

Abstract
As ligand-regulated transcription factors, the nuclear hormone receptors are nearly ideal drug targets, with internal pockets that bind to hydrophobic, drug-like molecules and well-characterized ligand-induced conformational changes that recruit transcriptional coregulators to promoter elements. Yet, due to the multitude of genes under the control of a single receptor, the major challenge has been the identification of ligands with gene-selective actions, impacting disease outcomes through a narrow subset of target genes and not across their entire gene-regulatory repertoire. Here, we summarize the concepts and work to date underlying the development of steroidal and nonsteroidal receptor ligands, including the use of crystal structures, high-throughput screens, and rational design approaches for finding useful therapeutic molecules. Difficulties in finding selective receptor modulators require a more complete understanding of receptor interdomain communications, posttranslational modifications, and receptor-protein interactions that could be exploited for target gene selectivity.
Keywords: nuclear receptor, drug discovery, protein-ligand interactions
INTRODUCTION
The nuclear receptors (NRs) are a family of transcription factors that bind and respond to certain steroids and other signaling molecules, such as vitamin D3, thyroid hormone, and retinoids (1–5). The NR family also encodes metabolic sensors for other cholesterol-related and diet-derived lipids, and in recent years, newly recognized and more diverse signaling molecules such as phospholipids and heme have been shown to be ligands for some members (6–11). Still, a distinct subset of the NRs remains “orphans,” lacking a defined endogenous ligand, and these receptors potentially could be regulated through other means such as post-translational modifications.
For the purposes of drug discovery, NRs have favorable attributes, with most containing protected internal cavities predisposed for occupation with hydrophobic molecules of volumes typical of drugs. Moreover, these transcription factors control central pathways impacting a wide range of pathophysiologies ranging from cancers to metabolic diseases. However, the main limitation in exploiting these receptors for drug discovery is their level of direct involvement in fairly complex pathways and the ability of a single NR to impact numerous genes. Therefore, the difficulty in fully exploiting these receptors for drug discovery has been identifying ligands that control the desired, limited subset of genes for therapeutic purposes, and not all the genes under their control. This review focuses on those human NRs in which substantial drug discovery efforts have been under way, and for which crystal structures have assisted our understanding of how small molecules and tissue-selective modulators impart NR activities in therapeutically beneficial ways.
EXPLOITING THE MOLECULAR PHYSIOLOGY OF THE NUCLEAR RECEPTORS FOR DRUG DISCOVERY
Nearly all NRs contain two well-defined structural domains: a DNA-binding domain (DBD) that resides approximately along the center of the polypeptides and a ligand-binding domain (LBD) positioned along the C-terminal 200–300 residues. The N-terminal regions of NRs are highly variable in both sequence and size and in some cases harbor potent transcriptional activation functions, known as activation function-1 (AF-1), independently of the LBD interactions with ligands. The DBD and LBD are linked via a hinge region that is also variable and often contains DNA minor groove binding residues positioned just C-terminal to the DBD, referred to as C-terminal extensions (CTEs) (12). The LBD has been the main focus for drug discovery, as it is the site on which both ligands bind and coactivator-derived motifs interact (13). LBDs in some cases can also form one of the homo- or heterodimerization surfaces in NRs (14, 15). Also within the LBD is the activation function-2 (AF-2) for most NRs; this function refers to the recruitment of transcriptional activators in a ligand-dependent manner. Some receptors have meaningful interactions with corepressors that are also ligand-dependent. Both types of coregulators link the NRs directly to chromatin modification factors. As such, the coregulators act very much as scaffolds on which the associated histone-modifying factors change the chromatin configuration to allow or to block gene expression (16, 17).
The physiological ligands used by NRs act simply as molecular messengers signaling the body’s physiological states. Living organisms must continuously adapt and respond to both their nutritional states and their energetic and metabolic needs. The availability of these ligands, whether through endocrine or nutritional signaling, allows their diffusion through cells and direct binding to NR LBDs. This is the first step toward obtaining genetic responses required to maintain normal physiology and homeostasis in response to these molecular signals. NR ligands are invariably lipophilic in nature and accordingly bind in a sheltered hydrophobic cavity deeply embedded within a highly α-helical LBD. In the second step of signal interpretation, the ligand binding inside the LBD causes allosteric changes that appear on the receptor surface at the crucial sites of receptor-coregulator interactions. The selectivity of the NRs for specific target-specific DNA response elements is mainly encoded by the DBDs, their DNA recognition helices, and the patterns of homo- and heterodimerization between NRs (12, 18). DNA response elements ultimately localize the physiological responses to ligands into a precise genetic program that is both temporally and spatially controlled.
NR LBD structures typically contain nearly 12 α helices that are arranged around a central hydrophobic pocket, with helices 3, 7, and 10 providing amino acid residues that shape the pocket (2, 19, 20). The sizes of these ligand pockets can range from nearly zero in volume (filled with the receptor’s own hydrophobic side chains) to larger than 1500 Å3 (19). Nevertheless, the most interesting finding over recent years has been that pocket shapes and sizes are highly inducible, both in size and in receptivity to molecules that are unrelated to endogenous ligands. The C-terminal-most helical segment, helix 12 (H12), is the major architectural feature associated with AF-2 function and can undergo dramatic shifts in position in response to the molecule in the pocket (2, 21). Other α helices in the LBDs also shift in positions in subtle but still meaningful ways that can impact receptor activation.
As shown in Figure 1, the ligand can behave as a switch for coactivator and corepressor binding (2). The transcriptional coactivators include more than 200 proteins at this time, with different cell and tissue distributions and enormously varied functions (16, 17, 22–24). The coregulators bind to NRs typically using one or more LXXLL motifs within their polypeptides and associate with H12 via a charge clamp that supports this motif’s binding as a short α helix to the surface of the LBD. Operationally, an agonist can be viewed as a molecule that enhances the interactions of LBDs with one or more coactivator LXXLL motifs and therefore leads to transcriptional activation in a cell-based assay. By contrast, antagonists position H12 to physically compete with and block the site of coactivators, or otherwise do not support this binding. This is most clearly seen in the case of the estrogen receptor (ER), in which H12 of the LBD competes via a set of residues homologous to the coactivator sequence (see Figure 1b). Aside from this way of defining antagonists, other molecules can be so designated by their ability to recruit corepressors, including NcoR and SMRT, to LBDs via their related leucine-rich motifs. The coactivator and corepressor regions typically adopt a two- and a three-helical segment, respectively, when bound to receptor LBDs (25) (see Figure 1c). Interestingly, whereas some ligands, such as rosiglitazone, directly contact and stabilize their receptor’s H12 [peroxisome proliferator–activated receptor γ (PPARγ)], placing it in an agonist conformation, a significant number of other receptors position their H12 into the same conformation using an indirect mechanism whereby H12 is not directly contacted by ligand (26, 27). An example of indirect H12 activation involving a so-called pication activation switch is shown in Figure 2.
Figure 1.

(a) The 12-helical canonical fold of nuclear receptor (NR) ligand-binding domains (LBDs) and the conformational changes in retinoid X receptor (RXR) associated with ligand binding and receptor activation. The unliganded RXR-LBD changes its protein conformation, including helix 12 (H12) (shown in red) positioning upon binding to an agonist (SR11237). The ligand-activated receptor uses H12, as well as additional features in H3 and H4, to promote the binding of short helical LXXLL (where L is leucine and X is any residue) elements from coactivator protein (shown in purple) (21). (b) H12 geometry and activation function-2 (AF-2) function is ligand dependent. NR agonists, such as the natural ligand 17β-estradiol (E2) in the estrogen receptor (ER), cause a conformation in the LBD, particularly in H12 (red), which supports the formation of a binding surface for the helical NR-box module of the coactivators. Antagonists such as raloxifene, through the extension group on the C-ring of their ligand, induce alternate conformations in the LBD in which H12 is displaced away from its agonist conformation where it normally supports NR-box binding. Instead, H12 moves into that site of coactivator motif, using its own helical LXXML sequence to mimic the interactions normally made by the LXXLL motifs (41, 42). (c) The ligand-mediated exchange of coactivators and corepressors on NRs. The peroxisome proliferator–activated receptor α (PPARα) binding to coactivator versus corepressor segments can be governed by specific ligands. Whereas agonist ligands such as GW731 recruit coactivator NR-box elements from coactivators (SRC-1, shown in purple) to PPARα LBD (PDB ID 2P54), some antagonist ligands, such as GW6471, prevent the AF-2 helix (H12, shown in red) from assuming the active conformation, resulting in a larger crevice that can accommodate a corepressor (SMRT, shown in blue) segment with the LXXXIXXXL motif (25).
Figure 2.

Ligand-induced effects on helix 12 (H12) through a pi-cation activation switch. A number of nuclear receptors, such as those shown here, use a conserved switch involving an electrostatic interaction between the indole ring histidine on H10/H11 and a single aromatic side chain, typically a trytophan residue, on H12. An oxygen group from each receptor’s endogenous ligand [6-ethyl-CDCA for farnesoid X receptor (FXR); 24-25S-epoxycholesterol for liver X receptor β (LXRβ); 1α,25-dihydroxyvitamin D3 with vitamin D receptor (VDR); T3 with thyroid hormone receptor β (TRβ)] directly interacts with the histidine on H10/H11 but never directly contacts H12, nevertheless stabilizing H12 in the agonist conformation (26, 27, 109, 164).
