

Abstract
A structure-activity relationship of the 3- and 6-positions of the pyrazolo[1,5-a]pyrimidine scaffold of the known BMP inhibitors dorsomorphin, 1, LDN193189, 2, and DMH1, 3, led to the identification of a potent and selective compound for ALK2 versus ALK3. The potency contributions of several 3-position substituents were evaluated with subtle structural changes leading to significant changes in potency. From these studies, a novel 5-quinoline molecule was identified and designated an MLPCN probe molecule, ML347, which shows >300-fold selectivity for ALK2 and presents the community with a selective molecular probe for further biological evaluation.
Keywords: ALK2 kinase; Bone morphogenetic receptor; Pyrazolo[1,5-a]pyrimidine; Selectivity; ML347
The bone morphogenetic protein (BMP) signaling pathway plays critical, diverse roles in embryonic pattern formation, and a number of disease processes.1 BMP ligands bind and activate the type-I and type-II BMP receptors, a family of serine-threonine kinases belonging to the TGF-β receptor superfamily, which then activate downstream mediators Smad1/5/8 by phosphorylation.2 Activated Smad1/5/8 translocate to the nucleus to turn on BMP target genes. Because there are more than 20 distinct BMP ligands, a number of extracellular antagonists, three type-II receptors (BMP type II receptor, BMPRII; Activin type II receptor; ActRIIa and ActIIb), and four BMP type-I receptors (activin-receptor like kinase 1, ALK1; ALK2, ALK3 and ALK6), the role of targeting an individual component in various signaling contexts is unclear.
Recently, in a chemical genetic screen for compounds that perturb zebrafish embryonic axis, we discovered dorsomorphin (DM), 1, the first small molecule inhibitor of the BMP pathway which directly targets the type-I receptor.3 DM, 1, and its analog LDN-193189, 2, have been instrumental in demonstrating the therapeutic potential of BMP inhibitors for anemia, Duchenne Muscular Dystrophy, atherosclerosis and heterotopic ossification syndromes.1,4 However, the early generation compounds DM, 1, LDN-193189, 2, and DMH1, 35, do not discriminate between ALK1, ALK2, ALK3 and ALK6 (Figure 1)5, and long-term consequences of pharmacological inhibition of all BMP signals are unknown. The issue of subtype selectivity is particularly germane to fibrodysplasia ossificans progressiva (FOP), a rare congenital disease of progressive soft tissue ossification, since it is caused by dysregulated BMP signaling due to a highly recurrent mutation (R206H) in ALK2.6,7 Although LDN-193189, 2, could blunt ectopic ossification in a mouse model expressing a constitutively active form of ALK2 (Q207D)8, inhibitors with greater subtype selectivity might be more desirable as lead compounds for FOP. Such inhibitors might also be useful chemical probes to interrogate the biology of BMP signaling at the subtype resolution. Moreover, because DM, 1, and LDN-193189, 2, have important off-target effects, including AMP-activated kinase (AMPK), platelet-derived growth factor receptor-β (PDGFRβ) and vascular endothelial growth factor type-II receptor (VEGFR-2/KDR), validity of some of their in vivo effects has been challenged.5,9 Therefore, we undertook a synthetic effort to develop compounds with greater subtype selectivity, centered around the central pyrazolo[1.5-a]pyrimidine scaffold which has culminated in the discovery of an ALK2 selective compound, ML347.
Figure 1.

Structures of previously disclosed BMP inhibitors, Dorsomorphin (DM), 1, LDN-193189, 2, and DMH1, 3.
