

Abstract
Purpose
Langerhans cells [LCs] are dendritic cells [DCs] localized to the epidermis. They should be the first antigen presenting cells to encounter squamous cell carcinoma (SCC). The aim of this study was to investigate the ability of LCs isolated from human SCC to induce T cell proliferation and polarization.
Experimental design
We investigated the ability of LCs from SCC and peritumoral skin to induce T-cell proliferation and polarization. We also studied the effect of SCC supernatant on the ability of LCs from normal skin, in vitro generated LCs, and DCs to activate and polarize T cells.
Results
LCs from SCC were stronger inducers of allogeneic CD4+ and CD8+ T-cell proliferation and interferon (IFN)-γ production than LCs from peritumoral skin. We found that tumor supernatants were rich in immunosuppressive cytokines; despite this, allogeneic CD4+ and CD8+ T-cell proliferation and IFN-γ induction by LCs were augmented by tumor supernatant. Moreover, tumor supernatant facilitated IFN-γ induction by in vitro generated LCs, but suppressed the ability of in vitro generated DCs to expand allogeneic CD4+ and CD8+ T cells.
Conclusions
We have demonstrated that LCs from SCC can induce type-1 immunity. Tumor supernatant induces IFN-γ induction by in vitro generated LCs. This contrasts greatly with prior studies showing that DCs from SCC cannot stimulate T cells. These data indicate that LCs may be superior to DCs for SCC immunotherapy and may provide a novel rationale to harnessing LCs for the treatment of cancer patients.
Keywords: Langerhans cells, dendritic cells, cutaneous squamous cell carcinoma, cancer immunotherapy, type 1 immunity
INTRODUCTION
Dendritic cells (DCs) are the most potent antigen-presenting cells (APCs), are comprised of a variety of subsets and serve as master regulators of adaptive immunity (Fricke and Gabrilovich 2006; Steinman and Banchereau 2007). Because of their potential to elicit tumor-specific T-cell responses, it is generally accepted that DCs play key roles in cancer immune surveillance (Gottfried et al. 2008). Numerous studies have demonstrated that DCs often infiltrate various human tumors (Vicari et al. 2002; Talmadge et al. 2007; Chaput et al. 2008). However, DCs from some cancers are functionally compromised (Pinzon-Charry et al. 2005; Fricke and Gabrilovich 2006; Bennaceur et al. 2008; Chaput et al. 2008; Gottfried et al. 2008). Many studies have demonstrated decreased ability of DCs from human cancer to stimulate T cells and induce interferon (IFN)-γ (Enk et al. 1997; Gabrilovich et al. 1997; Nestle et al. 1997; Troy et al. 1998; Curiel et al. 2003; Perrot et al. 2007). Hence, it has been postulated that dysfunction of DCs, induced by the tumor microenvironment, may allow tumors to escape immune surveillance (Pinzon-Charry et al. 2005).
The deficit of endogenous DCs frequently observed in cancer patients led to the use of ex vivo-generated DCs as carriers of cancer vaccines (Pinzon-Charry et al. 2005; Kalinski et al. 2009). Clinical trials using ex vivo-generated DCs have occasionally yielded significant tumor regression (Steinman and Banchereau 2007; Kalinski et al. 2009). However, objective clinical benefits are very limited, arguing for the need to improve the design of DC-based vaccines (Rosenberg et al. 2004; Banchereau and Palucka 2005). In particular, even the appropriate DC subset for clinical use remains undefined. Given the functional differences among DC subsets, further understanding of the biology of each DC subset in the complex tumor-associated environment is crucial (Shurin and Lotze 2009; Ueno et al. 2010).
The skin immune system harbors a rich network of DCs, mainly composed of epidermal Langerhans cells (LCs) and dermal myeloid DCs. We and others have demonstrated that dermal myeloid DCs, associated with cutaneous basal cell carcinoma (BCC) and squamous cell carcinoma (SCC), are deficient in their ability to stimulate T cells (Nestle et al. 1997; Bluth et al. 2009). We have previously shown that the number of LCs is significantly decreased in SCC, which corroborates previous observations by others (Galan and Ko 2007; Takahara et al. 2009). However, the functional significance of LCs in human cutaneous SCC remains undefined.
Since cutaneous SCC is a malignant proliferation of epidermal keratinocytes, LCs should be the first APCs to encounter tumor antigens. This prompted us to study the functional properties of LCs in cutaneous SCC. Here, we report novel observations relating to the potential value of using LCs for cancer immunotherapy.
Using human SCC specimens, we show that epidermal LCs, and in vitro-generated LCs, can be activated to elicit desirable type 1 immunity. This is in direct contrast to what we previously found for myeloid DCs in SCC, and thus demonstrates the unique biology of LCs which can be exploited for therapeutic advantage. Our findings provide a novel rationale for harnessing both in vivo and in vitro-generated LCs for cancer treatment. Manipulation of LCs generated from human blood precursors to drive antitumor response may be particularly useful in adoptive immunotherapy.
RESULTS
LCs from human SCC are more mature compared to LCs from peritumoral non-lesional skin
Since DC maturation is a key event in the induction of the immune responses, we first characterized the maturation status of LCs from SCC by analyzing the expression of maturation makers. HLA-DR+CD207+ cells from epidermal cell suspensions derived from SCC and peritumoral non-lesional (PTNL) skin were defined as LCs and evaluated for CD40, CD80, CD83, and CD86 expression by flow cytometry. SCC-derived LCs expressed higher levels of CD40, CD80, CD83, and CD86 compared to LCs from patient-matched PTNL skin (Figure 1). HLA-DR+CD207- cells were observed in 50% of cases. These cells did not express CD40, CD83, CD86, or common leukocyte antigen CD45 and were thus likely to be tumor cells.
Figure 1.
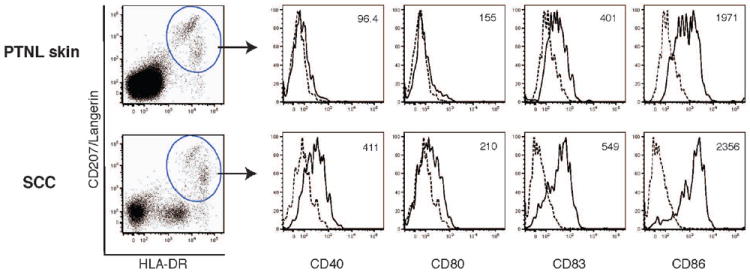
Flow cytometric analysis of maturation-associated molecules expressed on LCs from SCC and peritumoral non-lesional skin. HLA-DR+CD207+ LCs in the epidermal cell suspension from SCC and peritumoral non-lesional (PTNL) skin were subjected to the analysis of cell surface expression of CD40, CD80, CD83, and CD86. Dashed lines indicate isotype controls. Mean fluorescence intensity value is shown in the upper right hand of each histogram. Data are representative of three independent experiments.