The chemical structures and functionalities presented by the NR ligand direct the specificity for receptor binding and can be translated into a broad spectrum of agonist, partial agonist, and antagonist outputs in terms of gene responses. Moreover, for some NRs, ligands function as specific gene modulators. These molecules can activate a subset of the receptor’s target genes while remaining neutral or antagonistic on other target genes. The mechanism underlying this type of gene selectivity is believed to be cell specific and reflects the ratio of coactivators and corepressors, which codetermine whether a ligand is an on-switch or an off-switch. Partial agonists are typically the most promising candidates for development into gene-selective ligands and drugs and have the greatest promise for disease therapy with limited undesired side effects. As each of the 48 human NRs invariably controls a plethora of genes and occupies a central point of regulation in metabolic, reproductive, hormonal, and circadian control pathways, finding synthetic drug-like ligands with the right gene-selective profiles remains a low-probability event. The combination of large chemical libraries with hundreds of thousands of testable chemical entities, together with modern high-throughput screening facilities, helps defy the odds of finding the right molecules.
Once initial candidates are identified on the basis of an appropriate screen, each of their chemical scaffolds is evaluated and expanded in scope and functional group diversity through synthetic chemistry. The newly made and more focused libraries are then evaluated in secondary biochemical, cellular, and phenotypic assays, as well as specificity screens to evaluate if their targets might include any additional NRs. Crystal structures and structure-activity relationships among the variants of each scaffold are established through this work. The goals are to tune the chemical features of the ligand to produce greater specificity for the target receptor and the response.
Many other important factors must be considered in advancing molecules toward drug development, including the pharmacokinetic and pharmacodynamic properties of these molecules, their toxicities, and the ability to patent these compounds. Aside from finding drug candidates, a number of such molecules that have typically failed to advance have nonetheless proven to be useful reagents for NR research laboratories around the world, providing the first small-molecule tools with which to probe the physiology of receptors and to validate their roles in disease processes. For orphan receptors this type of pharmacological advance has not yet been proven possible. However, genetic approaches using either knockout or conditional knockout mice have yielded great insight into the developmental and physiological functions of these orphan receptors as exemplified by genetic studies on the orphan receptor COUP-TFII (28–30).
In addition to high-throughput screening methods to find receptor-regulating molecules, a rational design approach has also been demonstrated in some cases. Here one starts with the receptors’ endogenous ligands and builds on that chemical frame using crystallographic information at the outset. Crystal structures with the natural ligands provide information as to which portions lie next to the critical residues and secondary structure features that can allosterically affect the coregulator binding sites. Then, through synthetic chemistry, the portions of those natural ligands that face protein features controlling receptor function (such as H12) are adjusted and extended, and the outcome is monitored within a series of carefully designed receptor functional assays. This approach has been demonstrated to a large extent in the cases of the ER, the thyroid hormone receptor (TR), and the retinoic acid receptor (RAR) (21, 31–34). In some cases, the success has included identifying receptor-selective ligands, for example, those that differentially regulate the ERα versus ERβ, or the TRα versus TRβ, in which there is a very high level of amino acid conservation both within the LBD and within the hydrophobic pocket for the ligands. Each of these cases is described in detail below.
ESTROGEN RECEPTOR
Through the action of two distinct ERs, ERα and ERβ, targeted ligands that compete with the natural estrogen 17β-estradiol (E2) exert a remarkable set of effects on the growth, differentiation, and maintenance of many reproductive tissues as well as the heart, bone, and liver (35–40). As a result, ER targeting has been promising for therapeutic purposes such as treating breast cancer, osteoporosis, cardiovascular disease, obesity, as well as disorders of the central nervous system and immunity (40).
The first crystal structures of ER with agonists and antagonists shaped the NR field’s overall understanding about how ligands can set the conformation of H12 to affect receptor activation (see Figure 1b) (41–43). Important structure information revealed how binding of coactivator NR boxes with LXXLL motifs such as those found in SRC-2 is responsible for NR transcriptional activation (44). These studies paved the way for structural tools to greatly expand our knowledge of how a variety of other ER ligands interact within their pockets and to what extent the pocket was amenable to much more diverse chemical scaffolds (45–55).
The ERs do indeed bind to a wide variety of compounds with significant chemical diversity. But some features of these ligands seem to be universally conserved. Two amino acid residues located at the opposite ends of the receptor pockets are particularly well poised to recognize ligands via hydrogen bonding, so long as those ligands have the right functional groups available and pointed appropriately. One of these, Glu-353, provides recognition of the E2 hydroxyl moiety at position 3 on ring-A. At the other end of the pocket, His-524 provides the hydrogen bond with the 17β-OH on ring-D of the E2. The cavity size occupied by E2 is 450 Å3 in volume, significantly larger than the 250 Å3 associated with the ligand E2. Aside from the two hydrophilic residues at the extreme ends of the pocket, the remaining lining of the pocket is highly hydrophobic in character. Figure 3 shows that some of the features used by steroid receptors are very well conserved, yet there is generally a very high level of selectivity among the steroid receptors with respect to their cognate ligands. By contrast, nonsteroid receptors, including those for the retinoids, thyroid hormone, and heme, use very different features of their pockets for ligands (Figure 4).
Figure 3.

Close-up views of the ligand-binding interactions of nuclear receptors (NRs) that bind to steroidal molecules. Aside from farnesoid X receptor (FXR) binding to bile acids, the other receptors all position ring-A deep into the receptor pocket, where the 3-OH group forms hydrogen bonds with polar amino acid side chains. AR, androgen receptor; ER, estrogen receptor; GR, glucocorticoid receptor; LXR, liver X receptor; MR, mineralocorticoid receptor; PR, progesterone receptor; VDR, vitamin D receptor.
Figure 4.

Close-up views of the ligand binding interactions of nuclear receptors (NRs) that bind to nonsteroidal molecules, including all-trans retinoic acid (ATRA), 9-cis retinoic acid (9 cRA), triiodothryronine (T3), and iron-porphyrin IX (heme). RAR, retinoic acid receptor; RXR, retinoid X receptor; TR, thyroid hormone receptor.
Other well-characterized ER ligands span the range of partial agonists to agonists, including genistein, 4-hydroxytamoxifen, raloxifene, and ICI-164,384. These molecules generally bind across the ligand-binding cavity very similarly to the natural ligand E2 and the synthetic agonist diethylstilbestrol (DES). For all these molecules, the equivalent ring-A and the 3-hydroxyl groups occupy the same positions as this functional group of E2. The antagonists often contain extended groups stemming from the center of their molecules, which directly impact H12 positioning and displace it into the LXXLL-binding site, accounting for their blocking of coactivator interactions and transcriptional activation (41, 42, 45, 47, 40). Figure 5 summarizes this understanding of how agonists and antagonists can be inter-converted via their distinct functional groups and the directions in which these groups are positioned. More chemically diverse ER ligands include the natural products zearalenone and coumesterol, the pesticides o,p′-DD and kepone, the food components genestein and enterolactone, and commercial chemicals such as bisphenol A (56, 57). The ability of these highly distinct molecules to bind to the ER pocket clearly demonstrates substantial promiscuity in how NR pockets can be targeted.
Figure 5.

Principles of ligand design as agonists and antagonists for the estrogen receptor (ER). Estrogens (agonists) include the natural ligand E2 and the synthetic hormone diethylstilbestrol (DES). Tamoxifen/hydroxytamoxifen and raloxifene are antiestrogens and function more as SERMs (selective estrogen receptor modulators). Although mimicking some general features of the basic molecular structure in these ligands, including the central core, with an aromatic and phenolic group on either sides, additional moieties such as extensions along one of the rings can convert ligands from agonists to antagonists on the basis of the protrusion of this group disrupting the agonist conformation of H12. Close-up views of the pocket around these four ligands are provided at the bottom, showing that the conserved features of their chemical frame are recognized similarly within the ER pocket (165–167).
SERMs (selective estrogen receptor modulators) are a class of ER ligands that act in some tissues like the agonist E2 while acting in other tissues as an antagonist. SERMs fulfill our ideals of what a compound must achieve to be promising as a drug, with a targeted effect on gene regulation and with few side effects. Tamoxifen was among the first examples of a true SERM. Long-term tamoxifen administration for breast cancer initially came with concerns about the expected effects on bone mineral density and cardiovascular risk factors because agonism was known to be beneficial for these processes (58, 59). Tamoxifen was later shown to act as a partial agonist in skeletal tissue, maintaining bone mineral density (60, 61). At the same time, this drug proved to have a similar beneficial effect as E2 on lipid/cholesterol profiles of postmenopausal women (62). Thus, tamoxifen became the first ER ligand capable of showing a mixed agonist/estrogen antagonist character and at the same time tissue selectivity. This discovery proved to be the starting point for identifying additional SERMs, such as raloxifene, which also displayed the desired tissue selectivity and later was approved for the prevention of osteoporosis in postmenopausal women (58, 63).