The first round of SAR was designed by keeping the 6-(4-methoxyphenyl) moiety constant and varying the 3-position (R1). To that end, compounds 7a-m were synthesized as outlined in Scheme 1. Starting with the commercially available 2-(4-methoxyphenyl)malonaldehyde, 4, and condensing with 1H-pyrazol-5-amine, 5, under acidic conditions afforded the 6-(4-methoxyphenyl)pyrazolo[1,5-a]pyrimidine in 92% yield10, which was then iodinated (NIS, DMF) to give 6.11 The final compounds 7a-m were synthesized either by converting 6 to the boronate ester (diboronpinacol ester, Pd(dppf)Cl2*DCM, KOAc, DMF, 100 °C, 16 h) followed by Suzuki-Miyaura cross-coupling12 with the appropriate aryl halide (ArX, Pd(dppf)Cl2*DCM, K3PO4) or direct Suzuki-Miyaura cros-coupling with an appropriate aryl boronic acid (R1B(OH)2, Pd(dppf)Cl2*DCM, K3PO4).
Scheme 1.

Reactions and conditions: (a) AcOH, EtOH, 170 °C, 10 min., μW, 92%; (b) NIS, DMF; (c) diboronpinacol ester, Pd(dppf)Cl2·DCM, KOAc, DMF, 100 °C, 16 h; (d) R1X, Pd(dppf)Cl2·DCM, K3PO4, 1,4-dioxane, H2O, 120 °C, 30 min., μW, 17–27% (3 steps); (e) R1B(OH)2, Pd(dppf)Cl2·DCM, K3PO4, 1,4-dioxane, H2O, 120 °C, 30 min., μW, 10-65% (2 steps).
The SAR of the 3-position of the pyrazolo[1,5-a]pyrimidine scaffold is detailed in Table 1. Based on previous studies from our laboratories5, and others13, where it was shown that heterocycles with nitrogen in the 4-position were optimal, it was not surprising to see the substituted isoquinoline and 3- or 8-quinoline compounds inactive (7a-c, e, f). The 4-pyrazole compound (7d) was active (94.1 nM) as was the 5-quinoline (152 nM), which was not expected as this is the first compound without a nitrogen in the 4-position to show such potency. The most potent compound in the BMP4 cell assay was the 4-quinoline (7i, <1 nM), which is consistent with previous findings. Other nitrogen (7j and 7l) or sulfur compounds (7m) were inactive. However, subtle changes to the 3-postion substituent led to significant loss of activity. By changing the 4-quinoline (7i) to the 7-thieno[3,2-b]pyridine (7n) resulted in nearly complete erosion of activity (4571 nM) even though these compounds are similar in size and shape.
Table 1.
SAR of the 3-position of pyrazolo[1,5-a]pyrimidine scaffold (7a-m).
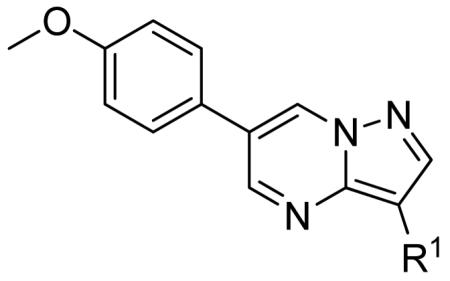
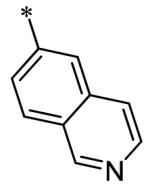
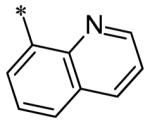
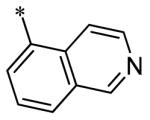
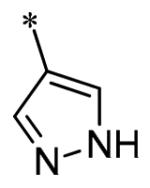
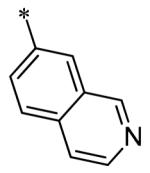
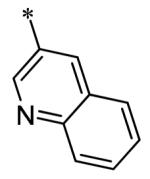
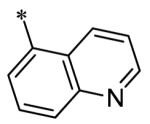
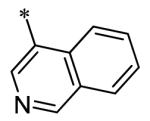
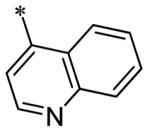
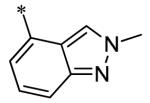