LCs from human SCC are more powerful stimulators of allogeneic CD4+ and CD8+ T-cell proliferation than those from peritumoral non-lesional skin
To explore whether the more mature phenotype of SCC-derived LCs was linked to their capacity to stimulate T-cell proliferation, we performed mixed leukocyte reactions. CFSE-labeled allogeneic total T cells from peripheral blood were cocultured with FACS-sorted HLA-DR+CD207+ LCs from either SCC tumor nests or patient-matched PTNL skin for 7 days; T-cell proliferation was then assessed by flow cytometry. We set gates on live CD3+CD4+ and CD3+CD8+ cells for analysis and evaluated T-cell proliferation by CFSE dilution. As shown in Figure 2A, LCs from SCC induced significantly higher levels of allogeneic CD4+ and CD8+ T-cell proliferation than those from PTNL skin. Representative FACS histograms are shown in Figure 2B.
Figure 2.
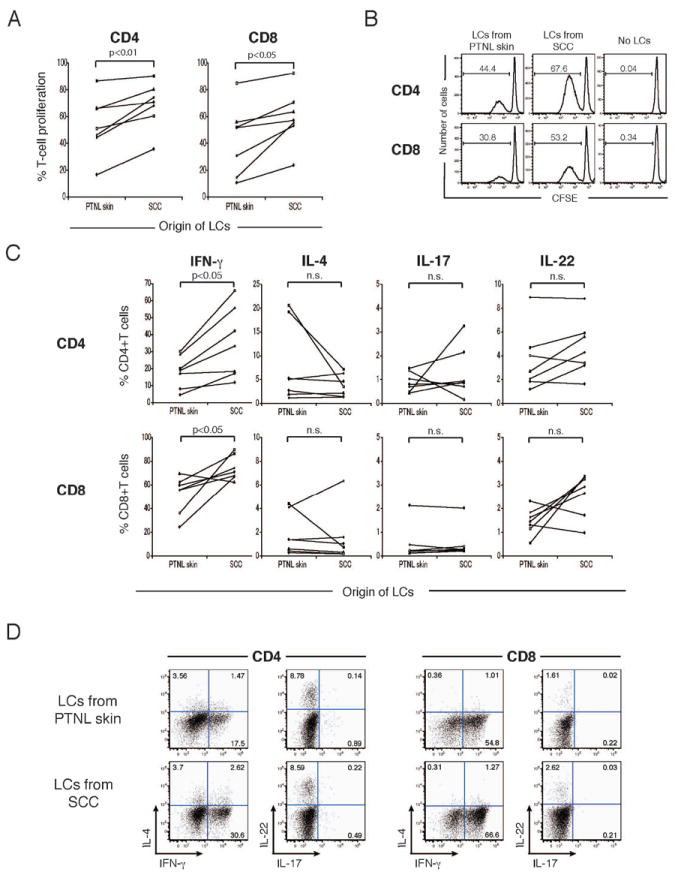
LCs from SCC stimulate proliferation of IFN-γ producing CD4+ and CD8+ T cells (A) CFSE labeled, allogeneic T were cultured with FACS-sorted LCs from SCC or PTNL. Percentages of proliferating cells within live CD3+CD4+ and CD3+CD8+ cells were determined by CFSE dilution (B) Representative FACS histograms of CFSE dilution assay using LCs form SCC and PTNL skin. Numbers show percentages of proliferating (CFSElow) cells. (C) Frequencies of cells positive for intracellular IFN-γ, IL-4, IL-17, and IL-22 detected in proliferating CD4+ and CD8+ T-cell populations following LC co-culture experiments. n.s., not significant. (D) Dot plot analysis of IFN-γ, IL-4, IL-17, and IL-22 expression in proliferating CD4+ and CD8+ T cells stimulated by LCs from SC vs. PTNL skin. Numbers indicate percent gated cells.
LCs from human SCC induce type 1 T-cell response more efficiently than those from PTNL skin
Next, we examined cytokine profiles of T cells activated by LCs from SCC and those from PTNL skin. We focused on the cytokines associated with specific T helper cell subtypes, including IFN-γ (Th1), IL-4 (Th2), IL-17 (Th17), and IL-22 (Th22). Total allogeneic T cells cultured with FACS-sorted LCs for 7 days were subsequently activated by PMA and ionomycin for 4 hours in the presence of brefeldin A, followed by intracellular cytokine staining. The percentages of T cells producing IFN-γ (Th1), IL-4 (Th2), IL-17 (Th17), and IL-22 (Th22) are shown in Figure 2C. LCs from SCC induced significantly greater percentages of IFN-γ-producing cells than those from PTNL skin in both CD4+ and CD8+ T cells. The frequencies of CD4+ and CD8+ T cells producing IL-4, IL-17, and IL-22 were not different between the two groups. Percentages of interferon producing T cells did not vary with numbers of LCs isolated per skin sample. Representative FACS plots of cytokine production by proliferating T cells are shown in Figure 2D.
LCs from SCC show a mixed gene expression profile with both immune activation and immune tolerance genes
We were interested in determining the genomic signature of SCC-associated LCs since they were unexpectedly more stimulatory than those from PTNL skin. Gene array analysis was performed to compare LCs isolated from SCC and those from patient-matched, PTNL skin with verification of FACS analysis (Supplementary Results and Supplementary Figure 1). We found that molecules associated with DC maturation and activation, including signal transducer and activator of transcription (STAT)-4, IL-15, LY75/CD205, CD80, IL-2RA/CD25, STAT-5B, and STAT-5A, were up-regulated exclusively in SCC-derived LC (Supplementary Table 1). Interestingly, molecules which can be involved in immune tolerance, such as CD200 and receptor activator of nuclear factor κB, were also up-regulated in LCs from SCC. Three probes corresponding to CLEC2D showed increased expression in SCC associated LCs (3.498 -4.768 fold, P < 0.0001 – 0.00.4, Supplemental Table 2). CLEC2D has been linked to the IFN-γ via interaction of with its receptor, CD161, which is expressed on T cells (Aldemir et al. 2005).
SCC supernatant enhances CD4+ and CD8+ T-cell proliferation induced by LCs
Since LCs should present tumor antigens to T cells in the context of the SCC-associated environment in vivo, we were also interested in how soluble factors released from the SCC microenvironment affect the interaction of LCs and T cells. To simulate the interaction of LCs and T cells in the SCC microenvironment, SCC culture supernatants were prepared and added to MLR experiments. FACS-sorted LCs from normal epidermis and CFSE-labeled allogeneic total T cells from peripheral blood were cocultured for 7 days in the presence or absence of 25% tumor supernatant from SCC; then T-cell proliferation was assessed as described above. Interestingly, as shown in Figure 3A, tumor supernatant applied to MLR assays significantly augmented the proliferation of both CD4+ and CD8+ T cells triggered by LCs. When T cells were cultured in the presence of tumor supernatant without LCs, their proliferation was not observed (data not shown). Representative FACS histograms are shown in Figure 3B.
