

SUMMARY
Using genetic data from an obesity candidate gene study of self-reported African Americans and European Americans, we investigated the number of Ancestry Informative Markers (AIMs) and candidate gene SNPs necessary to infer continental ancestry. Proportions of African and European ancestry were assessed with STRUCTURE (K=2), using 276 AIMs. These reference values were compared to estimates derived using 120, 60, 30, and 15 SNP subsets randomly chosen from the 276 AIMs and from 1144 SNPs in 44 candidate genes. All subsets generated estimates of ancestry consistent with the reference estimates, with mean correlations greater than 0.99 for all subsets of AIMs, and mean correlations of 0.99±0.003; 0.98± 0.01; 0.93±0.03; and 0.81± 0.11 for subsets of 120, 60, 30, and 15 candidate gene SNPs, respectively. Among African Americans, the median absolute difference from reference African ancestry values ranged from 0.01 to 0.03 for the four AIMs subsets and from 0.03 to 0.09 for the four candidate gene SNP subsets. Furthermore, YRI/CEU Fst values provided a metric to predict the performance of candidate gene SNPs. Our results demonstrate that a small number of SNPs randomly selected from candidate genes can be used to estimate admixture proportions in African Americans reliably.
Keywords: Ancestry Informative Markers, AIMs, African Americans, candidate genes, genetic ancestry, Structure, admixture, population stratification
INTRODUCTION
Genetic epidemiology studies seek to identify loci with statistically significant allele or genotype frequency differences between cases and controls. If case status covaries with differences in ancestry, many genetic loci can be expected to differ in their allele frequencies irrespective of etiology, potentially giving rise to spurious associations (Clayton et al., 2005; Price et al., 2008; Rosenberg and Nordborg, 2006; Tian et al., 2006; Tian et al., 2008b). To minimize the confounding effects of population substructure, quantitative estimates of individual ancestry need to be considered. Bayesian clustering or maximum likelihood methods are typically used to calculate these estimates (Alexander et al., 2009; Pritchard et al., 2000), or clusters are identified using Principal Components Analysis (PCA) or multidimensional scaling with genotyped genetic markers (Li and Yu, 2008; Novembre and Stephens, 2008; Price et al., 2006).
The number of genotyped markers sufficient to infer and to correct for ancestry depends on the genetic heterogeneity of the populations under study and the informativeness of the markers being used, with informativeness a function of allele frequency differences between the ancestral populations from which the study samples derive. The most commonly used markers for this purpose are termed Ancestry Informative Markers (AIMs), which are selected for their large allele frequency differences between ancestral populations (Akey et al., 2002; Halder et al., 2008; Nassir et al., 2009; Rosenberg et al., 2003; Shriver et al., 1997; Smith et al., 2004). However, genetic variants with relatively small allele frequency differences between populations can also be used to infer ancestry if they are genotyped in sufficient number. In large-scale candidate gene studies or in genome-wide association studies (GWAS), the sheer quantity of markers under study makes the use of independent AIMs unnecessary, even for assessing ancestry in highly homogenous populations. For example, using 960 cancer candidate gene SNPs, Sloan et al. (Sloan et al., 2009) were able to infer the European country of origin in immigrants to the US; and using over half a million SNPs, Novembre et al. (Novembre et al., 2008) were able to characterize substructure in a European population accurate to within a few hundred kilometers of geographic origin.
Although panels of AIMs have been assumed to be necessary in smaller candidate gene studies, the point at which they can be confidently bypassed, owing to a sufficient number of candidate SNPs, remains largely unexplored. Allocco et al. found that as few as 50 SNPs chosen randomly from the HapMap database can assign individuals to their ancestral continent of origin with an average accuracy of 95%, suggesting that AIMs may not be necessary even in studies with relatively few markers (Allocco et al., 2007). However, SNPs chosen randomly from throughout the genome via HapMap are likely to provide more independent information than SNPs chosen from a set of non-randomly distributed candidate genes, many of which may be in linkage disequilibrium. Moreover, many studies of admixed populations, such as African Americans, require an assessment of the proportion of admixture. These analyses demand more information from genotypic markers than when individuals need only be assigned to their continent of origin.
Herein, we present an analysis of 1300 self-reported African-American and 1247 self-reported European American subjects using 276 AIMs and 1144 obesity-related candidate gene SNPs to evaluate the number of AIMs and/or candidate gene SNPs necessary to characterize global ancestry adequately in similarly designed studies.
MATERIALS AND METHODS
Ethics Statement
The Southern Community Cohort Study (SCCS) participants provided written informed consent, and protocols were approved by the Vanderbilt University Human Research Program and Institutional Review Board and by the Meharry Medical College Institutional Review Board.
Study Population
The SCCS is a cohort study of cancer risk disparities related to ancestry and socioeconomic status among populations. Men and women aged 40–79 were recruited in person at community health centers and also by mail across 12 southeastern US states between 2002 and 2009 (Signorello et al., 2010; Signorello et al., 2005). Approximately 86,000 participants were enrolled, with African Americans comprising two-thirds of the study population (www.southerncommunitystudy.org).
For an obesity-related candidate gene study within the SCCS, 2157 female and 390 male participants (1300 of self-reported African ancestry and 1247 of self-reported European ancestry) were selected from among those who enrolled from March 2002–October 2004. Genomic DNA was extracted from blood samples using Qiagen’s DNA Purification kits (Qiagen, Valencia, CA) according to manufacturer’s instructions.