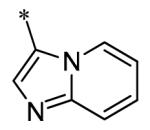
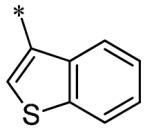
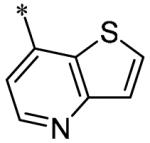
Having evaluated a number of 3-position substituents, we next looked at the 4-position of the 6-phenyl substituent. Previous results from our laboratory5 evaluated substituted alkyl chain substituents (such as those in 1 and 3) with much success and additional studies evaluated phenyl replacements (pyridinone16, unpublished results); however, any phenyl replacements led to inactive compounds. Thus, the 6-phenyl moiety has remained intact for this study. The synthesis of these analogs follows the outlined steps in Scheme 2. Starting with 2-bromomalonaldehyde, 8, and condensing with 1H-pyrazol-5-amine, 5, under acidic conditions led to 6-bromopyrazolo[1,5-a]pyrimidine, 9.10 Next, an appropriately substituted 4-phenylboronate ester was reacted with 9 under Suzuki-Miyaura cross-coupling conditions (Pd(dppf)Cl2*DCM, K3PO4) in good yields 56-73%. The resulting compound, 11, was then iodinated in the 3-position (NIS, DMF, 73%) and the final compounds (13a-r) were realized after a final Suzuki-Miyaura cross-coupling12 step with an appropriate boronic acid (R3B(OH)2, Pd(dppf)Cl2·DCM, K3PO4).
Scheme 2.

Reactions and conditions: (a) AcOH, EtOH, reflux; (b) Pd(dppf)Cl2·DCM, K3PO4, 1,4-dioxane, H2O, 150 °C, 30 min., μW, 56-73% (2 steps); (c) NIS, DMF, rt, 73%; (d) R3B(OH)2, Pd(dppf)Cl2·DCM, K3PO4, 1,4-dioxane, H2O, 120 °C, 30 min., μW, 14-52%.
The SAR of these compounds had two points of diversity on the molecule – with the 6-(4-phenyl)-position having molecules similar to that found in LDN193189, 2; namely six-membered heterocycloalkyl groups (piperazine in LDN-193189). The first R2 group evaluated was morpholine (13a-g) with the SAR tracking similarly to that seen in Table 1 and previously. Thus, the most active compounds were those containing heterocycles in the 4-position of the 3-substituent (4-pyrazole, 13a, 141 nM; 2-chloro-4-pyridine, 13c, 529 nM; 4-quinoline, 13d, <1.0 nM; 3-benzo[b]thiophene, 13g, 418 nM) (Table 2). These same R3 groups were active when the R2 group was changed to piperazine (13h-n) and 4-methylpiperazine (13o-r). In each case, the 4-quinoline or 4-pyrazole was the most potent of the R3 groups. Although the 3-benzo[b]thiophene was active in the morpholine groups, the activity significantly dropped off in the piperazine group (13n, 2636 nM) and was inactive in the 4-methylpiperazine group (13o). Notably, the 5-quinoline compound (13m) was equipotent to 7g. This SAR trends mimics that which was seen in our earlier work on this scaffold.5
Table 2.
SAR of the 6- and 3-position of the pyrazolo[1,5-a]pyrimidine scaffold (13a-r).