Figure 3.
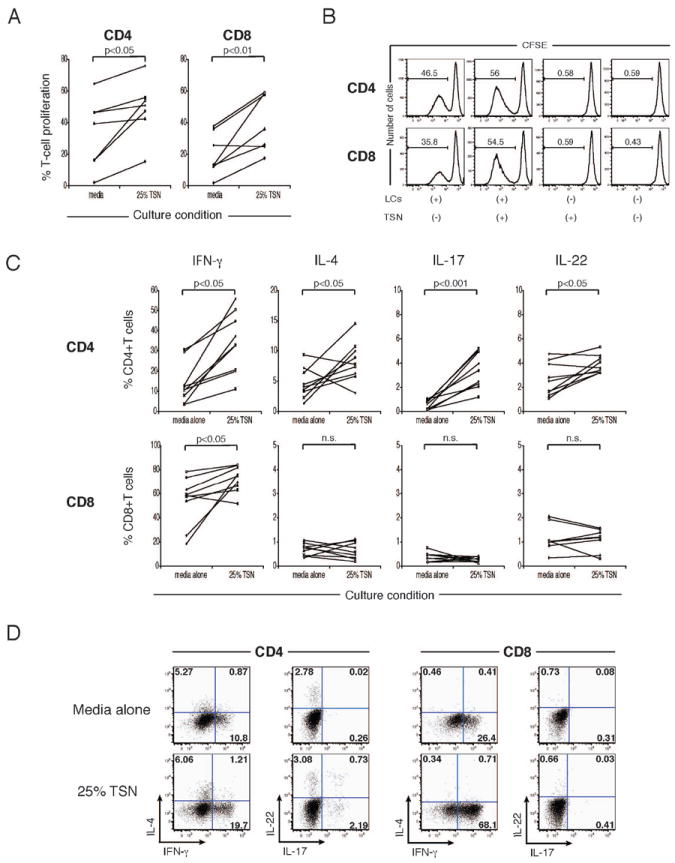
TSN enhances LC driven CD4+ and CD8+ T-cell proliferation, IFN-γ, IL-4, IL-17, and IL-22 production by CD4+cells, and, IFN-γ production by CD8+ cells. (A) LCs from normal skin were cultured with CFSE-labeled allogeneic T cells +/- 25% TSN. Percentages of proliferating cells within live CD3+CD4+ and CD3+CD8+ cells were determined by CSFE dilution. (B) FACS histograms of CFSE dilution assay using LCs and TSN. Numbers show percentages of proliferating cells. (C) Effect of TSN on production of IFN-γ, IL-4, IL-17, and IL-22 by CD4+ and CD8+ T cells. n.s., not significant. (D) Dot plot analysis of IFN-γ, IL-4, IL-17, and IL-22 expression in proliferating CD4+ and CD8+ T cells stimulated by LCs +/-TSN. Numbers show percent gated cells.
SCC supernatant facilitates the generation of broad range of CD4+ T cells as well as IFN-γ-producing CD8+ T cells among T cells expanded by LCs
We also investigated the cytokine profile of T cells stimulated by LCs in the presence or absence of tumor supernatant as described above. As shown in Figure 3C, the addition of tumor supernatant to MLR culture significantly increased the frequencies of the cells producing IFN-γ, IL-4, IL-17, and IL-22 among proliferating CD4+ T cells. With respect to CD8+ T cells, only the frequency of the cells producing IFN-γ was increased by tumor supernatant. Representative FACS plots of cytokine production by proliferating T cells are shown in Figure 3D.
SCC supernatant enhances the proliferation of CD4+ and CD8+ T cells induced by in vitro-generated LC-type DCs whereas it suppresses T-cell proliferation driven by monocyte-derived DCs
For in vitro-generated DC-based vaccine therapy for cancer patients, DCs are prepared by the culture of CD34+ HPCs in the presence of GM-CSF, TNF-α, and Flt3L or by culturing blood-derived monocytes in the presence of GM-CSF and IL-4 (El Marsafy et al. 2009). Of note, DCs derived from CD34+ HPCs contain CD1a+CD14- LC-type DCs. Therefore, we were further interested in whether CD34+ HPC-derived LC-type DCs were also capable of stimulating T cells effectively in the tumor-associated environment. We assessed the capability of the LC-type DCs to activate allogeneic T cells in the presence of SCC supernatant as above. We also used monocyte-derived DCs from the same donors for comparison. As shown in Supplementary Figure S2, we were able to obtain CD1a+CD14- cells with an LC phenotype by culturing CD34+ HPCs with GM-CSF, TNF-α, and Flt3L. Monocyte-derived DCs were obtained by culturing monocytes with GM-CSF and IL-4.
Consistent with our observations from normal epidermal LCs, the presence of tumor supernatant in MLR culture significantly enhanced the proliferation of both CD4+ and CD8+ T cells trigged by LC-type DCs (Figure 4A). On the other hand, the proliferation of both CD4+ and CD8+ T cells induced by monocyte-derived DCs was significantly suppressed by adding tumor supernatant to culture. Representative FACS histograms are shown in Figure 4B.
Figure 4.