Marker Selection and Genotyping
We selected AIMs from a list of 1,509 AIM SNPs from an Illumina-designed panel for ancestry estimation and an additional 360 SNPs with comparably large frequency differences between European (CEU) and African (YRI) samples in HapMap I. We selected AIMs using the following criteria: (1) AIMs were at least 5 MB from any of the 44 candidate gene boundaries to ensure independence from the candidate genes and (2) AIMS displayed the largest allele frequency differences between the CEU and YRI HapMap populations. From this list we chose 300 AIMs of which 292 passed the Illumina Scoring algorithm and were genotyped with the Illumina GoldenGate platform (Illumina Inc., San Diego, CA). All SNPs were assessed for deviations from Hardy-Weinberg equilibrium. Of the 292 AIMs, 276 were successfully genotyped with call rates greater than 95% and subsequently used to estimate African and European ancestry in the SCCS African Americans and European Americans.
An additional panel of genetic markers was selected from the obesity-related candidate genes, comprising 1244 SNPs. The candidate SNPs were selected for the obesity study using a tagSNP approach that combined tagSNPs from both the European (CEU) and Yoruba (YRI) HapMap 1 data (Thorisson et al., 2005). HapMap SNPs within each gene and an additional 10kb upstream and downstream of each gene were identified and evaluated by the Illumina scoring algorithm. SNPs that scored poorly or had minor allele frequencies below 0.05 in both CEU and YRI were excluded. LDSelect was then run separately for the CEU and YRI data using an r2 cutoff of 0.8 to partition SNPs into linkage disequilibrium (LD) bins for each population (Carlson et al., 2004). When multiple tagSNPs were in an LD bin, SNPs that tagged both populations were preferentially selected. Among equivalent tagSNPs of a given LD bin, one categorized as a candidate functional SNP or one previously employed on Illumina chips was preferentially selected for assay. The same quality control criteria were applied as for the AIMs, and resulted in the removal of 100 SNPs, yielding 1144 for ancestry estimation.
Analyses
African and European ancestry for each individual was estimated using STRUCTURE (version 2.2.3, http://pritch.bsd.uchicago.edu/structure.html), a software platform that uses a Bayesian clustering algorithm to identify groups of individuals with similar allele frequency profiles (Pritchard et al., 2000). The algorithm estimates the shared population ancestry of individuals based solely on their genotypes, under the assumptions of Hardy-Weinberg equilibrium and linkage equilibrium in ancestral populations. Individuals are assigned admixture estimates, proportions of ancestry summing to 1 across K clusters (K=2 ancestral populations for all analyses in this study). All runs of STRUCTURE were performed on the ACCRE supercomputing cluster at Vanderbilt University.
To create reference values of African and European ancestry proportions for all 2547 individuals, STRUCTURE was run 10 times (50,000 iterations after a burn-in of 50,000 iterations) using the 276 AIMs. CLUMPP was used to align multiple replicate analyses and to calculate the means of the 10 quantitative ancestry estimates per individual (Jakobsson and Rosenberg, 2007). This procedure was repeated using the 1144 candidate gene SNPs, and the Pearson correlation coefficient between the two sets of individual ancestry estimates was determined.
From each of the sets of 276 AIMs and 1144 candidate gene SNPs, 100 random subsets of 120, 60, 30, and 15 markers were selected. Markers were selected without replacement for each randomization. The number of genes represented in each random sample of SNPs was tabulated. STRUCTURE was run 10 times (50,000 iterations after a burn-in of 50,000 iterations) for each of the 800 randomized datasets (100 randomizations for each category of 120, 60, 30, and 15 AIMs and 120, 60, 30, and 15 candidate gene SNPs). The means of the 10 quantitative ancestry estimates per individual per randomization were calculated, generating 100 sets of 2547 mean ancestry estimates for each of the 8 categories (Figure 1S). The Pearson correlation coefficients between each of these sets of individual ancestry estimates and the reference vector were then calculated.
To assess how accurately a given individual’s ancestry could be estimated using only 120, 60, 30, or 15 AIMs or candidate gene SNPs, each individual’s reference estimate of African ancestry was subtracted from each of the individual’s 100 estimates of African ancestry per category of 120, 60, 30, or 15 AIMs and candidate gene SNPs. The frequency distributions of the 254,700 absolute values of differences per category were plotted, in addition to the frequency distributions for only the 1300 self-reported African Americans.
Mean Fst was calculated for each randomization of candidate gene SNPs, using allele frequency data from the YRI and CEU HapMap samples. For the 1300 self-reported African Americans, the association between Fst and the mean absolute difference of African ancestry estimates from reference values was assessed using linear regression for each category of candidate gene SNPs. The Weir and Cockerham algorithm was used to calculate Fst (Weir and Cockerham, 1984).
To determine whether the population differentiation of our SNPs was consistent with background levels of genetic distance between African Americans and European Americans, Fst values were first calculated for the 1144 candidate gene SNPs and 276 AIMs using genotype data from our study. These values were then compared to Fst values for 40 random draws of 1144 SNPs from unrelated HapMap phase III CEU and ASW (African Americans in the Southwest USA) samples, using the Kruskal–Wallis one-way analysis of variance test, a non-parametric method for determining whether samples originate from the same distribution. A two-sided significance probability of 0.05 was used to infer non-random influences.
Finally, the extent to which correlations with the reference estimates were impacted by the number of genes per randomization was determined. For each category of candidate gene SNPs, the associations between the number of genes represented in each randomized sample and the correlation of that sample’s ancestry estimates with the reference estimates was assessed using linear regression. Statistical analyses were done using R (http://cran.r-project.org/) and STATA 10.0.
RESULTS
Ancestry estimated from 276 AIMs and 1144 candidate gene SNPs
Reference values of ancestry for the 2547 individuals were calculated using 276 AIMs. The 1300 self-reported African-Americans had a mean proportion of 0.92 African ancestry and the 1247 self-reported European Americans a mean African ancestry of 0.01. Nine of the self-reported African Americans were found to have greater than 0. 88 proportion European ancestry, suggesting incorrect classification. The 1144 candidate gene SNPs yielded ancestry estimates that were highly correlated with the reference estimates derived from the 276 AIMs (r = 0.989).