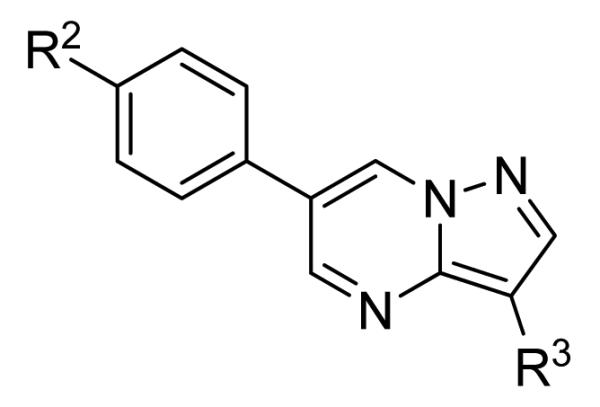
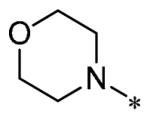
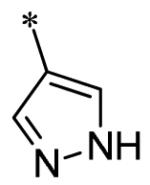
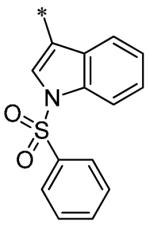
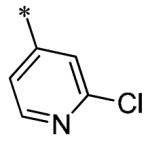
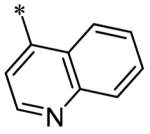
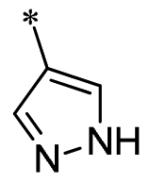
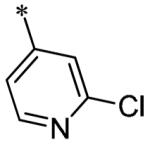
Having identified a number of potent inhibitors in the functional BMP4 cell based assay, which with certain structural classes can be difficult to interpret due to promiscuity, we next sent a number of compounds for kinase selectivity to Reaction Biology Corp. (Malvern, PA). We tested our active compounds against 10 kinases (Table 3) and each of the assays were run in 10-dose IC50 mode with 3-fold serial dilution starting at 100 μM. The reactions were carried out at 10 μM ATP. The three reference compounds have been previously run in these selectivity assays and are shown in Table 3. DM, 1, LDN-193189, 2, and DMH1, 3, are potent against ALK2 (ACVR1) however, all the compounds are equipotent or more potent against ALK3 (BMPR1A). In addition, these compounds have variable selectivity against the other kinases evaluated; however, DMH1, 3, shows the most selectivity within these three compounds. Across the board, all compounds tested were equipotent against ALK1 (ACVRL1) and ALK2 (ACVR1), however, there were two compounds identified that displayed selectivity against ALK3 (BMPR1A) and each compound contain a 5-quinoline R3 substituent (7g and 13m). The more potent (and selective) compound, 7g, has IC50’s of 46 and 32 nM, respectively, against ALK1 and ALK2; however, the IC50 against ALK3 is 10,800 nM, >300-fold selective over ALK3. In addition, 7g is completely inactive against all the other kinases tested (with weak activity against ALK6, 9830 nM and KDR (VEGFR2) 19,700 nM). It is interesting to note that it appears to be a combination of the 5-quinoline and 4-methoxyphenyl which gives rise to the selectivity profile, as 13m still retains significant ALK3 activity (539 nM). Due to the potency of 7g against the BMP4 cell assay, ALK1 and ALK2 and the significant selectivity against the other kinases, 7g, has been declared a probe molecule in the MLPCN and redesignated ML347.17
Table 3.
| Cmpd | IC50 (nM) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| ALK1/ ACVRL1 |
ALK2/ ACVR1 |
ALK3/ BMPR1A |
ALK4/ ACVR1B |
ALK5/ TGFBR1 |
ALK6/ BMPR1B |
BMPR2 | TGFBR2 | AMPK | KDR/ VEGFR2 |
|
| DM, 1 | 106.3 | 67.5 | 95 | 25740 | 17090 | 235 | 74 | 102.9 | 234.6 | 21.8 |
| LDN-193189, 2 | 13.3 | 40.7 | <5 | 1825 | 565 | 60 | 3845 | 140.4 | 1122 | 214.7 |
| DMH1, 3 | 27 | 107.9 | <5 | 9622 | Inactive | 47.6 | inactive | Inactive | Inactive | Inactive |
| 7d | 52.4 | 110 | 41 | Inactive | Inactive | 102 | 24 | 16 | 44 | 13 |
| 7g | 46 | 32 | 10800 | Inactive | Inactive | 9830 | Inactive | Inactive | Inactive | 19700 |
| 7i | 26.5 | 33.3 | 6.78 | 1840 | 33300 | 56.9 | Inactive | 236 | 7260 | 4300 |