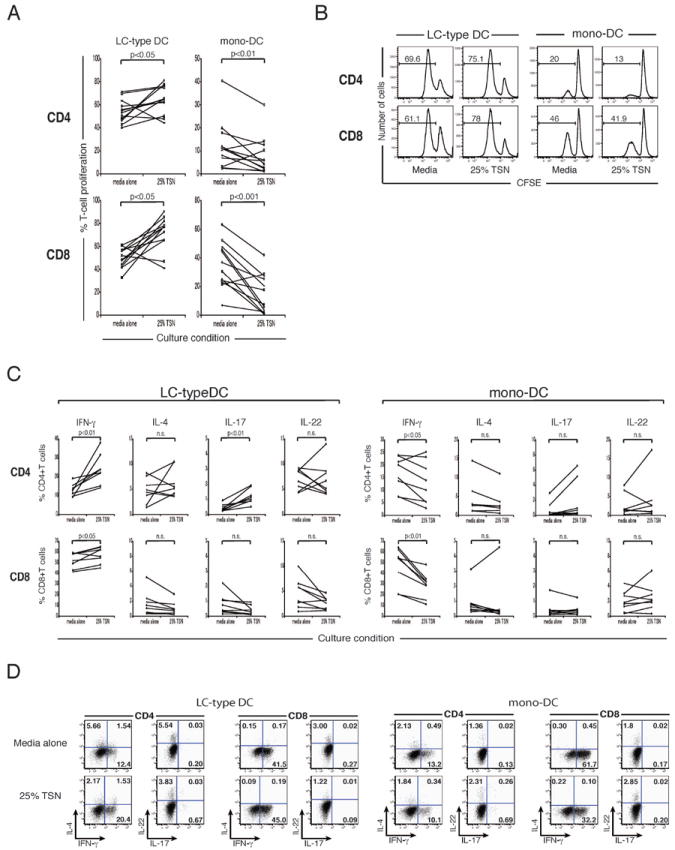
TSN enhances proliferation and IFN-γ response by CD4+ and CD8+ T cells induced by in vitro-generated LCs, but suppresses those by monocyte-derived DCs. (A) CD1a+CD14- LC-type DCs derived from CD34+ hematopoietic progenitors, or, monocyte-derived DCs were cultured with CFSE-labeled T cells +/- 25% TSN. Percentages of proliferating cells were determined by CFSE dilution. (B) FACS histograms of CFSE dilution assay. Numbers show percentages of proliferating cells. (C) Effect of TSN on in vitro generated LC or DC driven production of IFN-γ, IL-4, IL-17, or IL-22, by CD4+ and CD8+ T-cells. n.s., not significant. (D) Dot plot analysis of IFN-γ, IL-4, IL-17, and IL-22 expression in proliferating CD4+ and CD8+ T cells stimulated by LC-type DCs or mono-DCs, +/- TSN. Numbers show percent gated cells.
SCC supernatant enhances type 1 T-cell response elicited by in vitro-generated LC-type DCs, but suppresses that induced by monocyte-derived DCs
The cytokine profiles of T cells stimulated by LC-type DCs and monocyte-derived DCs in the presence or absence of SCC supernatant were examined as well. As shown in Figure 4C, the percentages of IFN-γ-producing cells among proliferating CD4+ and CD8+ T cells stimulated by LC-type DCs were significantly increased by the presence of tumor supernatant in culture. The addition of tumor supernatant also elevated the frequency of IL-17-producing CD4+ T cells activated by LC-type DCs. By contrast, the presence of tumor supernatant in culture significantly reduced the percentages of the cells expressing IFN-γ among proliferating CD4+ and CD8+ T cells stimulated by monocyte-derived DCs. Thus, type-1 immune response induced by LC-type DCs was promoted by tumor supernatant, whereas that induced by monocyte-derived DCs was attenuated. Representative FACS plots of cytokine production by proliferating T cells are shown in Figure 4D. Overall, Monocyte derived DCs were less able than LC to stimulate T cells than LCs with media alone. In another series of experiments, LCs or DCs, generated in vitro, were pretreated with TSN and extensively washed prior to culturing with allogeneic T cells. TSN pretreatment enhanced the ability of in vitro generated LCs to drive T cell proliferation and it suppressed the ability of in vitro generated DCs to drive allogeneic T cell proliferation.
SCC supernatants contain cytokines that suppress myeloid DCs
Abundant evidence indicates that immunosuppressive cytokines in the tumor microenvironment impair DC differentiation and function (Zou 2005; Fricke and Gabrilovich 2006). Indeed, in a previous study, we have shown high mRNA expression of IL-10, VEGF, and TGF-β1 in cutaneous SCC specimens (Bluth et al. 2009). To identify the cytokine milieu of cutaneous SCC in more detail, we screened tumor supernatants from SCC specimens for the production of cytokines. The amounts of IFN-γ, IL-1β, IL-2, IL-6, IL-8, IL-12p70, TNF-α, GM-CSF, IL-10, VEGF, and TGF-β1 in tumor supernatants were measured by an electrochemiluminescence-based system (n=12). As shown in Table 1, SCC supernatants contained significant levels of IFN-γ (9.8 ± 2.4 pg/ml; 4 out of 12 samples), IL-1β (22.5 ± 6.9 pg/ml; 11 out of 12 samples), IL-2 (4.1 ± 0.8 pg/ml; 10 out of 12 samples), IL-6 (450 ± 245 pg/ml; 11 out of 12 samples), IL-8 (7585 ± 1651 pg/ml; 12 out of 12 samples), IL-12p70 (1.0 ± 0.2 pg/ml; 7 out of 12 samples), TNF-α (23.4 ± 6.2 pg/ml; 5 out of 12 samples), GM-CSF (109 ± 49 pg/ml; 9 out of 12 samples), IL-10 (12.2 ± 2.7 pg/ml; 9 out of 12 samples), and VEGF (408 ± 120 pg/ml; 12 out of 12 samples; mean ± SEM for all). We could not detect these cytokines in culture media used for culturing tumor samples (n=3). Eleven of 12 tumor supernatant samples had higher concentrations of TGF-β1 (33.5 ± 4.4 pg/ml) than the media used for the culture (baseline level). Thus, tumor supernatants from cutaneous SCCs carry tumor immunosuppressive cytokines, such as VEGF, TGF-β1, IL-10, and IL-6.
Table 1.
Cytokines in the supernatants of whole-SCC tumor specimen culture
| Sample No. | IFN-γ (pg/ml) | IL-1β (pg/ml) | IL-2 (pg/ml) | IL-6 (pg/ml) | IL-8 (pg/ml) | TNF-α (pg/ml) | IL-12p70 (pg/ml) | GM-CSF (pg/ml) | IL-10 (pg/ml) | VEGF (pg/ml) | TGF-β1 (pg/ml) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| TSN 1 | 6.3 | 17.8 | 3.2 | 211.8 | 10985.6 | 15.8 | 1.2 | 39.7 | 13.6 | 164.9 | 38.8 |
| TSN 2 | N.D. | 76.1 | 5.9 | 162.9 | 11098.0 | 34.8 | N.D. | 43.9 | 25.1 | 545.2 | 31.5 |
| TSN 3 | N.D. | 43.8 | 1.8 | 2821.1 | 10704.9 | N.D. | 1.3 | 480.3 | 6.3 | 1489.2 | 67.7 |
| TSN 4 | N.D. | N.D. | N.D. | N.D. | 3.0 | N.D. | N.D. | N.D. | N.D. | 103.0 | 20.3 |
| TSN 5 | 8.0 | 2.8 | 0.1 | 9.6 | 282.9 | N.D. | N.D. | N.D. | N.D. | 231.5 | 46.7 |
| TSN 6 | 8.0 | 0.2 | N.D. | 0.9 | 145.4 | N.D. | N.D. | N.D. | N.D. | 94.2 | 11.0 |
| TSN 7 | N.D. | 1.1 | 5.1 | 108.6 | 14948.5 | N.D. | 0.5 | 62.9 | 5.6 | 763.6 | 21.9 |
| TSN 8 | N.D. | 26.3 | 4.8 | 510.7 | 12005.4 | 41.4 | 0.5 | 171.6 | 21.4 | 706.0 | 26.5 |
| TSN 9 | N.D. | 30.2 | 5.3 | 530.4 | 11517.7 | N.D. | N.D. | 64.1 | 20.1 | 171.2 | 20.9 |
| TSN 10 | N.D. | 25.8 | 6.2 | 107.8 | 6815.9 | 14.6 | 0.3 | 53.7 | 8.3 | 398.6 | 44.1 |
| TSN 11 | 16.8 | 3.1 | 0.7 | 17.8 | 479.0 | N.D. | 1.9 | 10.9 | 3.5 | 110.2 | 22.0 |
| TSN 12 | N.D. | 20.2 | 7.6 | 469.3 | 12037.7 | 10.2 | 1.1 | 49.9 | 6.2 | 117.1 | 28.1 |
| Media 1 | N.D. | N.D. | N.D. | N.D. | N.D. | N.D. | N.D. | N.D. | N.D. | N.D. | 11.0 |
| Media 2 | N.D. | N.D. | N.D. | N.D. | N.D. | N.D. | N.D. | N.D. | N.D. | N.D. | 10.1 |
| Media 3 | N.D. | N.D. | N.D. | N.D. | N.D. | N.D. | N.D. | N.D. | N.D. | N.D. | 11.1 |
Size comparable specimens of whole-tissue SCC specimens were cultured for 24 hours, and the contents of IFN-γ, IL-1β, IL-2, IL-6, IL-8, IL-12p70, TNF-α, GM-CSF, IL-10, VEGF, and TGF-β1 in the supernatants were analyzed using electrochemiluminescence-based method. Media 1-3 represent the media used for the culture of SCC specimens. TSN, tumor supernatant. N.D., Not detected.