Ancestry estimated from random subsets of 276 AIMs
For the full study population, the 15 AIM subsets generated ancestry estimates that were highly correlated with the reference values (r = 0.991) (Table 1). Of the total 254,700 ancestry estimates for all subjects generated using 15 AIMs, 93.8% fell within ± 0.15 of the corresponding reference estimates. The mean absolute difference from the reference values was 0.06 ± 0.04, and the median was less than 0.01 (Figure 1). For the self-reported African Americans, the mean absolute difference from the reference values was 0.06 ± 0.07, and the median was 0.03, with the great majority (89.1%) of estimates within ± 0.15 of the reference estimates (Figure 2). Using subsets of 30, 60, and 120 AIMs, the mean absolute difference from the reference values for the self-reported African Americans improved to 0.05 ± 0.06, 0.04 ± 0.04, and 0.02± 0.03, respectively, with medians of 0.03, 0.02, and 0.01 (Figure 2).
Table 1.
Correlations between sets of 2547 ancestry estimates derived using the full set of AIMs and those derived using subsets of SNPs.
| Mean r | S.E. | Min - Max | |
|---|---|---|---|
| Candidate gene SNP panel* | 0.989 | NA | NA |
| Random AIM subsets, n | |||
| 15 | 0.991 | 0.001 | 0.989 – 0.993 |
| 30 | 0.995 | <0.001 | 0.994 – 0.995 |
| 60 | 0.997 | <0.001 | 0.996 – 0.997 |
| 120 | 0.999 | <0.001 | 0.998 – 0.999 |
| Random candidate gene SNP subsets, n | |||
| 15 | 0.811 | 0.106 | 0.384 – 0.943 |
| 30 | 0.934 | 0.032 | 0.785 – 0.978 |
| 60 | 0.977 | 0.009 | 0.922 – 0.987 |
| 120 | 0.986 | 0.003 | 0.977 – 0.990 |
Candidate gene SNP panel: N=1144 SNPs
Mean r refers to mean of 100 correlations
S.E. = standard error
Figure 1. Variation in estimates of ancestry with AIM subsets.
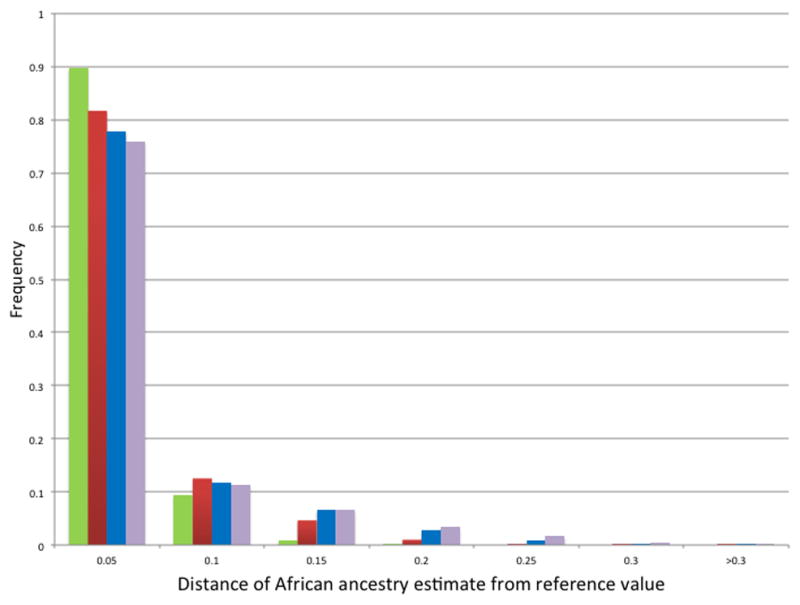
Distribution of absolute values of differences between reference estimates of African ancestry and corresponding estimates derived using random subsets of 120 (blue), 60 (red), 30 (green), and 15 (purple) AIMs, for all 2547 European American and African American study participants.
Figure 2. Variation in estimates of ancestry for self-reported African Americans only, using AIM subsets.
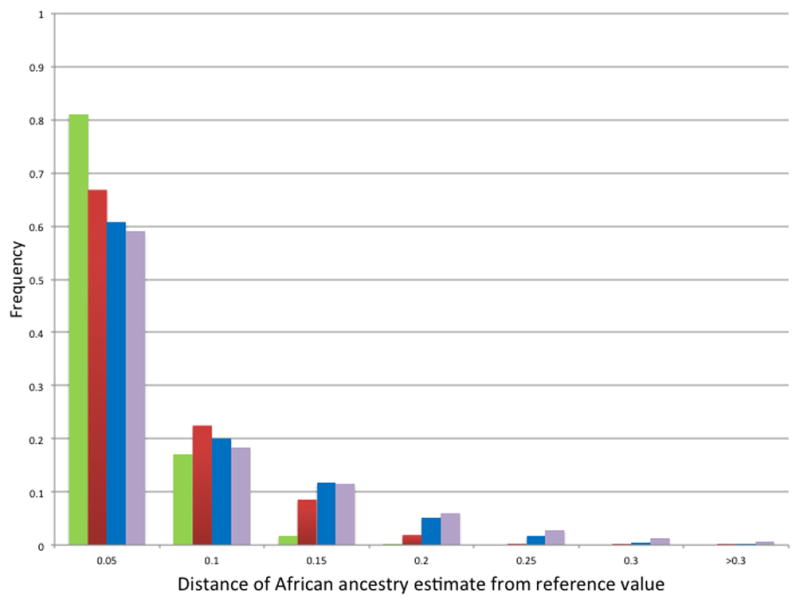
Distribution of absolute values of differences between the self-reported African Americans’ reference estimates of African ancestry and corresponding estimates derived using random subsets of 120 (blue), 60 (red), 30 (green), and 15 (purple) AIMs.