| 13a | 42.1 | 53.4 | 10.4 | 5930 | 10900 | 60.9 | 15.9 | 1.37 | 39.9 | 1.68 |
| 13d | <5 | <5 | <5 | 326 | 178 | <5 | 3640 | 26 | 2680 | 5330 |
| 13h | 55 | 43 | 55 | 3420 | 3320 | 129 | 59 | 38 | 82 | 14 |
| 13m | 14.4 | 26.2 | 539 | 4090 | 3960 | 92.9 | 695 | 327 | 2520 | 2040 |
| 13r | <5 | <5 | <5 | 183 | 75 | <5 | 2360 | 15 | 960 | 1520 |
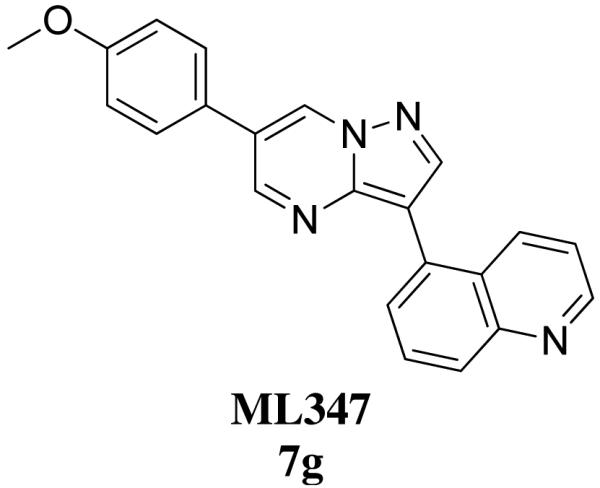
In order to further the BMP community as to the utility of ML347, we evaluated this molecule in our Tier 1 in vitro pharmacokinetic assays (Table 4). These studies are useful in order to evaluate the metabolic stability and predicted clearance in a number of species in order to inform on possible dosing routes. Utilizing rapid equilibrium dialysis, the protein binding of ML347 was determined in human, rat and mouse plasma. The results were similar in all three species with ML347 displaying high plasma protein biding (Fu ~0.01-0.015). ML347 was also assessed for its intrinsic clearance in hepatic microsomes. This measure will help predict the in vivo clearance in the same three species (CLHEP). ML347 was unstable to oxidative metabolism – possibly due to the labile methoxy group19 – and therefore was predicted to display high clearance in human and mouse, and moderate-to-high clearance in the rat. Going forward, the intrinsic clearance is predicting high clearance after oral dosing, a more appropriate dosing paradigm might be intraperitoneal dosing for this compound. Further in vivo experiments, including PK, will be reported in due course.
Table 4.
In vitro pharmacokinetic properties of ML347
| |||
|---|---|---|---|
| MW | 352.4 | ||
| cLogP | 4.00 | ||
| TPSA | 52.3 | ||
|
| |||
| In vitro PK Parameter | Human | Rat | Mouse |
|
| |||
| CLINT (mL/min/kg) | 516 | 148 | 617 |
| CLHEP (mL/min/kg) | 20.2 | 47.5 | 78.5 |
| PPB (% fu) | 0.9 | 1.4 | 1.4 |
|
| |||
| PBS Solubility | 4 μM | ||
In conclusion, SAR studies of the 3- and 6-positions of the pyrazolo[1,5-a]pyrimidine scaffold revealed a potent and selective inhibitor of ALK2 versus ALK3. These studies further validated that 4-phenyl substituents of the 6-position on the pyrazolo[1,5-a]pyrimidine scaffold allowed a wide range of substituents, from ethers to cycloheteroalkyl (morpholine, piperazine, 4-methylpiperazine). These studies also revealed that subtle changes of the 3-position substituents can drastically influence the BMP activity (e.g., 7i vs. 7n). These SAR studies culminated in the discovery of a highly selective ALK2 inhibitor, ML347, which shows >300-fold selectivity for ALK2 vs. ALK3. ML347 is potent in the BMP4 cell assay (152 nM) as well as the in vitro kinase assay for ALK1 (46 nM) and ALK2 (32 nM) and is devoid of activity in a number of related kinases. Further studies are planned for this selective inhibitor in a number of in vivo animal disease models, such as FOP, and results will be reported in due course.