DISCUSSION
The induction of effective anti-cancer response depends on innate and adaptive immunity coordinated by DCs (Pinzon-Charry et al. 2005). However, the dysfunction of DCs in tumor tissue has been noted in various types of cancer. Similar findings have also been reported in the blood-circulating DCs from cancer patients (Almand et al. 2000; Almand et al. 2001; Della Bella et al. 2003). These observations suggest that DC dysfunction may be a systemic event rather than a phenomenon simply confined to the local tumor microenvironment (Pinzon-Charry et al. 2005; Bennaceur et al. 2008). We previously showed that the T-cell stimulatory capacity of myeloid DCs from human cutaneous SCC is considerably impaired (Bluth et al. 2009). In this study, we evaluated the function of a different DC subset, epidermal LCs, isolated from cutaneous SCC. In sharp contrast to myeloid DCs, LCs from SCC were more powerful stimulators of allogeneic CD4+ and CD8+ T-cell proliferation than LCs from peritumoral non-lesional skin. This result was consistent with the more mature phenotype of SCC-derived LCs. Furthermore, LCs from SCC were superior to those from peritumoral non-lesional skin in polarizing CD4+ and CD8+ T cells to produce IFN-γ, a key cytokine required for anti-tumor immunity. Thus, contrary to cancer-associated myeloid DCs, LCs in SCC exhibited enhanced ability to stimulate T-cell proliferation and drive desirable type-1 T-cell responses.
In vitro, the proliferation of both CD4+ and CD8+ T cells driven by LCs from normal skin was augmented in the presence of tumor supernatant. Tumor supernatant was stimulatory; despite the fact that we found that it was rich in DC-suppressive cytokines. This is consistent with the results obtained from the comparison between SCC-derived LCs and those from peritumoral non-lesional skin. The effect of tumor supernatant was specific to IFN-γ induction by CD8+ T cells, and did not lead to induction of CD8+T cells producing IL-4 IL-17 or IL-22. The ability of epidermal LCs to both induce T-cell proliferation and drive IFN-γ response was enhanced even in the presence of tumor-derived soluble immunosuppressive mediators; this implies that T-cell proliferation and type 1 immune response based on LC-T cell interactions are not vulnerable to the immunosuppressive SCC microenvironment. This is consistent with the report that T-cell stimulatory ability of LCs is not affected by melanoma cell-derived factors (Berthier-Vergnes et al. 2001). LCs isolated from the lymph nodes of tumor-bearing mice are also shown to be fully competent in stimulating T-cell proliferation (Ishida et al. 1998). Taken together, these findings support the concept that epidermal LCs seem to be a unique DC subset which are resistant to a predominantly immunosuppressive tumor environment.
Nevertheless, patients’ immune systems fail to eradicate SCC tumors, despite the enhanced ability of SCC-derived LCs to induce strong type 1 immunity ex-vivo. This implies that tumor-specific immunity elicited by LCs is suboptimal in situ. One plausible explanation for this discrepancy is the relatively low number of LCs in the tumor nests. We and others have observed lower numbers of LCs in the SCC microenvironment (Galan and Ko 2007; Bluth et al. 2009; Takahara et al. 2009). We found fewer CD1a+ and Langerin+ cells infiltrating SCC tumor nests and even fewer Langerhans’ cells in adjacent non tumor bearing skin (Bluth et al. 2009). Other potential mechanisms include defective recognition, internalization, processing, and presentation of tumor antigens by LCs, as well as a deficit in their migration to gain access to T cells. Physical contact of LCs with immunosuppressive cells such as regulatory T cells and the presence of myeloid-derived suppressor cells in the tumor microenvironment should also be taken into consideration.
Since isolated LCs are immunologically active, they may have potential in LC-based SCC immunotherapy. One of the major strategies for DC-based cancer immunotherapy is the transfer of DCs generated from precursors under ex vivo tumor-free condition (Schuler et al. 2003; Steinman and Banchereau 2007; Banchereau et al. 2009). Most studies utilize DCs induced from peripheral blood monocytes (Nestle et al. 1998; Thurner et al. 1999; Berntsen et al. 2006; Palucka et al. 2006). DCs derived from CD34+ HPCs, containing LC-type DCs, have also been employed for this purpose (Banchereau et al. 2001; Di Nicola et al. 2004). We demonstrated that the ability of LC-type DCs to expand CD4+ and CD8+ T cells and drive IFN-γ response was significantly enhanced in the presence of SCC-derived factors; in contrast, the activity of monocytes-derived DCs was suppressed by SCC-derived factors. These results are consistent with previous reports that LC-type DCs or total DCs derived from CD34+ HPCs are superior to monocyte-derived DCs in the induction of antigen-specific CD8+ T cells (Mortarini et al. 1997; Ferlazzo et al. 1999; Ratzinger et al. 2004). Both SCC-derived LCs and LC-type DCs showed the ability to stimulate T-cell proliferation and promote desirable type 1 immune response. Since the tumor microenvironment may suppress myeloid DC activity (Vicari et al. 2002), our results support an idea that in vitro generated LCs may be suitable for cancer immunotherapy (Banchereau et al. 2001). Although the mechanisms underlying the observed differences between the two types of DC remain undefined, we believe our data lends a novel rationale to the use of LC-type DCs in cancer vaccine therapy.