Ancestry estimated from random subsets of 1144 candidate gene SNPs
Highly correlated ancestry estimates were also obtained when smaller subsets of the 1144 candidate genes were used for estimation (Table 1). With 120 candidate gene SNPs, the mean Pearson correlation coefficient with the reference estimates was 0.986 ± 0.003, indicating that little information was lost when roughly 10% of the full set of candidate gene SNPs was used. The smallest correlation obtained from the 100 randomizations of 120 candidate gene SNPs was 0.977 (Table 1 and Figure 3). Of the total 254,700 ancestry estimates generated using 120 randomly-chosen candidate gene SNPs, the mean absolute difference from the reference values was 0.04 ± 0.07, the median was 0.01, and 92.0% of estimates fell within ± 0.15 of their corresponding reference estimates (Figure 4). For the self-reported African Americans, the mean absolute difference from the reference estimates was 0.06 ± 0.08 and the median was 0.03; 86.5% of estimates fell within ± 0.15 of the reference estimates (Figure 5).
Figure 3. Correlations of ancestry estimates using candidate gene SNPs.
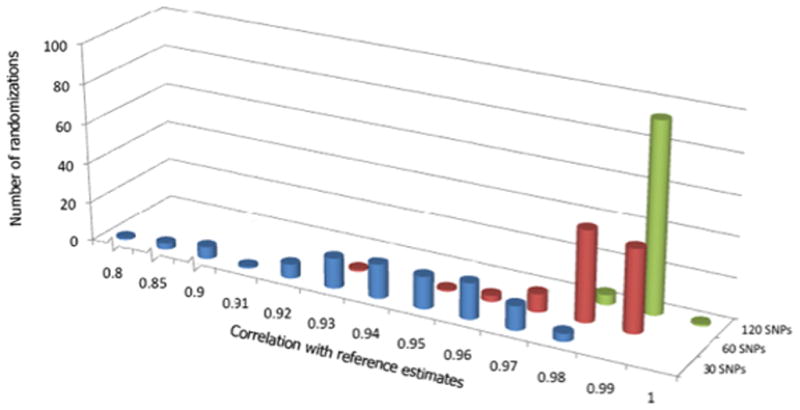
Distribution of correlations between the reference set of ancestry estimates for all 2547 study participants and 100 sets of corresponding estimates derived using 30 (blue), 60 (red), and 120 (green) random candidate gene SNPs. Data for 15 SNPs not shown; see Table 1.
Figure 4. Variation in estimates of ancestry with candidate gene SNP subsets.
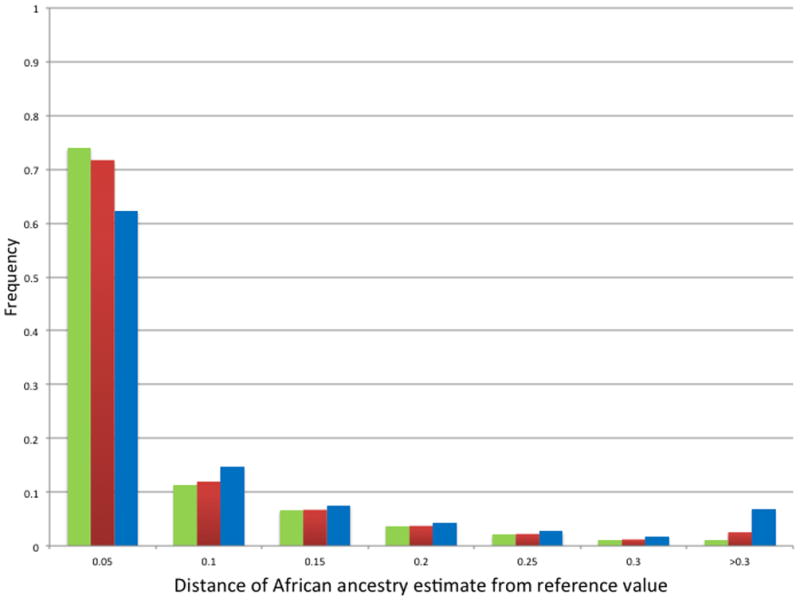
Distribution of absolute values of differences between reference estimates of African ancestry and corresponding estimates derived using random subsets of 120 (blue), 60 (red), and 30 (green) candidate gene SNPs, for all 2547 study participants.
Figure 5. Variation in estimates of ancestry for self-reported African Americans only, using candidate gene SNP subsets.
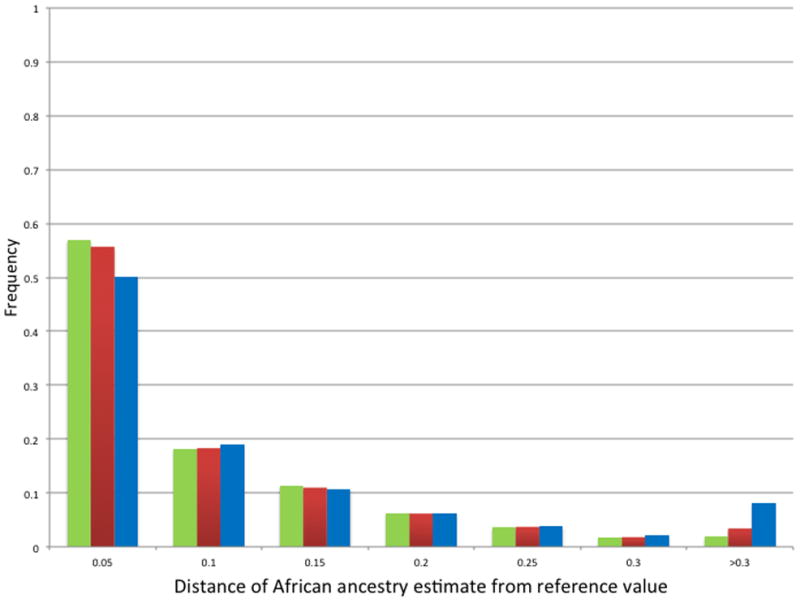
Distribution of absolute values of differences between the self-reported African Americans’ reference estimates of African ancestry and corresponding estimates derived using random subsets of 120 (blue), 60 (red), and 30 (green) candidate gene SNPs.