A more complete understanding of the pharmacology of natural and synthetic estrogens, antiestrogens, and SERMs requires the consideration of the two distinct ERs: ERα and ERβ, which regulate nonidentical subsets of target genes. Gene selectivity, therefore, can be achieved at least in part through selectivity for different ERs. The compound WAY-244 is an example of a subtype-selective ER ligand, demonstrating how, with only two residues differing in the ligand-binding pockets of these receptors, compounds with almost 100-fold β selectivity can nevertheless be identified (see Figure 6) (53). Besides receptor subtype selectivity, SERMs must take advantage of the different repertoires and concentrations of coregulatory proteins in various cells for their tissue-selective behaviors (64–69). In addition, it is now understood that both ERs employ two distinct protein regions for coactivator interactions and gene regulation. These receptor portions are the N-terminal AF-1 regions and the LBDs’ AF-2 region, which are shown to interact with the p160 coactivators GRIP-1 (gluco-corticoid receptor–interacting protein-1) and SRC-1 (steroid receptor coactivator-1) (70–74). The control of transcriptional activity by the ERs through both AF-1 and AF-2 sites is both cell-type and gene promoter–context specific, providing an additional mechanism for deriving SERM activities (72).
Figure 6.

Estrogen receptor (ER)α and β have different tissue/cell distributions and differential affinities for ligands and nonoverlapping genes that they regulate. As a result, targeting these receptors differentially can lead to selective gene activation profiles. Shown are the relationships of their amino acid sequences, including the level of conservation (in parentheses). Within the pocket, there are only two residues that differ, yet molecules such as WAY-244 can show a very high preference for ERβ (53). A–F designate the segments of the receptor polypeptides.
TARGETING OTHER STEROID RECEPTORS
Additional steroid receptors, such as the androgen receptor (AR), the mineralocorticoid receptor (MR), the glucocorticoid receptor (GR), and the progesterone receptor (PR), have also been intensely studied as drug targets.
The detailed binding interactions of each of these NRs with their endogenous ligands have been established through crystallographic studies and are shown in Figure 3. Like ER, the pockets are largely hydrophobic, and the two hydrophilic groups on the ligands are able to hydrogen bond to polar side chains at opposite ends of the pockets. AR, MR, GR, and PR, in the absence of bound ligands, are maintained in an inactive conformation by heat shock proteins in the cytoplasm. Upon ligand binding, they dissociate from their cytoplasmic chaperone complexes and become nuclear, potentially undergoing homodimerization in the process. In each case, the past decade has seen a heightened interest in the development and therapeutic potential of receptor modulators based on nonsteroid chemical frames, as these chemical entities are easier to synthesize and patent. Furthermore, the chemical diversity of these non-steroidal ligands is likely to increase both their tissue and gene selectivity.
For AR, the most potent endogenous ligand is 5α-dihydrotestosterone (DHT), derived from the conversion of the predominant circulating androgen, testosterone, via the actions of a 5α-reductase (75). These natural androgens exert nonselective anabolic and androgenic effects directly through the AR and are responsible for masculinization and gender-specific differences such as enhanced muscles and bones (anabolic effects). Initial crystal structures of AR-LBD focused on the agonists R1881 and DHT (76, 77). These studies were followed by crystal structures that showed how coactivator motifs and Phe- and Trp-rich motifs, mimicking those found in the N terminus of the AR protein, could interact with the LBD (78, 79). Crystal structures have also been completed with nonsteroidal antagonist ligands bound to the AR pocket, advancing our understanding of how drug-like molecules can be designed and improved (80, 81).
The field of nonsteroidal AR drug development and more specifically selective androgen receptor modulators (SARMs) has focused on the identification of selective tissue actions that promote muscle- and bone-building processes and spare the body from undesired effects of AR signaling and androgenic activity (82, 83). Pure AR antagonists, or antiandrogens have also been developed and used effectively in early prostate cancer treatment. In this clinical setting AR ligands must be devoid of any agonist activity to avoid the growth-promoting effects of androgens on prostate tissue. Antagonists and drug-like molecules currently used for prostate cancer include steroidal ligands such as cyproterone acetate and nonsteroidal ligands such as hydroxyflutamide, nilutamide, and bicalutamide (82). Prostate cancer therapy often starts with a strong AR antagonist such as bicalutamide, along with a GnRH superagonist, such as leuprollide, which blocks the hypothalamic-pituitary-gonadal axis to effectively shut down all gonadal production of androgens.
More diverse SARMs are now being advanced through the clinic for a variety of other uses that relate to their anabolic potential (83). These indications include age-related decline in lean body mass (known as sarcopenia); more acute conditions that result in rapid loss of muscle, often seen in patients with AIDS, cancer, and kidney diseases; and osteoporosis (83). The AR is widely distributed among a wide range of reproductive and nonreproductive tissues, including the prostate, skin, testis, ovary, cartilage, sebaceous glands, hair follicles, and cardiac and smooth muscle. Again it is predicted that the specific distribution and varying levels of receptor coregulators in these tissues would result in gene-selective profiles with partial agonists. As such, SARMs would in principle best be derived from partial agonists that take advantage of tissue-specific coregulator distributions. Our understanding of how AR target genes are activated and repressed has been substantially enhanced through chromatin immunoprecipitation (ChIP) studies, which have shown that AR agonists recruit coregulators in a time-dependent and cyclical manner to various response elements (84).
The MR is responsible for the maintenance of electrolyte homeostasis and blood pressure through effects on the distal nephron (85). Aldosterone is the main physiological ligand, although the receptor also is quite capable of binding to the glucocorticoid cortisol. Although binding of aldosterone and cortisol to MR can take place with similar affinity, there is some basis for selective and tissue-specific effects of these molecules with MR. In plasma, glucocorticoid concentrations exceed those of aldosterone by up to three orders of magnitude, suggesting that MR is normally occupied by cortisol or corticosterone. In part, preferential binding of aldosterone to MR is achieved in epithelial target tissues because of 11β-hydroxysteroid dehydrogenase type 2 (11β HSD2), which converts cortisol into the inactive metabolite cortisone, which is no longer able to bind MR.
A series of MR LBD structures with different ligands has assisted drug discovery efforts on this receptor (86–89). In the structure of MR-LBD with bound progesterone, which acts as an MR antagonist, the ligand occupies the pocket but fails to make the expected contacts with helices H3, H5, and H12 and thus fails to achieve the expected agonist conformation seen with aldosterone (88). At present, the best-studied MR antagonists are the steroidal compounds spironolactone and eplerenone. These molecules have demonstrated potential in improving patient outcomes in both mortality and morbidity in terms of heart failure (90, 91). Given the role of MR in both the kidney and the heart, recent attempts at drug discovery have focused on identifying ligands that dissociate the actions of MR in these two tissues, allowing for therapeutic benefits in the heart without impacting the kidney. One remaining challenge in MR drug discovery is to find ligands that do not bind to or activate other steroid receptors.
GR has been the focus of intense studies due to its important roles in a number of physiological processes (92). The multiple roles of glucocorticoids in human physiology include mediating immune function, inflammation, glucose balance (from which the name glucocorticoid is derived), the stress response, fat distribution, and normal growth and development (93, 94). Unfortunately, given the multitude of genes regulated by GR, the potential number of side effects is significant, thus rendering the search for selective gene modulators particularly difficult (95). A major undesired side effect of glucocorticoids involves the feedback suppression of the hypothalamus-pituitary-adrenal axis that eventually results in adrenal atrophy and loss of endogenous glucocorticoid synthesis. Long-term usage of synthetic glucocorticoids is associated with osteoporosis, decreased muscle mass and strength, hypertension, and insulin resistance (96).
In the case of the GR, there has been a widespread focus to identify so-called dissociated ligands, which will not globally activate transcription but instead will act to dissociate GR’s unique manner of gene activation versus repression (96). In this regard, it is believed that GR-mediated transcriptional activation and repression can occur on genes within the same cells, due to specific factors that associate and physically interact with GR on those promoter-regulatory sites. For example, the interactions between c-Jun N-terminal kinase and GR negatively impact AP-1 activity and are important for inflammation (97). Moreover, the tethering interactions between the GR and AP-1 along certain promoters further block transcription from selective genes (98, 99). Producing GR ligands for activation/repression dissociation was initially based on several steroidal compounds [RU24858, RU40066, and RU24782 (100)] and the nonsteroidal ligand ZK216385 (101) and has now been extended to other compounds (96, 102–104).
One promising feature of the GR for drug discovery is its highly adaptable ligand pocket, which accepts a surprising variety of steroid-derived ligands and nonsteroidal synthetic compounds (see Figure 7). The initial structures of the GR LBD with dexamathasone (DEX) and the antagonist RU486 showed the first glimpses of how the pocket and H12 interact with these opposing ligands (105, 106). These were followed by the structure bound to deacylcortivazol (DAC), which induced an expansion of the pocket to more than 1000 Å3, from the volume of 540 Å3 associated with the DEX-occupied ligand. Additional comparisons with fluticasone furoate (FF) show that the pocket is expandable in two directions beyond what was originally observed with DEX (107). More recently, the structure of the GR LBD with a nonsteroidal ligand was reported, showing how amino-pyrazole 2,6-dichloro-N-ethyl benzamide as a selective GR agonist uses both possibilities for pocket extension while leaving the position of H12 in a conformation that still allows for the association of a coactivator LXXLL motif (108).
Figure 7.
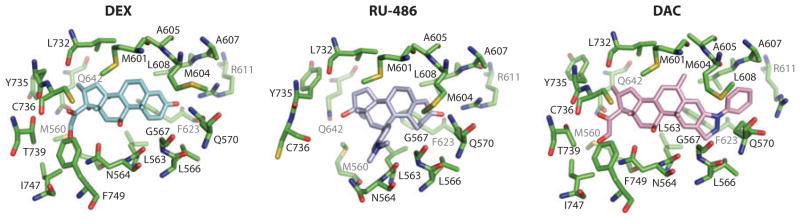
Demonstration of how ligands can stretch and significantly reshape receptor ligand-binding domain (LBD) pockets. The three structures of the glucocorticoid receptor (GR) LBD pocket with dexamathasone (DEX), RU-486, and DAC indicate how the residues surrounding the ligands can undergo both side-chain and main-chain movements to nearly double the size of the pocket to fit with each of these steroidal molecules (106–108).