Another promising strategy for DC-based cancer immunotherapy is the targeted delivery of tumor antigens to in vivo DCs, either by using anti-DC antibodies such as CD205 and CD207 or by simple and direct application on to barrier-disrupted skin (Steinman and Banchereau 2007; Flacher et al. 2009; Romani et al. 2010; Stoitzner et al. 2010). Skin DCs, especially LCs, are targeted in this approach because of relatively easy access. Indeed, the pivotal role of LCs in the protecting effect of epicutaneous immunization in tumor-bearing mice has been shown using an LC depletion model (Stoitzner et al. 2008). Considering that myeloid DCs can be systemically impaired in cancer patients, our observation that epidermal LCs in tumor-associated environment acted as even more powerful APCs, also supports the validity of this second maneuver exploiting in vivo LCs.
The immune microenvironment associated with cutaneous SCC is a highly complex milieu comprised of opposing forces driving activation of one DC type and suppression of another. The observed contrasting effects of tumor supernatant may be the result of the combined effect of a wide range of soluble mediators that are both stimulatory and inhibitory. The simultaneous upregulated gene expression of both immunostimulatory and immunosuppressive molecules in SCC-derived LCs might reflect this complicated tumor microenvironment as well. Our results demonstrate that the cancer microenvironment can affect distinct DC subtypes in totally different ways.
One potential explanation for our findings may reside in the CLEC2D/CD161 axis. CLEC2D is a C-lectin that has been linked to IFN-γ induction via interaction with CD161 on T cells (Aldemir et al. 2005). Maggi, et al, showed that CD161 is expressed Th1, Th2, Th0 and Th17 cells and is inducible by RORC (Maggi et al. 2010). We found that CLEC2D was overexpressed in LCs from SCC (3.5 to 4.8 fold, P < 0.004 -0.0001) based on expression of probes 220132-s, 233500_x, and 23522 (Supplemental Table 1). Thus it may be that LC expression of CLEC2D may be key in initiating Type 1 anti-tumor immunity. The mechansisms governing these processes will be the focus of ongoing work by our group.
In conclusion, we have shown the evidence that human LCs act as powerful inducers of type 1 T-cell response in the cancer microenvironment. Our findings provide a novel rationale to the use of LCs for DC-based cancer immunotherapy, and thus pave the way to novel cancer immunotherapy based on LC function.
MATERIALS AND METHODS
The study was approved by the Institutional Review Board of Weill Cornell Medical College and The Rockefeller University. Written informed consent was obtained before enrolling patients to participate in this study. The study was performed in adherence with the Declaration of Helsinki Principles.
Skin sample preparation for flow cytometry
Tumors of cutaneous SCC and site-matched peritumoral non-lesional (PTNL) skin were obtained at surgery, and normal skin was obtained as the discarded product of dermatologic and plastic surgery. All SCC were primary stage 1 squamous cell carcinomas from sun exposed areas of the head and neck. Epidermal single cell suspensions were prepared using Dispase II (Roche Diagnostics, Mannheim, Germany) and trypsin-ethylenediamine tetraacetic acid (Invitrogen, Carlsbad, CA) as described previously (Fujita et al. 2009).
Flow cytometry
Fluorescence-activated cell sorting (FACS) of epidermal HLA-DR+CD207+ LCs, as well as FACS phenotyping of epidermal LCs and in vitro-generated LC-type DCs, were performed as previously described (Fujita et al. 2009). Antibodies used are outlined in Supplementary Table S2.
Generation of LC-type DCs in vitro
LC-type DCs were obtained by culturing CD34+ hematopoietic progenitor cells (HPCs) with granulocyte-macrophage colony-stimulating factor (GM-CSF) (100 ng/ml; R&D Systems, Minneapolis, MN), fms-like tyrosine kinase 3 ligand (Flt3L) (100 ng/ml; R&D Systems), and tumor necrosis factor (TNF)-α (10 ng/ml; R&D Systems) for 8 days as previously described (Fujita et al. 2009). On day 8, CD1a+CD14- LC-type DCs were sorted on a FACSAria (BD Biosciences).
Generation of monocyte-derived conventional DCs
Monocyte-derived DCs were obtained by culturing peripheral blood mononuclear cells with 25 ng/ml interleukin (IL)-4 and 100 ng/ml GM-CSF (both from R&D Systems) for 8 days as previously described (Fujita et al. 2009). On day 8, live and forward scatter- and side scatter-high cells were sorted on a FACSAria for purification of DCs.
Mixed Leukocyte Reaction Assay
The Mixed Leukocyte Reaction (MLR) assay was performed as previously described (Fujita et al. 2009). For the evaluation of cell proliferation, T cells were labeled with carboxyfluorescein diacetate succinimidyl ester (CFSE) using the Vybrant CFDA SE Cell Tracer Kit (Invitrogen/Molecular Probes) as previously described (Fujita et al. 2009).
Intracellular cytokine staining
T cells stimulated with allogeneic LCs/DCs for 7 days were restimulated for 4 hours with 25 ng/ml phorbol myristate acetate (PMA) and 2 mg/ml ionomycin in the presence of 10 mg/ml brefeldin A (all from Sigma Aldrich) at 37°C. Then intracellular cytokine staining was performed as previously described (Fujita et al. 2009). Antibodies used are outlined in Supplementary Table S1. Expression of each molecule was analyzed in activated (forward scatter- and side scatter-high) T cells.
Collection of tumor supernatants
Freshly excised whole-tissue specimens of cutaneous SCC of uniform size (~1.5 cm2) were cut into small pieces with a scalpel and incubated at 37°C in 6-well plate in 2 ml of complete medium. Medium were harvested after 24 h of culture and centrifuged at 1,500 rpm for 5 min, then supernatants were collected and run over a 0.22 μm Millex-GP filter (Millipore, Bedford, MA) and stored at -20°C. The supernatants were added to the MLR culture at a 1:4 dilution as described previously (Enk et al. 1997).
Measurement of cytokines in SCC supernatants
Levels of vascular endothelial growth factor (VEGF), transforming growth factor (TGF)-β1, and pro-inflammatory cytokines including IFN-γ, IL-1β, IL-2, IL-6, IL-8, IL-12p70, TNF-α, GM-CSF, and IL-10 in the SCC supernatants were measured using an electrochemiluminescence-based Human VEGF Tissue Culture Assay, Human TGF-β1 Assay, or Human Pro-inflammatory 9-Plex Tissue Culture Kit (Meso Scale Discovery, Gaithersburg, MD). Samples were analyzed in duplicate and compared with control culture media used for culture. Plates were read in a SECTOR Imager 2400 instrument (Meso Scale Discovery).