Results remained consistent when 60 candidate gene SNPs were used. The mean correlation with the reference estimates was 0.977 ± 0.009, with only 2 of 100 randomizations yielding correlations less than 0.95 (0.922 and 0.947) (Table 1 and Figure 3). The mean absolute difference from the reference values was 0.05 ± 0.09, and the median was 0.01; only 9.6% of all ancestry estimates differed from corresponding reference values by more than ±0.15 (Figure 4). For the self-reported African Americans, the mean difference from the reference estimates was 0.07 ± 0.10 and the median was 0.04; 85.0% of estimates fell within ± 0.15 of the reference values (Figure 5).
Random subsets of 30 candidate gene SNPs generated ancestry estimates less consistent with the reference values, with correlations ranging from 0.78 to 0.95. However, 92% of the 30 SNP randomizations yielded correlations greater than 0.90 (Table 1 and Figure 3). For the self-reported African Americans, the mean absolute difference from the reference values was 0.10 ± 0.15, the median was 0.05, and 80.0% of estimates fell within ± 0.15 of the reference values (Figure 5). Random subsets of 15 candidate gene SNPs performed markedly worse than the other subsets (Table 1), especially with respect to inferring the ancestry of the self-reported African Americans: the mean absolute difference from the reference values was 0.18 ± 0.21 and the median was 0.09, with 34.9% of estimates missing the reference by more than 0.15.
Performance of candidate gene SNP subsets based on YRI/CEU Fst
An inverse relationship existed between the YRI/CEU Fst values of candidate gene SNP randomizations and their accuracy in estimating African ancestry in the 1300 self-reported African Americans (as measured by mean absolute value of differences from reference values). This relationship was stronger in smaller SNP subsets, and significant in all subsets except the 120 SNP data (Figure 6; Table 1S). For the 120 SNP subsets, the r2 = 0.02 (p = 0.16).
Figure 6. Relationship between Fst and accuracy of African ancestry estimation.
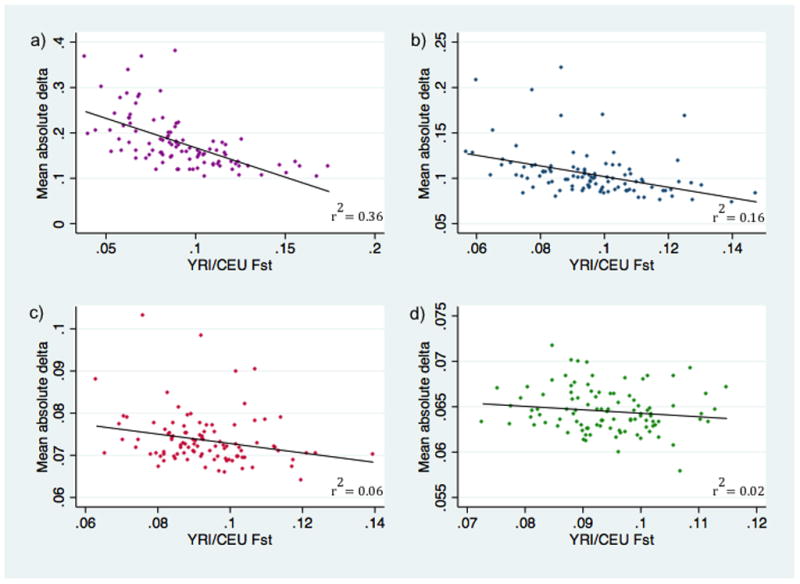
For each category of 15 (panel a), 30 (panel b), 60 (panel c), and 120 (panel d) candidate gene SNPs, randomized subsets are mapped by mean YRI/CEU Fst (horizontal axis) and mean absolute difference of African ancestry estimates from corresponding reference estimates for the 1300 self-reported African Americans (vertical axis). The slope of the linear fit was significantly different from zero for 15, 30, and 60 SNPs (p<0.0001, p<0.0001, p=0.017, respectively). Horizontal and vertical axes vary among the panels.
Genetic differentiation of AIMs, 1144 candidate gene SNPs, and HapMap data
The median Fst value between self-reported African Americans and self-reported European Americans for the 1144 candidate gene SNPs was 0.059, which was slightly higher than the median Fst value calculated for 40 draws of 1144 random SNPs taken from the HapMap CEU and ASW samples (0.050). The mean Fst for the 1144 candidate gene SNPs (0.087) was slightly lower than that for the 40 random draws from HapMap (0.091). As expected, these Fst estimates were much smaller than the mean and median Fst for the 276 AIMs (both were 0.51) (Figure 7). Forty percent (16/40) of the Fst distribution comparisons between the ASW/CEU simulations and the candidate gene SNPs were not statistically significant at p = 0.05.
Figure 7. Distribution of Fst values.