THYROID HORMONE RECEPTOR
TR, through a pair of endogenous ligands, thyronine (T4) and triiodothyronine (T3), regulates development and a wide variety of critical cellular functions including basal metabolic rate and metabolism of protein, fat, and carbohydrate. Thyroid hormones (T3 and T4) are the only known molecules in terrestrial organisms that have covalently incorporated iodine atoms, with T3 being the more active but shorter-lived thyroid hormone. The most common thyroid endocrine disorders include hyperthyroidism (e.g., Graves’s disease) and hypothyroidism (e.g., Hashimoto’s disease). Treatment of hyperthyroidism includes the use of antithyroid drugs that target peripheral deiodinases and radioactive iodine therapy, whereas hyperthyroidism is treated simply with thyroid hormone replacement. The crystallographic study of the TRα with ligand was one of the first detailed visualizations of any NR LBDs (109). A close-up view of this pocket and its interactions with ligands is shown in Figure 4. The drug discovery efforts that followed this structural solution represent some of the first and most elegant rational approaches to design novel NR ligands using the so-called extension hypothesis. This hypothesis suggested that addition of bulky groups on a known agonist that point to and dislodge H12 from its normal position would destabilize coactivator binding (34).
More recent efforts at developing TR targeting have focused on lowering LDL levels, cholesterol, and triglycerides from serum (110). As therapeutic molecules, TR agonists are potentially promising in preventing atherosclerosis, promoting fat loss, and treating diabetes. However, a major goal of TR-targeting efforts is to minimize the undesired effects of thyroid hormone mimetics on the heart, bone, and muscle (111, 112). This goal is further complicated by the selective actions of any developed TR ligands on two TR subtypes (TRα and TRβ), which elicit very different physiological responses. In addition, one has to consider the promoter selectivity of various TR-regulated genes as well as the differential tissue-selective uptake (heart versus muscle versus bone) when developing synthetic TR ligands in order to achieve the desired therapeutic profiles (112).
The development of TR subtype–selective ligands has proceeded, despite the fact that the two ligand-binding pockets are markedly similar with only a single amino acid difference (Asn versus Ser) between the LBDs of TRα and TRβ. TR LBD subtypes also show differences in a loop intervening between H1 and H3 that can affect ligand recognition and, potentially, coregulator interactions (113–115). Two TR modulators have been developed so far through a structure-guided approach; these include GC-1 and GC-24, which bear close structural similarity to the natural thyroid hormones. GC-1 has a 4–10-fold selectivity toward TRβ, as compared with T3, which is nonselective (113). Furthermore, GC-1 can lower cholesterol and triglyceride levels in mice without adversely stimulating the heart (111). The TR crystal structures with GC-1 show this ligand makes differential use of the same arginine residue in the two TR subtypes, partially accounting for its selectivity (111, 113).
GC-24 provides selectivity through a bulky 3′ benzyl side group in its chemical frame that does not support equally favorable binding in the T3α pocket (115, 116). Other TR-directed molecules have been described that reduce body weight and adiposity in obese models of rats, without obvious loss of muscle. These results with TR subtype– and gene-selective compounds are promising and at the same time fuel further searches for therapeutic molecules directed at these two receptors (110, 117).
TARGETING NUCLEAR RECEPTORS THAT FUNCTION AS METABOLIC AND LIPID SENSORS
Cholesterol is an essential component of living cells and their membranes and a necessary precursor for all steroid and bile acid production. However, excessive levels of cholesterol also promote the development of atherosclerosis and other cardiovascular diseases. Transcriptional control of key cholesterol and bile acid metabolic genes is mediated in part by two liver X receptor (LXR) isotypes, LXRα and LXRβ, identified as oxysterol sensors, and the farnesoid X receptor (FXR), which acts as the bile acid sensor. These NRs function in each case as heterodimers with the retinoid X receptor (RXR) and work reciprocally in controlling several key lipid- and bile acid–regulated genes. LXRs function as cholesterol/oxysterol sensors, regulating genes associated with the absorption, transport, efflux, and elimination of cholesterol. FXR is bound by bile acids, which are the major end products of cholesterol in the liver and have a role in the solubilization, absorption, and entrahepatic circulation of cholesterol and other lipophilic molecules. These endogenous ligands are able to bind and activate LXRs and FXR receptors in a concentration range of 1–50 μM, which matches their free concentration levels in certain cells. These binding and activation properties are in stark contrast to the steroid and thyroid hormone receptors that typically bind and are activated by their cognate ligands in the 0.1–1.0-nM concentration range.
The pocket interactions of LXRβ and FXR with their cognate ligands are shown in Figure 3. Despite the differences in binding constants, Figure 3 shows that the pocket features of LXRs, FXRs, and the classic steroid hormone receptors have certain features in common, including a highly hydrophobic content of residues that form van der Walls interactions with these molecules. Interestingly, bile acids like chenodeoxycholic acid (CDCA) bind to the FXR pocket in the reverse orientation as do LXR ligands and other steroid ligands, with the bile acids’ carboxylate extensions beyond ring-D and on the 24 position facing the receptor site that normally accepts ring-A in all the other steroid receptors. The reason for this ligand reversal is nicely explained by the unusual cis juncture connecting ring-A with ring-B, ring-C, and ring-D, a feature unique to bile acids. Two crystal structures with CDCA-related ligands show precisely how this unusual shape for bile acids, not common to the other steroids, is effectively used as a recognition feature in the FXR LBD (26). Crystal structures of LXRβ with oxysterols and the synthetic agonist T-0901317 have been accompanied with an LXRα-RXR LBD heterodimer structure (27, 118).
The activation of LXRs via some ligands can lead to an altered reverse cholesterol transport and increased circulating high-density lipoprotein (HDL) levels, which are beneficial. In many ways, LXRs act as master regulators of hepatic lipid metabolism. As such, synthetic LXR agonists appear to induce lipogenesis, boosting plasma triglyceride concentrations. Given that dyslipidemia is a risk factor for atherosclerosis and cardiovascular diseases, activation of the LXR pathway as a potential target for therapeutic intervention is undesired, despite the beneficial cholesterol-lowering effects. However, the opposing effects of LXR ligands on cholesterol versus serum triglycerides could be accounted for by two different LXR subtypes and their tissue distributions (119). It has also been shown that LXR agonists attenuate the development of atherosclerosis in mice (120–124). Therefore, synthetic LXR agonists may prove promising for antiatherosclerotic effects, whereas the potentially adverse lipogenic effects could in theory be averted through receptor subtype selectivity. Two nonsteroidal LXR ligands, T0901317 and GW3965, show interesting differences in their biological activities (125, 126), and these differences are described in full elsewhere (127).
FXR has emerged as a key modulator of a variety of metabolic processes related to liver cholesterol and bile acid balance. Selective and potent FXR agonists, such as 6-ECDCA, GW4064, and fexaramine, have been described (128–130). These compounds have allowed identification of a subset of FXR target genes implicated in cholesterol and bile acid homeostasis, suggesting an interrelationship between bile acid metabolism, triglyceride metabolism, and insulin resistance (131–137). As such, FXR is a potential therapeutic target for diseases including liver and bile acid disorders, hyperlipidemia, and obesity.
Other examples of NRs that recognize lipids and that could act as physiological lipid sensors include SF-1 and LRH-1, which appear to bind to phospholipids such as phosphotidyl glycerol, phosphotidyl ethanolamine, and phosphatidyl inositols. In some cases, the initial discoveries of these molecules as potential ligands were suggested entirely from crystallographic structures in which the molecules were serendipitously trapped during protein purification and later identified by their electron densities and follow-up mass spectrometry. Those crystal structures and ligand-binding interactions have been recently reviewed elsewhere (7). For both SF-1 and LRH-1, the broader functional implications of phospholipids as NR signaling molecules still remain to be evaluated for potential drug therapy (138).
The diversity of NR natural ligands and activators has also been extended to fatty acids, as suggested by the recently described crystal structure of the PPARγ LBD binding covalently to one or more oxidized fatty acids, including 9-HODE and 13-HODE (8). PPARγ is a master regulator of adipogenesis and controls lipid and glucose metabolism (139). Several unsaturated fatty acids, such as oleate, linoleate, eicosapentaenoic, and arachidonic acids as well as the prostanoid 15-deoxy-δ-12,14-prostaglandin J2, were previously described as PPARγ ligands (139). Figure 8 shows the side-by-side comparisons of all the lipid-binding interactions described thus far for LRH-1 (liver receptor homolog-1), SF-1 (steroidogenic factor-1), HNF4-α (hepatocyte nuclear factor-4α), and PPARγ, showing how their respective modes of lipid recognition compare. Owing to the fact that the thiazoladinedione class of PPARγ agonist ligands has already been used in diabetic patients for their insulin-sensitizing and glucose control benefits, there have been considerable efforts aimed at further drug development for this PPAR subclass of receptors. A class of fibrate drugs, long in use for therapeutic purposes, is believed to target the PPARα protein, accounting for the ability of these molecules to reduce triglycerides and raise HDL lipids. This receptor subtype also remains a major target for drug development for cardiovascular indications (140, 141). More recently, PPARγ ligands are being evaluated for their therapeutic potential in carcinogenesis and Alzheimer’s disease (142–147).