Supplementary Material
ACKNOWLEDGMENTS
The authors would like to thank Katrina Brewer and Ryan Morrison for technical assistance with the PK experiments. Vanderbilt is a member of the MLPCN and houses the Vanderbilt Specialized Chemistry Center for Accelerated Probe Development. This work was generously supported by the NIH/MLPCN grant U54 MH084659 (C.W.L.), and VA Merit Award (C.C.H.), NIH R01HL104040 (CCH), and the Developmental Grants from the Center for Research in Fibrodysplasia Ossificans Progressiva and Related Disorders (C.R.H, C.C.H).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES AND NOTES
- 1.Hong CC, Yu PB. Cytokine & Growth Factor Rev. 2009;20:409. doi: 10.1016/j.cytogfr.2009.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Feng X-H, Derynck R. Ann. Rev. Cell. Dev. Biol. 2005;21:659. doi: 10.1146/annurev.cellbio.21.022404.142018. [DOI] [PubMed] [Google Scholar]
- 3.Yu PB, Hong CC, Sachidanandan C, Babitt JL, Deng DY, Hoyng SA, Lin HY, Bloch KD, Peterson RT. Nature Chem. Biol. 2008;4:33. doi: 10.1038/nchembio.2007.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.(a) Steinbicker AU, Bartnikas TB, Lohmeyer LK, Leyton P, Mayeur C, Kao SM, Pappas AE, Peterson RT, Bloch DB, Yu PB, Fleming MD, Bloch KD. Blood. 2011;118:4224. doi: 10.1182/blood-2011-03-339952. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Steinbicker AU, Sachidanandan C, Vonner AJ, Yusuf RZ, Deng DY, Lai CS, Rauwerdink KM, Winn JC, Saez B, Cook CM, Szekely BA, Roy CN, Seehra JS, Cuny GD, Scadden DT, Peterson RT, Bloch KD, Yu PB. Blood. 2011;117:4915. doi: 10.1182/blood-2010-10-313064. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Derwall M, Malhotra R, Lai CS, Beppu Y, Aikawa E, Seehra JS, Zapol WM, Bloch KD, Yu PB. Arterioscler. Thromb. Vasc. Biol. 2012;32:613. doi: 10.1161/ATVBAHA.111.242594. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Saeed O, Otsuka F, Polavarapu R, Karmali V, Weiss D, Davis T, Rostad B, Pachura K, Adams L, Elliott J, Taylor WR, Narula J, Kolodgie F, Virmani R, Hong CH, Finn AV. Arterioscler. Thromb. Vasc. Biol. 2012;32:299. doi: 10.1161/ATVBAHA.111.240101. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Theurl I, Schroll A, Sonnweber T, Nairz M, Theurl M, Willenbacher W, Eller K, Wolf D, Seifert M, Sun CC, Babitt JL, Hong CC, Menhall T, Gearing P, Lin HY, Weiss G. Blood. 2011;118:4977. doi: 10.1182/blood-2011-03-345066. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Shi S, Hoogaars WMH, de Gorter DJJ, van Heiningen SH, Lin HY, Hong CC, Kemaladewi DU, Aartsma-Rus A, ten Dijke P, ’t Hoen PAC. Neurobiol. Dis. 2011;41:353. doi: 10.1016/j.nbd.2010.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hao J, Ho JN, Lewis JA, Karim KA, Daniels RN, Gentry PR, Hopkins CR, Lindsley CW, Hong CC. ACS Chem. Biol. 2010;5:245. doi: 10.1021/cb9002865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shore EM, Xu M, Feldman GJ, Fenstermacher DA, Cho T-J, Choi IH, Connor JM, Delai P, Glaser DL, LeMerrer M, Morhart R, Rogers JG, Smith R, Triffitt JT, Urtizberea JA, Zasloff M, Brown MA, Kaplan FS. Nature Genet. 2006;38:525. doi: 10.1038/ng1783. [DOI] [PubMed] [Google Scholar]
