to Toll-like receptor 9 activation. J Exp Med. 2006;203:1999–2008. doi: 10.1084/jem.20060401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Honda K, et al. Spatiotemporal regulation of MyD88-IRF-7 signalling for robust type-I interferon induction. Nature. 2005;434:1035–1040. doi: 10.1038/nature03547. [DOI] [PubMed] [Google Scholar]
- 79.Ewald SE, et al. The ectodomain of Toll-like receptor 9 is cleaved to generate a functional receptor. Nature. 2008;456:658–662. doi: 10.1038/nature07405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Park B, et al. Proteolytic cleavage in an endolysosomal compartment is required for activation of Toll-like receptor 9. Nat Immunol. 2008;9:1407–1414. doi: 10.1038/ni.1669. References 79 and 80 demonstrate the cleavage requirement for TLR9 activation by CpG. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Asagiri M, et al. Cathepsin K-dependent toll-like receptor 9 signaling revealed in experimental arthritis. Science. 2008;319:624–627. doi: 10.1126/science.1150110. [DOI] [PubMed] [Google Scholar]
- 82.Matsumoto F, et al. Cathepsins are required for Toll-like receptor 9 responses. Biochem Biophys Res Commun. 2008;367:693–699. doi: 10.1016/j.bbrc.2007.12.130. [DOI] [PubMed] [Google Scholar]
- 83.Sepulveda FE, et al. Critical role for asparagine endopeptidase in endocytic Toll-like receptor signaling in dendritic cells. Immunity. 2009;31:737–748. doi: 10.1016/j.immuni.2009.09.013. This is the first demonstration of a role for asparagine endopeptidase (AEP) in TLR9 activation in primary DCs. [DOI] [PubMed] [Google Scholar]
- 84.Ewald SE, et al. Nucleic acid recognition by Toll-like receptors is coupled to stepwise processing by cathepsins and asparagine endopeptidase. J Exp Med. 2011 doi: 10.1084/jem.20100682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Saitoh T, et al. Antiviral protein Viperin promotes Toll-like receptor 7- and Toll-like receptor 9-mediated type I interferon production in plasmacytoid dendritic cells. Immunity. 34:352–363. doi: 10.1016/j.immuni.2011.03.010. [DOI] [PubMed] [Google Scholar]
- 86.Gerold G, et al. A Toll-like receptor 2-integrin beta3 complex senses bacterial lipopeptides via vitronectin. Nat Immunol. 2008;9:761–768. doi: 10.1038/ni.1618. [DOI] [PubMed] [Google Scholar]
- 87.Kagan JC, Medzhitov R. Phosphoinositide-mediated adaptor recruitment controls Toll-like receptor signaling. Cell. 2006;125:943–955. doi: 10.1016/j.cell.2006.03.047. [DOI] [PubMed] [Google Scholar]
- 88.Gantner BN, Simmons RM, Canavera SJ, Akira S, Underhill DM. Collaborative induction of inflammatory responses by dectin-1 and Toll-like receptor 2. J Exp Med. 2003;197:1107–1117. doi: 10.1084/jem.20021787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Nagai Y, et al. The radioprotective 105/MD-1 complex links TLR2 and TLR4/MD-2 in antibody response to microbial membranes. J Immunol. 2005;174:7043–7049. doi: 10.4049/jimmunol.174.11.7043. [DOI] [PubMed] [Google Scholar]
- 90.Divanovic S, et al. Negative regulation of Toll-like receptor 4 signaling by the Toll-like receptor homolog RP105. Nat Immunol. 2005;6:571–578. doi: 10.1038/ni1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Blumenthal A, et al. RP105 facilitates macrophage activation by Mycobacterium tuberculosis lipoproteins. Cell Host Microbe. 2009;5:35–46. doi: 10.1016/j.chom.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Uccellini MB, et al. Autoreactive B cells discriminate CpG-rich and CpG-poor DNA and this response is modulated by IFN-alpha. J Immunol. 2008;181:5875–5884. doi: 10.4049/jimmunol.181.9.5875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Viglianti GA, et al. Activation of autoreactive B cells by CpG dsDNA. Immunity. 2003;19:837–847. doi: 10.1016/s1074-7613(03)00323-6. [DOI] [PubMed] [Google Scholar]
- 94.Lau CM, et al. RNA-associated autoantigens activate B cells by combined B cell antigen receptor/Toll-like receptor 7 engagement. J Exp Med. 2005;202:1171–1177. doi: 10.1084/jem.20050630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Chaturvedi A, Dorward D, Pierce SK. The B cell receptor governs the subcellular location of Toll-like receptor 9 leading to hyperresponses to DNA-containing antigens. Immunity. 2008;28:799–809. doi: 10.1016/j.immuni.2008.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Avalos AM, et al. Differential cytokine production and bystander activation of autoreactive B cells in response to CpG-A and CpG-B oligonucleotides. J Immunol. 2009;183:6262–6268. doi: 10.4049/jimmunol.0901941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hori O, et al. The receptor for advanced glycation end products (RAGE) is a cellular binding site for amphoterin. Mediation of neurite outgrowth and co-expression of rage and amphoterin in the developing nervous system. J Biol Chem. 1995;270:25752–25761. doi: 10.1074/jbc.270.43.25752. [DOI] [PubMed] [Google Scholar]
- 98.Bierhaus A, et al. Understanding RAGE, the receptor for advanced glycation end products. J Mol Med. 2005;83:876–886. doi: 10.1007/s00109-005-0688-7. [DOI] [PubMed] [Google Scholar]
- 99.Boullier A, et al. The binding of oxidized low density lipoprotein to mouse CD36 is mediated in part by oxidized phospholipids that are associated with both the lipid and protein moieties of the lipoprotein. J Biol Chem. 2000;275:9163–9169. doi: 10.1074/jbc.275.13.9163. [DOI] [PubMed] [Google Scholar]
- 100.Coraci IS, et al. CD36, a class B scavenger receptor, is expressed on microglia in Alzheimer’s disease brains and can mediate production of reactive oxygen species in response to beta-amyloid fibrils. Am J Pathol. 2002;160:101–112. doi: 10.1016/s0002-9440(10)64354-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Jimenez-Dalmaroni MJ, et al. Soluble CD36 ectodomain binds negatively charged diacylglycerol ligands and acts as a co-receptor for TLR2. PLoS One. 2009;4:e7411. doi: 10.1371/journal.pone.0007411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Yang RB, Mark MR, Gurney AL, Godowski PJ. Signaling events induced by lipopolysaccharide-activated toll-like receptor 2. J Immunol. 1999;163:639–643. [PubMed] [Google Scholar]
- 103.Muta T, Takeshige K. Essential roles of CD14 and lipopolysaccharide-binding protein for activation of toll-like receptor (TLR)2 as well as TLR4 Reconstitution of TLR2- and TLR4-activation by distinguishable ligands in LPS preparations. Eur J Biochem. 2001;268:4580–4589. doi: 10.1046/j.1432-1327.2001.02385.x. [DOI] [PubMed] [Google Scholar]