Figure 8.

Nuclear receptor interactions with lipid molecules. LRH-1 is shown with phosphotidyl glycerol, SF-1 with di-palmitoyl-3-SN-phosphotidylethanolamine, HNF4-α with myristic acid, and peroxisome proliferator–activated receptor γ (PPARγ) with 9-HODE (7, 8). LRH-1, liver receptor homolog-1; HNF-4, hepatocyte nuclear factor-4; SF-1, steroidogenic factor-1.
Two other recently studied human NRs appear to bind to their ligands in the micromolar range of equilibrium dissociation constants, suggesting the possibility that they also function as physiological sensors for signaling molecules. In the case of the Rev-erb-α and -β receptors, iron porphyrin IX has been shown by three groups to act as the endogenous ligands, regulating the corepressor binding and gene repression activities of these receptors (9–11). The crystal structure of the Rev-erb-β LBD with heme has also been completed, showing the basis for the recognition of heme as an exchangeable ligand and potentially allowing for gas-responsive functions (11). A previous crystallographic study of the unliganded Rev-Erb-β LBD had suggested that there was no available pocket for any ligand and that the repression function of Rev-Erb-β would be constitutive (148). The finding that a ligand as large as heme can create its own pocket underscores further the flexibility of the internal sites within NRs and shows how endogenous and synthetic ligands can create their own appropriately shaped pockets. This may be an important lesson for other NRs, such as nuclear receptor–related protein 1 (Nurr1) and estrogen-related receptor γ (ERRγ), for which crystallographic reports have suggested an absent or insufficient space for ligand binding (13, 149). In the case of the Rev-Erb receptors, heme has a bonafide role in regulating the circadian rhythm through the receptors’ target genes and also appears to directly regulate genes important for metabolism (9, 10).
Finally, a recent report has identified retinoic acid as a potential ligand for the COUP-TFII orphan receptor (150). These authors also used a combination of the unliganded crystal structure of this receptor’s LBD and mutagenesis to show how this receptor is able to achieve both constitutive transcriptional activity and an auto-repressed conformation with H10 bent into the receptor pocket and the AF-2 region blocking the coactivator binding site, respectively. Although the role of retinoic acid is not firmly established in this receptor, related molecules have long been known to activate the RXRs and RARs and at the same time to exert significant developmental functions through those receptors. Both of those receptors are well-established targets for cancer therapy (21, 151). In the case of COUP-TFII, the finding that a cavity can be created inside the LBD is highly promising, even though the binding affinity for retinoids is quite weak. Nevertheless, the unexpected finding of a ligand binding cavity in some NRs suggests that further drug development could target the various physiological functions of these receptors, including angiogenesis, neuronal development, metabolic homeostasis, and the circadian rhythm (28–30).
FULL-LENGTH STRUCTURAL INFORMATION AND DOMAIN-DOMAIN INTERACTIONS
Despite more than 20 years of structural efforts focused on NR LBDs and DBDs in isolation, the full-length crystal structure of an intact receptor had remained an unfulfilled goal. The difficulties in crystallizing a full-length receptor have largely stemmed from general difficulties in their protein expression and isolation and also from the dynamic nature of the A/B, hinge, and DBD regions, which prevents these polypeptides from forming ordered arrays in crystal packing. Nevertheless, there remains a significant need to visualize those NRs already being explored for drug development, as a series of published findings has largely questioned the previous notions that domains function autonomously. For example, characteristic features of some steroid receptors are the interactions of their AF-1 in the N-terminal domain and AF-2 in the LBD, and these domains further drive interactions with coregulators (78, 152–156). The extent to which the DBD and LBD cooperate in each other’s respective functions has also not been addressed. In other cases, portions to the C terminus of the 12-helical LBD (known as the F-region) are known to impact ligand binding and modulate the activity of the ligands but have yet to be visualized (156).
Our laboratory recently reported the structure of the intact PPARγ-RXRα heterodimer bound to its idealized DNA site, with coactivator peptides and ligands of both receptors (see Figure 9) (157). The structure showed that there are three distinct heterodimerization domains, two of which are DNA dependent and had not been previously known. Importantly, the geometry and spacing of the DNA response elements predispose most of the domain-domain interactions that occur both inter- and intramolecularly. The PPARγ ligand pocket is highly adaptable and can provide a Y-shaped space to accommodate different synthetic ligands (see Figure 10). Nevertheless, structures of the complex in which three PPARγ ligands range from a full agonist (rosiglitazone) to a partial agonist and an antagonist did not alter the interdomain contacts overall. Surfaces on the PPARγ LBD that directly impacted the DNA binding of both receptors were identified, contradicting the long-held view that the NR LBD and DBD maintain autonomous and separable functions. Given the contribution of the PPAR LBD to DNA binding, and the likelihood that ligands that impact DNA binding through the LBD can be identified, it is now conceivable that selective modulators can be identified on the basis of their ability to target subsets of DNA response elements, instead of through differential binding to coregulators. The concept of the DNA response element acting as an allosteric regulator of ligand binding and receptor activation is not new but has yet to be exploited as a drug discovery paradigm (158–163).
Figure 9.
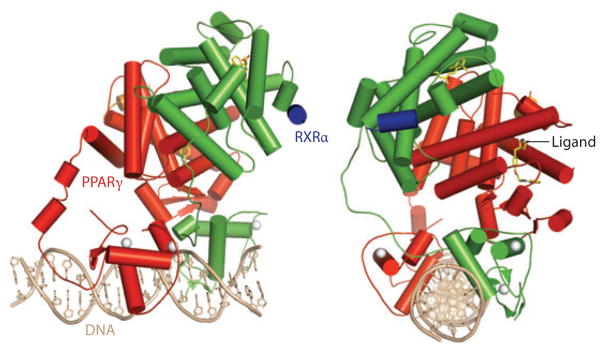
Overviews of the intact structures of the PPARγ-RXRα heterodimer on PPAR response element DNA. The PPARγ is in red, RXRα in blue, DNA in copper, coactivator LXXLL motifs in blue and orange, and the ligands in yellow. The PPARγ LBD makes interdomain contacts with all the other ordered domains in both polypeptides. An unexpected interface between the PPARγ LBD and both DBDs suggests that ligands at PPARγ may modulate DNA site affinities (157). DBD, DNA-binding domain; LBD, ligand-binding domain; PPARγ, peroxisome proliferator–activated receptor γ; RXR, retinoid X receptor.
Figure 10.
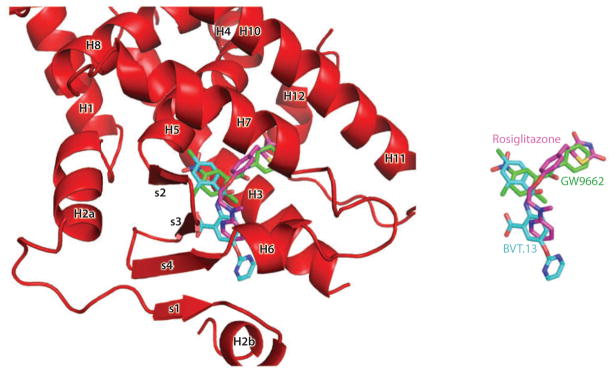
Superposition of the binding modes of three peroxisome proliferator–activated receptor γ (PPARγ) synthetic ligands. Rosiglitazone is a full agonist, BVT.13 is a partial agonist, and GW9662 is a suicide inhibitor that binds to the receptor pocket. Together, the superposition shows the Y-shaped inducible fit possible in this receptor pocket (157).
FUTURE DIRECTIONS
It is important to advance other crystallographic efforts to examine full-length NRs, as the domain-domain interactions that become revealed will certainly impact our understanding of the physiology and pharmacology of these receptors and alter our views of how drug discovery screens should best be designed. Because the unique cognate DNA response elements will no doubt foster significant interdomain communications in a receptor, the PPAR-RXR complex is not adequate in providing predictive models of how any of the other NR complexes are configured and assembled. For each receptor, unique and important lessons are likely to be derived about whether new drug-interacting sites become available at the juncture of domain-domain communications. Alternatively, if the LBD pocket can be screened, can it be done in a manner that targets distant sites responsible for coregulator or DNA binding in an allosteric fashion?
Future studies should also focus on the role of phosphorylation, sumolyation, and acetylation in the NRs and how the modifications that occur in cells impact ligand binding, receptor activation, and ultimately drug discovery. It is plausible that some of these phosphorylations themselves give rise to promoter-selective activities of NRs, and thus screening for drugs using such phosphostates of NRs may assist in the identification of gene-selective molecules. Another area that needs further attention is that of the orphan receptors, in which there are as yet no endogenous ligand candidates or even chemical tools to unravel their gene-regulatory functions. At the same time, there are a considerable number of drugs on the market, some dating back decades since their introduction, that have no known targets of action. A comprehensive examination of the 48 human NRs with these molecules may reveal important new information about which orphan receptors may already be major therapeutic targets and at the same time may provide important chemical tools to the biologists. Finally, the physical interactions of the NRs with other promoter-binding factors have yet to be investigated by crystallographic methods or probed by chemical screening. The implicit abilities of the NRs to interact with other gene-regulatory factors largely govern how promoter-selective responses are born, and understanding the details of these interactions promises to provide new paradigms for finding gene-selective and dissociating small-molecule drugs.
SUMMARY POINTS.