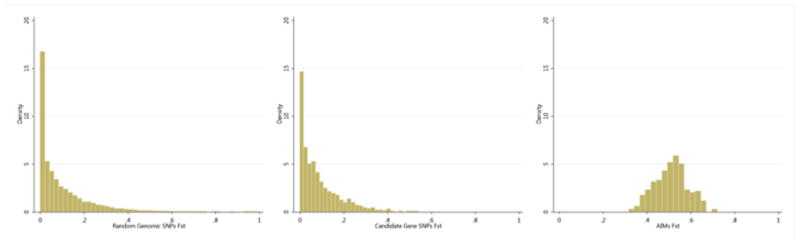
The distribution of Fst values between the CEU and ASW HapMap populations using 40 iterations of 1144 randomly selected SNPs (left), and the Fst distributions of the 1144 candidate gene SNPs (center) and 276 AIMs (right) for the self-reported African American and European American study participants.
Effect of number of genes used to estimate ancestry
The candidate gene SNPs were sampled from a total of 44 candidate genes. The mean number of candidate genes represented in the subsets of 120, 60, 30, and 15 SNPs was 34.3 ± 2.0, 27.1 ± 2.1, 18.9 ± 1.9, and 11.8 ± 1.4, respectively. For a given number of SNPs (120, 60, 30, or 15), correlations did not vary significantly with the number of candidate genes represented in the randomized samples (Figure 2S; p > 0.28 for all subsets).
DISCUSSION
The need to adjust genetic association studies for differences in ancestry between cases and controls is now well recognized. Differentially distributed continental ancestry, in particular, increases the risk of type 1 error, because the fraction of SNPs with >40% allele frequency differences between continental populations is an order of magnitude greater than the fraction within continental sub-populations (Tian et al., 2008a). Because smaller candidate gene studies and follow-up studies to GWAS assess only a limited number of markers, panels of AIMs are often genotyped to address these issues (Seldin and Price, 2008). Recent studies suggest that ancestry, and especially continental ancestry, can be characterized with fewer AIMs than originally thought (Risch et al., 2002; Ruiz-Narvaez et al., 2011; Sampson et al., 2011; Tsai et al., 2005). For example, a subset of 24 AIMs from a set of 128 adequately distinguished European and West African ancestry (Kosoy et al., 2009).
Using a large number of AIMs to estimate ancestry for our reference measure and a rigorous re-sampling approach, our study confirms that as few as 15 AIMs provide excellent correlation with reference estimates, indicating that a small number of AIMs is sufficient to differentiate continental ancestry. The accuracy of the estimates tended to be lower among African Americans than European Americans, but with only 15 AIMs, 89.1% of the ancestry estimates for the 1300 self-reported African Americans fell within ± 0.15 of reference estimates. When 30, 60, and 120 AIMs were used, the percent of estimates falling within ± 0.15 of the reference values in African Americans improved to 92.6%, 97.9%, and 99.9%, respectively. Thus, the practical utility of genotyping more than 15 AIMs for some types of studies, including those distinguishing African Americans from European Americans, would appear to be marginal. However, studies requiring increased accuracy, including those distinguishing moderate from high African ancestry among self-reported African Americans in the context of small effect sizes (Reich et al., 2004), could more prudently use approximately 60 AIMs, assuming that the level of accuracy we observed in this study is sufficient to minimize any threats to internal validity.
The history of candidate gene studies indicates that most interrogated markers do not associate with the phenotypes under study. Because we expect a far greater proportion of selected markers to associate significantly with continental ancestry than with any particular phenotype, the necessity of using AIMs to infer ancestry in medium- to large-scale candidate gene studies with a large number of unassociated markers is not clear. The numerical threshold at which ordinary SNPs perform as well as AIMs in this respect has been largely unexplored. Allocco et al. provided evidence that as few as 50 randomly selected HapMap SNPs can assign individuals to their continent of origin, but to our knowledge, our study is the first to systematically investigate the minimum number of non-independently drawn SNPs (e.g. candidate gene SNPs) sufficient to estimate proportions of admixture in a mixed study population. Our approach allowed us to evaluate the point at which genotyping AIMs may become superfluous, not only as a theoretical matter, but also as a practical guideline for minimizing expense in future candidate gene studies, deep sequencing analyses, and GWAS replications.
We found that as few as 60 SNPs drawn from 22–31 genes generated ancestry estimates that correlated well with reference ancestry estimates in our total sample (mean r=0.977). Within self-reported African Americans, 15.0% of the ancestry estimates deviated from the reference estimates by more than 0.15 when 60 candidate gene SNPs were used, not appreciably different than when 120 candidate gene SNPs were used (13.6%). The number of genes from which a given number of random SNPs were drawn did not significantly influence the correlations. Although not directly tested in our study, it is probable that admixture proportions can be well estimated with even fewer genes, as long as the number of independent SNPs from those genic regions (i.e., tagSNPs) is similar to the number that we tested.
To determine whether our particular candidate gene SNPs influenced these results, we calculated Fst values for the 1144 SNPs and compared them to Fst values for 1144 SNPs drawn randomly forty times from CEU and ASW HapMap samples. A moderate inflation in the differences between populations was expected, because we selected our candidate gene SNPs to tag both African American and European American samples. The median Fst of our candidate gene SNPs (0.059) was slightly higher than that of the random draws (0.050) and the mean was slightly lower (0.087 vs. 0.091), but both were much lower than the AIMs’ mean (0.51) and median (0.51), indicating that the variation in our candidate gene SNPs was not atypical of background levels of genetic variation between the two populations.