Supplementary Material
Acknowledgments
We thank Dr. Ralph M. Steinman (Laboratory of Cellular Physiology and Immunology, The Rockefeller University) for his review of the manuscript.
Financial support: This study was in part supported by the Empire State Stem Cell Fund through NYSDOH Contract #C023046. Opinions expressed here are solely those of the authors and do not necessarily reflect those of the Empire State Stem Cell Fund, the NYSDOH, or the State of New York.
Footnotes
Conflict of Interest: The authors declare no conflict of interest.
References
- Aldemir H, Prod’homme V, Dumaurier MJ, et al. Cutting edge: lectin-like transcript 1 is a ligand for the CD161 receptor. Journal of immunology. 2005;175(12):7791–7795. doi: 10.4049/jimmunol.175.12.7791. [DOI] [PubMed] [Google Scholar]
- Almand B, Clark JI, Nikitina E, et al. Increased production of immature myeloid cells in cancer patients: a mechanism of immunosuppression in cancer. J Immunol. 2001;166(1):678–689. doi: 10.4049/jimmunol.166.1.678. [DOI] [PubMed] [Google Scholar]
- Almand B, Resser JR, Lindman B, et al. Clinical significance of defective dendritic cell differentiation in cancer. Clin Cancer Res. 2000;6(5):1755–1766. [PubMed] [Google Scholar]
- Banchereau J, Klechevsky E, Schmitt N, et al. Harnessing human dendritic cell subsets to design novel vaccines. Ann N Y Acad Sci. 2009;1174:24–32. doi: 10.1111/j.1749-6632.2009.04999.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banchereau J, Palucka AK. Dendritic cells as therapeutic vaccines against cancer. Nat Rev Immunol. 2005;5(4):296–306. doi: 10.1038/nri1592. [DOI] [PubMed] [Google Scholar]
- Banchereau J, Palucka AK, Dhodapkar M, et al. Immune and clinical responses in patients with metastatic melanoma to CD 34(+) progenitor-derived dendritic cell vaccine. Cancer Res. 2001;61(17):6451–6458. [PubMed] [Google Scholar]
- Bennaceur K, Chapman J, Brikci-Nigassa L, et al. Dendritic cells dysfunction in tumour environment. Cancer Lett. 2008;272(2):186–196. doi: 10.1016/j.canlet.2008.05.017. [DOI] [PubMed] [Google Scholar]
- Berntsen A, Geertsen PF, Svane IM. Therapeutic dendritic cell vaccination of patients with renal cell carcinoma. Eur Urol. 2006;50(1):34–43. doi: 10.1016/j.eururo.2006.03.061. [DOI] [PubMed] [Google Scholar]
- Berthier-Vergnes O, Gaucherand M, Peguet-Navarro J, et al. Human melanoma cells inhibit the earliest differentiation steps of human Langerhans cell precursors but failed to affect the functional maturation of epidermal Langerhans cells. Br J Cancer. 2001;85(12):1944–1951. doi: 10.1054/bjoc.2001.2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bluth MJ, Zaba LC, Moussai D, et al. Myeloid dendritic cells from human cutaneous squamous cell carcinoma are poor stimulators of T-cell proliferation. J Invest Dermatol. 2009;129(10):2451–2462. doi: 10.1038/jid.2009.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaput N, Conforti R, Viaud S, et al. The Janus face of dendritic cells in cancer. Oncogene. 2008;27(45):5920–5931. doi: 10.1038/onc.2008.270. [DOI] [PubMed] [Google Scholar]
- Curiel TJ, Wei S, Dong H, et al. Blockade of B7-H1 improves myeloid dendritic cell-mediated antitumor immunity. Nat Med. 2003;9(5):562–567. doi: 10.1038/nm863. [DOI] [PubMed] [Google Scholar]
- Della Bella S, Gennaro M, Vaccari M, et al. Altered maturation of peripheral blood dendritic cells in patients with breast cancer. Br J Cancer. 2003;89(8):1463–1472. doi: 10.1038/sj.bjc.6601243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Nicola M, Carlo-Stella C, Mortarini R, et al. Boosting T cell-mediated immunity to tyrosinase by vaccinia virus-transduced, CD 34(+)-derived dendritic cell vaccination: a phase I trial in metastatic melanoma. Clin Cancer Res. 2004;10(16):5381–5390. doi: 10.1158/1078-0432.CCR-04-0602. [DOI] [PubMed] [Google Scholar]
- El Marsafy S, Bagot M, Bensussan A, et al. Dendritic cells in the skin--potential use for melanoma treatment. Pigment Cell Melanoma Res. 2009;22(1):30–41. doi: 10.1111/j.1755-148X.2008.00532.x. [DOI] [PubMed] [Google Scholar]
- Enk AH, Jonuleit H, Saloga J, et al. Dendritic cells as mediators of tumor-induced tolerance in metastatic melanoma. Int J Cancer. 1997;73(3):309–316. doi: 10.1002/(sici)1097-0215(19971104)73:3<309::aid-ijc1>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- Ferlazzo G, Wesa A, Wei WZ, et al. Dendritic cells generated either from CD34+ progenitor cells or from monocytes differ in their ability to activate antigen-specific CD8+ T cells. J Immunol. 1999;163(7):3597–3604. [PubMed] [Google Scholar]
- Flacher V, Sparber F, Tripp CH, et al. Targeting of epidermal Langerhans cells with antigenic proteins: attempts to harness their properties for immunotherapy. Cancer Immunol Immunother. 2009;58(7):1137–1147. doi: 10.1007/s00262-008-0563-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fricke I, Gabrilovich DI. Dendritic cells and tumor microenvironment: a dangerous liaison. Immunol Invest. 2006;35(3-4):459–483. doi: 10.1080/08820130600803429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita H, Nograles KE, Kikuchi T, et al. Human Langerhans cells induce distinct IL-22-producing CD4+ T cells lacking IL-17 production. Proc Natl Acad Sci U S A. 2009;106(51):21795–21800. doi: 10.1073/pnas.0911472106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabrilovich DI, Corak J, Ciernik IF, et al. Decreased antigen presentation by dendritic cells in patients with breast cancer. Clin Cancer Res. 1997;3(3):483–490. [PubMed] [Google Scholar]
- Galan A, Ko CJ. Langerhans cells in squamous cell carcinoma vs. pseudoepitheliomatous hyperplasia of the skin. J Cutan Pathol. 2007;34(12):950–952. doi: 10.1111/j.1600-0560.2007.00741.x. [DOI] [PubMed] [Google Scholar]
- Gottfried E, Kreutz M, Mackensen A. Tumor-induced modulation of dendritic cell function. Cytokine Growth Factor Rev. 2008;19(1):65–77. doi: 10.1016/j.cytogfr.2007.10.008. [DOI] [PubMed] [Google Scholar]