Nuclear receptors (NRs) are ideal drug development targets because they bind to hydrophobic pockets inside a protected cavity and in response regulate genes involved in a variety of disease processes.
Crystal structures have been determined for a large number of receptor ligand-binding domains (LBDs), including both endogenous ligands and synthetic molecules and small portions of coregulators, and this type of information has guided our notions of how ligands can impact the ability of receptors to activate or repress genes.
The major goal of drug discovery has been to identify molecules, known as selective modulators, that act in a receptor- and gene-selective fashion and do not globally regulate all receptor genes in the same way.
Selective modulators can be obtained in some cases by taking advantage of compounds that distinguish their binding between highly related subtypes of a given NR or allow different coregulators to be recruited depending on the tissue distributions and availability of these coregulators.
There is only one example of a crystal structure of full-length NRs, and it is highlighting how our assumptions about the LBDs’ functions are oversimplistic. Taking into account the allosteric communications involving LBDs may assist with the identification of selective modulators.
Other areas that need to be explored, and that could assist the search for selective modulators, include the effects of posttranslational modifications and protein-protein interactions that are promoter selective.
Glossary
- NR
nuclear receptor
- DBD
DNA-binding domain
- LBD
ligand-binding domain
- AF-1
activation function-1
- AF-2
activation function-2
- ER
estrogen receptor
- PPAR
peroxisome proliferator–activated receptor
- TR
thyroid hormone receptor
- RAR
retinoic acid receptor
- E2
estradiol
- SERM
selective estrogen receptor modulator
- GRIP-1
glucocorticoid receptor–interacting protein-1
- SRC-1
steroid receptor coactivator-1
- AR
androgen receptor
- MR
mineralocorticoid receptor
- GR
glucocorticoid receptor
- PR
progesterone receptor
- SARM
selective androgen receptor modulator
- DEX
dexamathasone
- T4
thyronine
- T3
triiodothyronine
- RXR
retinoid X receptor
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
Contributor Information
Pengxiang Huang, Email: ph6v@virginia.edu.
Vikas Chandra, Email: vc6e@virginia.edu.
Fraydoon Rastinejad, Email: fr9c@virginia.edu.
LITERATURE CITED
- 1.McEwan IJ. Nuclear receptors: one big family. Methods Mol Biol. 2009;505:3–18. doi: 10.1007/978-1-60327-575-0_1. [DOI] [PubMed] [Google Scholar]
- 2.Nagy L, Schwabe JW. Mechanism of the nuclear receptor molecular switch. Trends Biochem Sci. 2004;29:317–24. doi: 10.1016/j.tibs.2004.04.006. [DOI] [PubMed] [Google Scholar]
- 3.Sonoda J, Pei L, Evans RM. Nuclear receptors: decoding metabolic disease. FEBS Lett. 2008;582:2–9. doi: 10.1016/j.febslet.2007.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mangelsdorf DJ, Thummel C, Beato M, Herrlich P, Schutz G, et al. The nuclear receptor superfamily: the second decade. Cell. 1995;83:835–39. doi: 10.1016/0092-8674(95)90199-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mangelsdorf DJ, Evans RM. The RXR heterodimers and orphan receptors. Cell. 1995;83:841–50. doi: 10.1016/0092-8674(95)90200-7. [DOI] [PubMed] [Google Scholar]
- 6.Chawla A, Repa JJ, Evans RM, Mangelsdorf DJ. Nuclear receptors and lipid physiology: opening the X-files. Science. 2001;294:1866–70. doi: 10.1126/science.294.5548.1866. [DOI] [PubMed] [Google Scholar]
- 7.Ingraham HA, Redinbo MR. Orphan nuclear receptors adopted by crystallography. Curr Opin Struct Biol. 2005;15:708–15. doi: 10.1016/j.sbi.2005.10.009. [DOI] [PubMed] [Google Scholar]
- 8.Itoh T, Fairall L, Amin K, Inaba Y, Szanto A, et al. Structural basis for the activation of PPARγ by oxidized fatty acids. Nat Struct Mol Biol. 2008;15:924–31. doi: 10.1038/nsmb.1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Raghuram S, Stayrook KR, Huang P, Rogers PM, Nosie AK, et al. Identification of heme as the ligand for the orphan nuclear receptors REV-ERBα and REV-ERBβ. Nat Struct Mol Biol. 2007;14:1207–13. doi: 10.1038/nsmb1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yin L, Wu N, Curtin JC, Qatanani M, Szwergold NR, et al. Rev-erbα, a heme sensor that coordinates metabolic and circadian pathways. Science. 2007;318:1786–89. doi: 10.1126/science.1150179. [DOI] [PubMed] [Google Scholar]
- 11.Pardee KI, Xu X, Reinking J, Schuetz A, Dong A, et al. The structural basis of gas-responsive transcription by the human nuclear hormone receptor REV-ERBβ. PLoS Biol. 2009;7:e43. doi: 10.1371/journal.pbio.1000043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rastinejad F. Retinoid X receptor and its partners in the nuclear receptor family. Curr Opin Struct Biol. 2001;11:33–38. doi: 10.1016/s0959-440x(00)00165-2. [DOI] [PubMed] [Google Scholar]
- 13.Greschik H, Wurtz JM, Sanglier S, Bourguet W, van Dorsselaer A, et al. Structural and functional evidence for ligand-independent transcriptional activation by the estrogen-related receptor 3. Mol Cell. 2002;9:303–13. doi: 10.1016/s1097-2765(02)00444-6. [DOI] [PubMed] [Google Scholar]
- 14.Moras D, Gronemeyer H. The nuclear receptor ligand-binding domain: structure and function. Curr Opin Cell Biol. 1998;10:384–91. doi: 10.1016/s0955-0674(98)80015-x. [DOI] [PubMed] [Google Scholar]
- 15.Bourguet W, Vivat V, Wurtz JM, Chambon P, Gronemeyer H, Moras D. Crystal structure of a heterodimeric complex of RAR and RXR ligand-binding domains. Mol Cell. 2000;5:289–98. doi: 10.1016/s1097-2765(00)80424-4. [DOI] [PubMed] [Google Scholar]
- 16.O’Malley BW, Qin J, Lanz RB. Cracking the coregulator codes. Curr Opin Cell Biol. 2008;20:310–15. doi: 10.1016/j.ceb.2008.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wolf IM, Heitzer MD, Grubisha M, DeFranco DB. Coactivators and nuclear receptor transactivation. J Cell Biochem. 2008;104:1580–86. doi: 10.1002/jcb.21755. [DOI] [PubMed] [Google Scholar]
- 18.Khorasanizadeh S, Rastinejad F. Nuclear-receptor interactions on DNA-response elements. Trends Biochem Sci. 2001;26:384–90. doi: 10.1016/s0968-0004(01)01800-x. [DOI] [PubMed] [Google Scholar]
- 19.Li Y, Lambert MH, Xu HE. Activation of nuclear receptors: a perspective from structural genomics. Structure. 2003;11:741–46. doi: 10.1016/s0969-2126(03)00133-3. [DOI] [PubMed] [Google Scholar]
- 20.Wurtz JM, Bourguet W, Renaud JP, Vivat V, Chambon P, et al. A canonical structure for the ligand-binding domain of nuclear receptors. Nat Struct Biol. 1996;3:87–94. doi: 10.1038/nsb0196-87. [DOI] [PubMed] [Google Scholar]
- 21.de Lera AR, Bourguet W, Altucci L, Gronemeyer H. Design of selective nuclear receptor modulators: RAR and RXR as a case study. Nat Rev Drug Discov. 2007;6:811–20. doi: 10.1038/nrd2398. [DOI] [PubMed] [Google Scholar]
- 22.Kininis M, Kraus WL. A global view of transcriptional regulation by nuclear receptors: gene expression, factor localization, and DNA sequence analysis. Nucl Recept Signal. 2008;6:e005. doi: 10.1621/nrs.06005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lonard DM, O’Malley BW. Nuclear receptor coregulators: judges, juries, and executioners of cellular regulation. Mol Cell. 2007;27:691–700. doi: 10.1016/j.molcel.2007.08.012. [DOI] [PubMed] [Google Scholar]
- 24.Lonard DM, Lanz RB, O’Malley BW. Nuclear receptor coregulators and human disease. Endocr Rev. 2007;28:575–87. doi: 10.1210/er.2007-0012. [DOI] [PubMed] [Google Scholar]
- 25.Xu HE, Stanley TB, Montana VG, Lambert MH, Shearer BG, et al. Structural basis for antagonist-mediated recruitment of nuclear co-repressors by PPARα. Nature. 2002;415:813–17. doi: 10.1038/415813a. [DOI] [PubMed] [Google Scholar]
- 26.Mi LZ, Devarakonda S, Harp JM, Han Q, Pellicciari R, et al. Structural basis for bile acid binding and activation of the nuclear receptor FXR. Mol Cell. 2003;11:1093–100. doi: 10.1016/s1097-2765(03)00112-6. [DOI] [PubMed] [Google Scholar]