- 7.Kaplan FS, Glaser DL, Pignolo RJ, Shore EM. Expert Opin. Biol. Ther. 2007;7:705. doi: 10.1517/14712598.7.5.705. [DOI] [PubMed] [Google Scholar]
- 8.Yu PB, Deng DY, Lai CS, Hong CC, Cuny GD, Bouxsein ML, Hong DW, McManus PM, Katagiri T, Sachidanandan C, Kamiya N, Fukuda T, Mishina Y, Peterson RT, Bloch KD. Nat. Med. 2008;14:1363. doi: 10.1038/nm.1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vogt J, Traynor R, Sapkota GP. Cell. Signal. 2011;23:1831. doi: 10.1016/j.cellsig.2011.06.019. [DOI] [PubMed] [Google Scholar]
- 10.Daniels RN, Kim K, Lebois EP, Muchalski H, Hughes M, Lindsley CW. Tetrahedron Lett. 2008;49:305. [Google Scholar]
- 11.Berdini V, Besong GE, Callaghan O, Carr MG, Congreve MS, Gill AL, Griffiths-Jones CM, Madin A, Murray CW, Nijjar RK, O’Brien MA, Pike A, Saxty G, Taylor RD, Vickerstaffe E. WO2008/078100. 2008
- 12.Miyaura N, Suzuki A. Chem. Rev. 1995;95:2457. [Google Scholar]
- 13.Cuny GD, Yu PB, Laha JK, Xing X, Liu J-F, Lai CS, Deng DY, Sachidanandan C, Bloch KD, Peterson RT. Bioorg. Med. Chem. Lett. 2008;18:4388. doi: 10.1016/j.bmcl.2008.06.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.BMP-responsive luciferase reporter assays. BMP-responsive C2C12BRA cells, stably transformed with the Id1 promoter-firefly luciferase reporter; kind gift of D, Rifkin, NYU Medical Center) were seeded in 96-well plates, and incubated overnight with the compounds and BMP4 (50 ng/mL). The cells were then lysed, and cell extracts were then subjected to the firefly luciferase assay using Steady-Glo luciferase assay kit (Promega). The results were normalized to cell titers, as measured using Cell Titer-Glo luminescence assay (Promega).
- 15.Zilberberg L, ten Dijke P, Sakai LY, Rifkin DB. BMC Cell Biol. 2007;8:41. doi: 10.1186/1471-2121-8-41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fraley ME, Rubino RS, Hoffman WF, Hambaugh SR, Arrington KL, Hungate RW, Bilodeau MT, Tebben AJ, Rutledge RZ, Kendall RL, McFall RC, Huckle WR, Coll KE, Thomas KA. Bioorg. Med. Chem. Lett. 2002;12:3537. doi: 10.1016/s0960-894x(02)00827-2. [DOI] [PubMed] [Google Scholar]
- 17.7g, VU0469381 (ML347) has been declared a probe via the Molecular Libraries Probe Production Centers Network (MLPCN) and is available through the network, see: http://mli.nih.gov.
- 18.Kinase assay. All kinase assays were conducted by Reaction Biology Corp (Malvern, PA), as previously reported. In brief, compounds were tested at 10 concentrations by 3-fold serial dilutions starting at 100 μM. In vitro kinase reactions were carried out in the presence of 10 μM (33P)γATP. Eleven human kinases tested were the BMP type-I receptors ALK-1/ACVRL1, ALK2/ACRV1, ALK3/BMPR1A and ALK6/BMPR1B, the TGFβ type-I receptors ALK4/ACVR1B and ALK5/TGFβR1, the BMP type-II receptor (BMPR2), the TGFβ type-II receptor (TGFβR2), VEGF type-II receptor (KDR/VEGFR2), AMP-activated protein kinase (AMPK-A1/B1/G1) and the human platelet-derived growth factor receptor-β (PDGFRβ).
- 19.LDN-193189, 2, which has replaced the labile methoxy group with piperazine (similar to 13h-n) was shown to be much more stable in liver microsome assay (see Ref. 13).
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.