One way to assess the utility of candidate gene SNPs as ancestry estimators in studies with African Americans is to determine if the mean Fst value of a set of SNPs can predict their performance using Fst values calculated from pre-existing data, such as the YRI/CEU allele frequencies from HapMap. We found a significant linear relationship between mean YRI/CEU Fst and accuracy (measured as the mean absolute difference of estimates from reference values) for subsets of 15, 30 and 60 SNPs. The magnitude of correlation between mean Fst and accuracy decreased as the number of SNPs increased and the range of the mean absolute differences from references narrowed. For the 120 SNP category, where the range of differences in estimated ancestry was 0.058 to 0.072, the linear relationship between mean Fst and accuracy was not statistically significant, even though mean Fst varied by as much as 50% across different randomizations. This probably reflects the fact that the increasing ancestry information provided by the increased number of SNPs significantly outweighed the contribution of average differences in mean allele frequency somewhere between 60 and 120 SNPs. This analysis provides a metric with which to judge the likelihood that candidate gene SNPs will estimate ancestry well, and allows investigators to define their own tolerance for error for smaller sets of candidate gene SNPs.
Our data indicate that small numbers of AIMs and a moderately larger number of candidate gene SNPs can be effective in estimating continental ancestry. While larger studies with greater sample sizes should require less precision of individual assignments, if more precision is sought, mixing a few AIMs (e.g., 15) with the candidate gene SNPs will likely be adequate to correct for population stratification while still providing substantial cost savings. Selection of markers in this way can be a practical and cost-effective approach to estimating global genetic ancestry in admixed population studies.
Supplementary Material
Figure 1S: Flowchart of analytical approach. Sets of 15, 30, 60, and 120 markers were drawn randomly 100 times from the full set of AIMs and candidate gene SNPs, respectively. Each randomized set of markers was then used to derive estimates of African ancestry that were compared to the reference estimates.
Figure 2S: Correlations between reference values and candidate gene SNP analyses as a function of gene number. For each randomized set of 30 (blue), 60 (red) and 120 (green) candidate gene SNPs, the horizontal axis depicts the number of genes from which the SNPs were drawn, and the vertical axis depicts the correlation of that set’s ancestry estimates with the reference estimates.
Table 1S: The YRI/CEU Fst of candidate gene SNPs versus accuracy of African ancestry estimates for the 1300 self-reported African Americans
Acknowledgments
This project was supported in part by the Komen for the Cure grant OP05-0927-DR1 and by NIH grants R01CA092447 and 3T32GM080178-03S1. DNA sample preparation was conducted at the Survey and Biospecimen Shared Resource that is supported in part by the Vanderbilt-Ingram Cancer Center (P30 CA68485). Analysis was conducted in part using the resources of the Advanced Computing Center for Research and Education at Vanderbilt University, Nashville TN. We wish to thank Regina Courtney for DNA sample preparation.
References
- Akey JM, Zhang G, Zhang K, Jin L, Shriver MD. Interrogating a high-density SNP map for signatures of natural selection. Genome research. 2002;12:1805–1814. doi: 10.1101/gr.631202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander DH, Novembre J, Lange K. Fast model-based estimation of ancestry in unrelated individuals. Genome Res. 2009;19:1655–1664. doi: 10.1101/gr.094052.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allocco DJ, Song Q, Gibbons GH, Ramoni MF, Kohane IS. Geography and genography: prediction of continental origin using randomly selected single nucleotide polymorphisms. BMC genomics. 2007;8:68. doi: 10.1186/1471-2164-8-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlson CS, Eberle MA, Rieder MJ, Yi Q, Kruglyak L, Nickerson DA. Selecting a maximally informative set of single-nucleotide polymorphisms for association analyses using linkage disequilibrium. Am J Hum Genet. 2004;74:106–120. doi: 10.1086/381000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton DG, Walker NM, Smyth DJ, Pask R, Cooper JD, Maier LM, Smink LJ, Lam AC, Ovington NR, Stevens HE, et al. Population structure, differential bias and genomic control in a large-scale, case-control association study. Nature genetics. 2005;37:1243–1246. doi: 10.1038/ng1653. [DOI] [PubMed] [Google Scholar]
- Halder I, Shriver M, Thomas M, Fernandez JR, Frudakis T. A panel of ancestry informative markers for estimating individual biogeographical ancestry and admixture from four continents: utility and applications. Human mutation. 2008;29:648–658. doi: 10.1002/humu.20695. [DOI] [PubMed] [Google Scholar]
- Jakobsson M, Rosenberg NA. CLUMPP: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics. 2007;23:1801–1806. doi: 10.1093/bioinformatics/btm233. [DOI] [PubMed] [Google Scholar]
- Kosoy R, Nassir R, Tian C, White PA, Butler LM, Silva G, Kittles R, Alarcon-Riquelme ME, Gregersen PK, Belmont JW, et al. Ancestry informative marker sets for determining continental origin and admixture proportions in common populations in America. Hum Mutat. 2009;30:69–78. doi: 10.1002/humu.20822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li QZ, Yu K. Improved correction for population stratification in genome-wide association studies by identifying hidden population structures. Genetic Epidemiology. 2008;32:215–226. doi: 10.1002/gepi.20296. [DOI] [PubMed] [Google Scholar]