- 27.Williams S, Bledsoe RK, Collins JL, Boggs S, Lambert MH, et al. X-ray crystal structure of the liver X receptor beta ligand binding domain: regulation by a histidine-tryptophan switch. J Biol Chem. 2003;278:27138–43. doi: 10.1074/jbc.M302260200. [DOI] [PubMed] [Google Scholar]
- 28.Pereira FA, Qiu Y, Zhou G, Tsai MJ, Tsai SY. The orphan nuclear receptor COUP-TFII is required for angiogenesis and heart development. Genes Dev. 1999;13:1037–49. doi: 10.1101/gad.13.8.1037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhou C, Qiu Y, Pereira FA, Crair MC, Tsai SY, Tsai MJ. The nuclear orphan receptor COUP-TFI is required for differentiation of subplate neurons and guidance of thalamocortical axons. Neuron. 1999;24:847–59. doi: 10.1016/s0896-6273(00)81032-6. [DOI] [PubMed] [Google Scholar]
- 30.Zhou C, Tsai SY, Tsai M. From apoptosis to angiogenesis: new insights into the roles of nuclear orphan receptors, chicken ovalbumin upstream promoter-transcription factors, during development. Biochim Biophys Acta. 2000;1470:M63–68. doi: 10.1016/s0304-419x(00)00005-6. [DOI] [PubMed] [Google Scholar]
- 31.Greene GL, Shiau AK, Nettles KW. A structural explanation for ERalpha/ERβ SERM discrimination. Ernst Schering Res Found Workshop. 2004:33–45. doi: 10.1007/978-3-662-05386-7_3. [DOI] [PubMed] [Google Scholar]
- 32.Nettles KW, Bruning JB, Gil G, O’Neill EE, Nowak J, et al. Structural plasticity in the oestrogen receptor ligand-binding domain. EMBO Rep. 2007;8:563–68. doi: 10.1038/sj.embor.7400963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shiau AK, Barstad D, Radek JT, Meyers MJ, Nettles KW, et al. Structural characterization of a subtype-selective ligand reveals a novel mode of estrogen receptor antagonism. Nat Struct Biol. 2002;9:359–64. doi: 10.1038/nsb787. [DOI] [PubMed] [Google Scholar]
- 34.Webb P, Nguyen NH, Chiellini G, Yoshihara HA, Cunha Lima ST, et al. Design of thyroid hormone receptor antagonists from first principles. J Steroid Biochem Mol Biol. 2002;83:59–73. doi: 10.1016/s0960-0760(02)00270-4. [DOI] [PubMed] [Google Scholar]
- 35.Walter P, Green S, Greene G, Krust A, Bornert JM, et al. Cloning of the human estrogen receptor cDNA. Proc Natl Acad Sci USA. 1985;82:7889–93. doi: 10.1073/pnas.82.23.7889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Green S, Walter P, Kumar V, Krust A, Bornert JM, et al. Human oestrogen receptor cDNA: sequence, expression and homology to v-erb-A. Nature. 1986;320:134–39. doi: 10.1038/320134a0. [DOI] [PubMed] [Google Scholar]
- 37.Kuiper GG, Lemmen JG, Carlsson B, Corton JC, Safe SH, et al. Interaction of estrogenic chemicals and phytoestrogens with estrogen receptor beta. Endocrinology. 1998;139:4252–63. doi: 10.1210/endo.139.10.6216. [DOI] [PubMed] [Google Scholar]
- 38.Ogawa S, Inoue S, Watanabe T, Hiroi H, Orimo A, et al. The complete primary structure of human estrogen receptor β (hERβ) and its heterodimerization with ERα in vivo and in vitro. Biochem Biophys Res Commun. 1998;243:122–26. doi: 10.1006/bbrc.1997.7893. [DOI] [PubMed] [Google Scholar]
- 39.Muramatsu M, Inoue S. Estrogen receptors: How do they control reproductive and nonreproductive functions? Biochem Biophys Res Commun. 2000;270:1–10. doi: 10.1006/bbrc.2000.2214. [DOI] [PubMed] [Google Scholar]
- 40.Ascenzi P, Bocedi A, Marino M. Structure-function relationship of estrogen receptor α and β: impact on human health. Mol Aspects Med. 2006;27:299–402. doi: 10.1016/j.mam.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 41.Brzozowski AM, Pike AC, Dauter Z, Hubbard RE, Bonn T, et al. Molecular basis of agonism and antagonism in the oestrogen receptor. Nature. 1997;389:753–58. doi: 10.1038/39645. [DOI] [PubMed] [Google Scholar]
- 42.Shiau AK, Barstad D, Loria PM, Cheng L, Kushner PJ, et al. The structural basis of estrogen receptor/coactivator recognition and the antagonism of this interaction by tamoxifen. Cell. 1998;95:927–37. doi: 10.1016/s0092-8674(00)81717-1. [DOI] [PubMed] [Google Scholar]
- 43.Tanenbaum DM, Wang Y, Williams SP, Sigler PB. Crystallographic comparison of the estrogen and progesterone receptor’s ligand binding domains. Proc Natl Acad Sci USA. 1998;95:5998–6003. doi: 10.1073/pnas.95.11.5998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Warnmark A, Treuter E, Gustafsson JA, Hubbard RE, Brzozowski AM, Pike AC. Interaction of transcriptional intermediary factor 2 nuclear receptor box peptides with the coactivator binding site of estrogen receptor α. J Biol Chem. 2002;277:21862–68. doi: 10.1074/jbc.M200764200. [DOI] [PubMed] [Google Scholar]
- 45.Pike AC, Brzozowski AM, Hubbard RE, Bonn T, Thorsell AG, et al. Structure of the ligand-binding domain of oestrogen receptor β in the presence of a partial agonist and a full antagonist. EMBO J. 1999;18:4608–18. doi: 10.1093/emboj/18.17.4608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pike AC, Brzozowski AM, Walton J, Hubbard RE, Bonn T, et al. Structural aspects of agonism and antagonism in the oestrogen receptor. Biochem Soc Trans. 2000;28:396–400. [PubMed] [Google Scholar]
- 47.Pike AC, Brzozowski AM, Walton J, Hubbard RE, Thorsell AG, et al. Structural insights into the mode of action of a pure antiestrogen. Structure. 2001;9:145–53. doi: 10.1016/s0969-2126(01)00568-8. [DOI] [PubMed] [Google Scholar]
- 48.Gangloff M, Ruff M, Eiler S, Duclaud S, Wurtz JM, Moras D. Crystal structure of a mutant hERα ligand-binding domain reveals key structural features for the mechanism of partial agonism. J Biol Chem. 2001;276:15059–65. doi: 10.1074/jbc.M009870200. [DOI] [PubMed] [Google Scholar]
- 49.Henke BR, Consler TG, Go N, Hale RL, Hohman DR, et al. A new series of estrogen receptor modulators that display selectivity for estrogen receptor β. J Med Chem. 2002;45:5492–505. doi: 10.1021/jm020291h. [DOI] [PubMed] [Google Scholar]
- 50.Renaud J, Bischoff SF, Buhl T, Floersheim P, Fournier B, et al. Selective estrogen receptor modulators with conformationally restricted side chains. Synthesis and structure-activity relationship of ERα-selective tetrahydroisoquinoline ligands. J Med Chem. 2005;48:364–79. doi: 10.1021/jm040858p. [DOI] [PubMed] [Google Scholar]
- 51.Kim S, Wu JY, Birzin ET, Frisch K, Chan W, et al. Estrogen receptor ligands. II Discovery of benzoxathiins as potent, selective estrogen receptor α modulators. J Med Chem. 2004;47:2171–75. doi: 10.1021/jm034243o. [DOI] [PubMed] [Google Scholar]
- 52.Malamas MS, Manas ES, McDevitt RE, Gunawan I, Xu ZB, et al. Design and synthesis of aryl diphenolic azoles as potent and selective estrogen receptor-β ligands. J Med Chem. 2004;47:5021–40. doi: 10.1021/jm049719y. [DOI] [PubMed] [Google Scholar]
- 53.Manas ES, Unwalla RJ, Xu ZB, Malamas MS, Miller CP, et al. Structure-based design of estrogen receptor-β selective ligands. J Am Chem Soc. 2004;126:15106–19. doi: 10.1021/ja047633o. [DOI] [PubMed] [Google Scholar]
- 54.Wu YL, Yang X, Ren Z, McDonnell DP, Norris JD, et al. Structural basis for an unexpected mode of SERM-mediated ER antagonism. Mol Cell. 2005;18:413–24. doi: 10.1016/j.molcel.2005.04.014. [DOI] [PubMed] [Google Scholar]
- 55.Hsieh RW, Rajan SS, Sharma SK, Guo Y, DeSombre ER, et al. Identification of ligands with bicyclic scaffolds provides insights into mechanisms of estrogen receptor subtype selectivity. J Biol Chem. 2006;281:17909–19. doi: 10.1074/jbc.M513684200. [DOI] [PubMed] [Google Scholar]
- 56.Turner JV, Agatonovic-Kustrin S, Glass BD. Molecular aspects of phytoestrogen selective binding at estrogen receptors. J Pharm Sci. 2007;96:1879–85. doi: 10.1002/jps.20987. [DOI] [PubMed] [Google Scholar]
- 57.Roy JR, Chakraborty S, Chakraborty TR. Estrogen-like endocrine disrupting chemicals affecting puberty in humans—a review. Med Sci Monit. 2009;15:RA137–45. [PubMed] [Google Scholar]
- 58.Jordan VC. Antiestrogens and selective estrogen receptor modulators as multifunctional medicines. 2 Clinical considerations and new agents. J Med Chem. 2003;46:1081–111. doi: 10.1021/jm020450x. [DOI] [PubMed] [Google Scholar]