- Ishida T, Oyama T, Carbone DP, et al. Defective function of Langerhans cells in tumor-bearing animals is the result of defective maturation from hemopoietic progenitors. J Immunol. 1998;161(9):4842–4851. [PubMed] [Google Scholar]
- Kalinski P, Urban J, Narang R, et al. Dendritic cell-based therapeutic cancer vaccines: what we have and what we need. Future Oncol. 2009;5(3):379–390. doi: 10.2217/FON.09.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maggi L, Santarlasci V, Capone M, et al. CD161 is a marker of all human IL-17-producing T-cell subsets and is induced by RORC. European journal of immunology. 2010;40(8):2174–2181. doi: 10.1002/eji.200940257. [DOI] [PubMed] [Google Scholar]
- Mortarini R, Anichini A, Di Nicola M, et al. Autologous dendritic cells derived from CD34+ progenitors and from monocytes are not functionally equivalent antigen-presenting cells in the induction of melan-A/Mart-1(27-35)-specific CTLs from peripheral blood lymphocytes of melanoma patients with low frequency of CTL precursors. Cancer Res. 1997;57(24):5534–5541. [PubMed] [Google Scholar]
- Nestle FO, Alijagic S, Gilliet M, et al. Vaccination of melanoma patients with peptide- or tumor lysate-pulsed dendritic cells. Nat Med. 1998;4(3):328–332. doi: 10.1038/nm0398-328. [DOI] [PubMed] [Google Scholar]
- Nestle FO, Burg G, Fah J, et al. Human sunlight-induced basal-cell-carcinoma-associated dendritic cells are deficient in T cell co-stimulatory molecules and are impaired as antigen-presenting cells. Am J Pathol. 1997;150(2):641–651. [PMC free article] [PubMed] [Google Scholar]
- Palucka AK, Ueno H, Connolly J, et al. Dendritic cells loaded with killed allogeneic melanoma cells can induce objective clinical responses and MART-1 specific CD8+ T-cell immunity. J Immunother. 2006;29(5):545–557. doi: 10.1097/01.cji.0000211309.90621.8b. [DOI] [PubMed] [Google Scholar]
- Perrot I, Blanchard D, Freymond N, et al. Dendritic cells infiltrating human non-small cell lung cancer are blocked at immature stage. J Immunol. 2007;178(5):2763–2769. doi: 10.4049/jimmunol.178.5.2763. [DOI] [PubMed] [Google Scholar]
- Pinzon-Charry A, Maxwell T, Lopez JA. Dendritic cell dysfunction in cancer: a mechanism for immunosuppression. Immunol Cell Biol. 2005;83(5):451–461. doi: 10.1111/j.1440-1711.2005.01371.x. [DOI] [PubMed] [Google Scholar]
- Ratzinger G, Baggers J, de Cos MA, et al. Mature human Langerhans cells derived from CD34+ hematopoietic progenitors stimulate greater cytolytic T lymphocyte activity in the absence of bioactive IL-12p70, by either single peptide presentation or cross-priming, than do dermal-interstitial or monocyte-derived dendritic cells. J Immunol. 2004;173(4):2780–2791. doi: 10.4049/jimmunol.173.4.2780. [DOI] [PubMed] [Google Scholar]
- Romani N, Thurnher M, Idoyaga J, et al. Targeting of antigens to skin dendritic cells: possibilities to enhance vaccine efficacy. Immunol Cell Biol. 2010;88(4):424–430. doi: 10.1038/icb.2010.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg SA, Yang JC, Restifo NP. Cancer immunotherapy: moving beyond current vaccines. Nat Med. 2004;10(9):909–915. doi: 10.1038/nm1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuler G, Schuler-Thurner B, Steinman RM. The use of dendritic cells in cancer immunotherapy. Curr Opin Immunol. 2003;15(2):138–147. doi: 10.1016/s0952-7915(03)00015-3. [DOI] [PubMed] [Google Scholar]
- Shurin MR, Lotze MT. Dendritic cells in Cancer: Emergence of the Discipline. In: Shurin MR, Salter RD, editors. Dendritic Cells in Cancer. New York: Springer; 2009. pp. 11–30. [Google Scholar]
- Steinman RM, Banchereau J. Taking dendritic cells into medicine. Nature. 2007;449(7161):419–426. doi: 10.1038/nature06175. [DOI] [PubMed] [Google Scholar]
- Stoitzner P, Green LK, Jung JY, et al. Tumor immunotherapy by epicutaneous immunization requires langerhans cells. J Immunol. 2008;180(3):1991–1998. doi: 10.4049/jimmunol.180.3.1991. [DOI] [PubMed] [Google Scholar]
- Stoitzner P, Sparber F, Tripp CH. Langerhans cells as targets for immunotherapy against skin cancer. Immunol Cell Biol. 2010;88(4):431–437. doi: 10.1038/icb.2010.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahara M, Chen S, Kido M, et al. Stromal CD10 expression, as well as increased dermal macrophages and decreased Langerhans cells, are associated with malignant transformation of keratinocytes. J Cutan Pathol. 2009;36(6):668–674. doi: 10.1111/j.1600-0560.2008.01139.x. [DOI] [PubMed] [Google Scholar]
- Talmadge JE, Donkor M, Scholar E. Inflammatory cell infiltration of tumors: Jekyll or Hyde. Cancer Metastasis Rev. 2007;26(3-4):373–400. doi: 10.1007/s10555-007-9072-0. [DOI] [PubMed] [Google Scholar]
- Thurner B, Haendle I, Roder C, et al. Vaccination with mage-3A1 peptide-pulsed mature, monocyte-derived dendritic cells expands specific cytotoxic T cells and induces regression of some metastases in advanced stage IV melanoma. J Exp Med. 1999;190(11):1669–1678. doi: 10.1084/jem.190.11.1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troy AJ, Summers KL, Davidson PJ, et al. Minimal recruitment and activation of dendritic cells within renal cell carcinoma. Clin Cancer Res. 1998;4(3):585–593. [PubMed] [Google Scholar]
- Ueno H, Schmitt N, Klechevsky E, et al. Harnessing human dendritic cell subsets for medicine. Immunol Rev. 2010;234(1):199–212. doi: 10.1111/j.0105-2896.2009.00884.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicari AP, Caux C, Trinchieri G. Tumour escape from immune surveillance through dendritic cell inactivation. Semin Cancer Biol. 2002;12(1):33–42. doi: 10.1006/scbi.2001.0400. [DOI] [PubMed] [Google Scholar]
- Zou W. Immunosuppressive networks in the tumour environment and their therapeutic relevance. Nat Rev Cancer. 2005;5(4):263–274. doi: 10.1038/nrc1586. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.