- Nassir R, Kosoy R, Tian C, White PA, Butler LM, Silva G, Kittles R, Alarcon-Riquelme ME, Gregersen PK, Belmont JW, et al. An ancestry informative marker set for determining continental origin: validation and extension using human genome diversity panels. BMC genetics. 2009;10:39. doi: 10.1186/1471-2156-10-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novembre J, Johnson T, Bryc K, Kutalik Z, Boyko AR, Auton A, Indap A, King KS, Bergmann S, Nelson MR, et al. Genes mirror geography within Europe. Nature. 2008;456:98–101. doi: 10.1038/nature07331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novembre J, Stephens M. Interpreting principal component analyses of spatial population genetic variation. Nature genetics. 2008;40:646–649. doi: 10.1038/ng.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price AL, Butler J, Patterson N, Capelli C, Pascali VL, Scarnicci F, Ruiz-Linares A, Groop L, Saetta AA, Korkolopoulou P, et al. Discerning the ancestry of European Americans in genetic association studies. Plos Genet. 2008;4:e236. doi: 10.1371/journal.pgen.0030236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nature genetics. 2006;38:904–909. doi: 10.1038/ng1847. [DOI] [PubMed] [Google Scholar]
- Pritchard JK, Stephens M, Donnelly P. Inference of population structure using multilocus genotype data. Genetics. 2000;155:945–959. doi: 10.1093/genetics/155.2.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reich D, Freedman ML, Penney KL, McDonald GJ, Mignault AA, Patterson N, Gabriel SB, Topol EJ, Smoller JW, Pato CN, et al. Assessing the impact of population stratification on genetic association studies. Nat Genet. 2004;36:388–393. doi: 10.1038/ng1333. [DOI] [PubMed] [Google Scholar]
- Risch N, Burchard E, Ziv E, Tang H. Categorization of humans in biomedical research: genes, race and disease. Genome Biol. 2002;3 doi: 10.1186/gb-2002-3-7-comment2007. comment 2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg NA, Li LM, Ward R, Pritchard JK. Informativeness of genetic markers for inference of ancestry. Am J Hum Genet. 2003;73:1402–1422. doi: 10.1086/380416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg NA, Nordborg M. A general population-genetic model for the production by population structure of spurious genotype-phenotype associations in discrete, admixed or spatially distributed populations. Genetics. 2006;173:1665–1678. doi: 10.1534/genetics.105.055335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz-Narvaez EA, Rosenberg L, Wise LA, Reich D, Palmer JR. Validation of a small set of ancestral informative markers for control of population admixture in African Americans. Am J Epidemiol. 2011;173:587–592. doi: 10.1093/aje/kwq401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampson JN, Kidd KK, Kidd JR, Zhao H. Selecting SNPs to identify ancestry. Ann Hum Genet. 2011;75:539–553. doi: 10.1111/j.1469-1809.2011.00656.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seldin MF, Price AL. Application of ancestry informative markers to association studies in European Americans. Plos Genet. 2008;4 doi: 10.1371/journal.pgen.0040005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shriver MD, Smith MW, Jin L, Marcini A, Akey JM, Deka R, Ferrell RE. Ethnic-affiliation estimation by use of population-specific DNA markers. American journal of human genetics. 1997;60:957–964. [PMC free article] [PubMed] [Google Scholar]
- Signorello LB, Hargreaves MK, Blot WJ. The Southern Community Cohort Study: investigating health disparities. Journal of health care for the poor and underserved. 2010;21:26–37. doi: 10.1353/hpu.0.0245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Signorello LB, Hargreaves MK, Steinwandel MD, Zheng W, Cai Q, Schlundt DG, Buchowski MS, Arnold CW, McLaughlin JK, Blot WJ. Southern community cohort study: establishing a cohort to investigate health disparities. Journal of the National Medical Association. 2005;97:972–979. [PMC free article] [PubMed] [Google Scholar]
- Sloan CD, Andrew AD, Duell EJ, Williams SM, Karagas MR, Moore JH. Genetic population structure analysis in New Hampshire reveals Eastern European ancestry. PloS one. 2009;4:e6928. doi: 10.1371/journal.pone.0006928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MW, Patterson N, Lautenberger JA, Truelove AL, McDonald GJ, Waliszewska A, Kessing BD, Malasky MJ, Scafe C, Le E, et al. A high-density admixture map for disease gene discovery in african americans. Am J Hum Genet. 2004;74:1001–1013. doi: 10.1086/420856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorisson GA, Smith AV, Krishnan L, Stein LD. The International HapMap Project Web site. Genome Res. 2005;15:1592–1593. doi: 10.1101/gr.4413105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian C, Gregersen PK, Seldin MF. Accounting for ancestry: population substructure and genome-wide association studies. Hum Mol Genet. 2008a;17:R143–R150. doi: 10.1093/hmg/ddn268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian C, Hinds DA, Shigeta R, Kittles R, Ballinger DG, Seldin MF. A genomewide single-nucleotide-polymorphism panel with high ancestry information for African American admixture mapping. American journal of human genetics. 2006;79:640–649. doi: 10.1086/507954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian C, Plenge RM, Ransom M, Lee A, Villoslada P, Selmi C, Klareskog L, Pulver AE, Qi L, Gregersen PK, et al. Analysis and application of European genetic substructure using 300 K SNP information. PLoS genetics. 2008b;4:e4. doi: 10.1371/journal.pgen.0040004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai HJ, Choudhry S, Naqvi M, Rodriguez-Cintron W, Burchard EG, Ziv E. Comparison of three methods to estimate genetic ancestry and control for stratification in genetic association studies among admixed populations. Hum Genet. 2005;118:424–433. doi: 10.1007/s00439-005-0067-z. [DOI] [PubMed] [Google Scholar]
- Weir BS, Cockerham CC. Estimating F-Statistics for the Analysis of Population-Structure. Evolution. 1984;38:1358–1370. doi: 10.1111/j.1558-5646.1984.tb05657.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure 1S: Flowchart of analytical approach. Sets of 15, 30, 60, and 120 markers were drawn randomly 100 times from the full set of AIMs and candidate gene SNPs, respectively. Each randomized set of markers was then used to derive estimates of African ancestry that were compared to the reference estimates.
Figure 2S: Correlations between reference values and candidate gene SNP analyses as a function of gene number. For each randomized set of 30 (blue), 60 (red) and 120 (green) candidate gene SNPs, the horizontal axis depicts the number of genes from which the SNPs were drawn, and the vertical axis depicts the correlation of that set’s ancestry estimates with the reference estimates.
Table 1S: The YRI/CEU Fst of candidate gene SNPs versus accuracy of African ancestry estimates for the 1300 self-reported African Americans