- 59.Nilsson S, Koehler KF. Oestrogen receptors and selective oestrogen receptor modulators: molecular and cellular pharmacology. Basic Clin Pharmacol Toxicol. 2005;96:15–25. doi: 10.1111/j.1742-7843.2005.pto960103.x. [DOI] [PubMed] [Google Scholar]
- 60.Jordan VC, Fritz NF, Tormey DC. Long-term adjuvant therapy with tamoxifen: effects on sex hormone binding globulin and antithrombin III. Cancer Res. 1987;47:4517–19. [PubMed] [Google Scholar]
- 61.Love RR, Barden HS, Mazess RB, Epstein S, Chappell RJ. Effect of tamoxifen on lumbar spine bone mineral density in postmenopausal women after 5 years. Arch Intern Med. 1994;154:2585–88. [PubMed] [Google Scholar]
- 62.Love RR, Wiebe DA, Feyzi JM, Newcomb PA, Chappell RJ. Effects of tamoxifen on cardiovascular risk factors in postmenopausal women after 5 years of treatment. J Natl Cancer Inst. 1994;86:1534–39. doi: 10.1093/jnci/86.20.1534. [DOI] [PubMed] [Google Scholar]
- 63.Riggs BL, Hartmann LC. Selective estrogen-receptor modulators—mechanisms of action and application to clinical practice. N Engl J Med. 2003;348:618–29. doi: 10.1056/NEJMra022219. [DOI] [PubMed] [Google Scholar]
- 64.Bramlett KS, Wu Y, Burris TP. Ligands specify coactivator nuclear receptor (NR) box affinity for estrogen receptor subtypes. Mol Endocrinol. 2001;15:909–22. doi: 10.1210/mend.15.6.0649. [DOI] [PubMed] [Google Scholar]
- 65.Paige LA, Christensen DJ, Gron H, Norris JD, Gottlin EB, et al. Estrogen receptor (ER) modulators each induce distinct conformational changes in ERα and ERβ. Proc Natl Acad Sci USA. 1999;96:3999–4004. doi: 10.1073/pnas.96.7.3999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wijayaratne AL, Nagel SC, Paige LA, Christensen DJ, Norris JD, et al. Comparative analyses of mechanistic differences among antiestrogens. Endocrinology. 1999;140:5828–40. doi: 10.1210/endo.140.12.7164. [DOI] [PubMed] [Google Scholar]
- 67.Chang CY, Norris JD, Jansen M, Huang HJ, McDonnell DP. Application of random peptide phage display to the study of nuclear hormone receptors. Methods Enzymol. 2003;364:118–42. doi: 10.1016/s0076-6879(03)64007-3. [DOI] [PubMed] [Google Scholar]
- 68.Huang HJ, Norris JD, McDonnell DP. Identification of a negative regulatory surface within estrogen receptor α provides evidence in support of a role for corepressors in regulating cellular responses to agonists and antagonists. Mol Endocrinol. 2002;16:1778–92. doi: 10.1210/me.2002-0089. [DOI] [PubMed] [Google Scholar]
- 69.Iannone MA, Simmons CA, Kadwell SH, Svoboda DL, Vanderwall DE, et al. Correlation between in vitro peptide binding profiles and cellular activities for estrogen receptor-modulating compounds. Mol Endocrinol. 2004;18:1064–81. doi: 10.1210/me.2003-0432. [DOI] [PubMed] [Google Scholar]
- 70.Berry M, Metzger D, Chambon P. Role of the two activating domains of the oestrogen receptor in the cell-type and promoter-context dependent agonistic activity of the anti-oestrogen 4-hydroxytamoxifen. EMBO J. 1990;9:2811–18. doi: 10.1002/j.1460-2075.1990.tb07469.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Barkhem T, Carlsson B, Nilsson Y, Enmark E, Gustafsson J, Nilsson S. Differential response of estrogen receptor α and estrogen receptor β to partial estrogen agonists/antagonists. Mol Pharmacol. 1998;54:105–12. doi: 10.1124/mol.54.1.105. [DOI] [PubMed] [Google Scholar]
- 72.Metivier R, Penot G, Flouriot G, Pakdel F. Synergism between ERα transactivation function 1 (AF-1) and AF-2 mediated by steroid receptor coactivator protein-1: requirement for the AF-1α-helical core and for a direct interaction between the N- and C-terminal domains. Mol Endocrinol. 2001;15:1953–70. doi: 10.1210/mend.15.11.0727. [DOI] [PubMed] [Google Scholar]
- 73.Webb P, Nguyen P, Shinsako J, Anderson C, Feng W, et al. Estrogen receptor activation function 1 works by binding p160 coactivator proteins. Mol Endocrinol. 1998;12:1605–18. doi: 10.1210/mend.12.10.0185. [DOI] [PubMed] [Google Scholar]
- 74.Tremblay A, Tremblay GB, Labrie F, Giguere V. Ligand-independent recruitment of SRC-1 to estrogen receptor β through phosphorylation of activation function AF-1. Mol Cell. 1999;3:513–19. doi: 10.1016/s1097-2765(00)80479-7. [DOI] [PubMed] [Google Scholar]
- 75.Roy AK, Chatterjee B. Androgen action. Crit Rev Eukaryot Gene Expr. 1995;5:157–76. doi: 10.1615/critreveukargeneexpr.v5.i2.30. [DOI] [PubMed] [Google Scholar]
- 76.Matias PM, Donner P, Coelho R, Thomaz M, Peixoto C, et al. Structural evidence for ligand specificity in the binding domain of the human androgen receptor. Implications for pathogenic gene mutations. J Biol Chem. 2000;275:26164–71. doi: 10.1074/jbc.M004571200. [DOI] [PubMed] [Google Scholar]
- 77.Sack JS, Kish KF, Wang C, Attar RM, Kiefer SE, et al. Crystallographic structures of the ligand-binding domains of the androgen receptor and its T877A mutant complexed with the natural agonist dihydrotestosterone. Proc Natl Acad Sci USA. 2001;98:4904–9. doi: 10.1073/pnas.081565498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.He B, Gampe RT, Jr, Kole AJ, Hnat AT, Stanley TB, et al. Structural basis for androgen receptor interdomain and coactivator interactions suggests a transition in nuclear receptor activation function dominance. Mol Cell. 2004;16:425–38. doi: 10.1016/j.molcel.2004.09.036. [DOI] [PubMed] [Google Scholar]
- 79.Hur E, Pfaff SJ, Payne ES, Gron H, Buehrer BM, Fletterick RJ. Recognition and accommodation at the androgen receptor coactivator binding interface. PLoS Biol. 2004;2:E274. doi: 10.1371/journal.pbio.0020274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Bohl CE, Wu Z, Miller DD, Bell CE, Dalton JT. Crystal structure of the T877A human androgen receptor ligand-binding domain complexed to cyproterone acetate provides insight for ligand-induced conformational changes and structure-based drug design. J Biol Chem. 2007;282:13648–55. doi: 10.1074/jbc.M611711200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gao W, Bohl CE, Dalton JT. Chemistry and structural biology of androgen receptor. Chem Rev. 2005;105:3352–70. doi: 10.1021/cr020456u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mohler ML, Bohl CE, Jones A, Coss CC, Narayanan R, et al. Nonsteroidal selective androgen receptor modulators (SARMs): dissociating the anabolic and androgenic activities of the androgen receptor for therapeutic benefit. J Med Chem. 2009;25:3597–617. doi: 10.1021/jm900280m. [DOI] [PubMed] [Google Scholar]
- 83.Narayanan R, Mohler ML, Bohl CE, Miller DD, Dalton JT. Selective androgen receptor modulators in preclinical and clinical development. Nucl Recept Signal. 2008;6:e010. doi: 10.1621/nrs.06010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Wang Q, Carroll JS, Brown M. Spatial and temporal recruitment of androgen receptor and its coactivators involves chromosomal looping and polymerase tracking. Mol Cell. 2005;19:631–42. doi: 10.1016/j.molcel.2005.07.018. [DOI] [PubMed] [Google Scholar]
- 85.Fuller PJ, Young MJ. Mechanisms of mineralocorticoid action. Hypertension. 2005;46:1227–35. doi: 10.1161/01.HYP.0000193502.77417.17. [DOI] [PubMed] [Google Scholar]
- 86.Fagart J, Huyet J, Pinon GM, Rochel M, Mayer C, Rafestin-Oblin ME. Crystal structure of a mutant mineralocorticoid receptor responsible for hypertension. Nat Struct Mol Biol. 2005;12:554–55. doi: 10.1038/nsmb939. [DOI] [PubMed] [Google Scholar]
- 87.Li Y, Suino K, Daugherty J, Xu HE. Structural and biochemical mechanisms for the specificity of hormone binding and coactivator assembly by mineralocorticoid receptor. Mol Cell. 2005;19:367–80. doi: 10.1016/j.molcel.2005.06.026. [DOI] [PubMed] [Google Scholar]
- 88.Bledsoe RK, Madauss KP, Holt JA, Apolito CJ, Lambert MH, et al. A ligand-mediated hydrogen bond network required for the activation of the mineralocorticoid receptor. J Biol Chem. 2005;280:31283–93. doi: 10.1074/jbc.M504098200. [DOI] [PubMed] [Google Scholar]
- 89.Huyet J, Pinon GM, Fay MR, Fagart J, Rafestin-Oblin ME. Structural basis of spirolactone recognition by the mineralocorticoid receptor. Mol Pharmacol. 2007;72:563–71. doi: 10.1124/mol.107.036459. [DOI] [PubMed] [Google Scholar]
- 90.Pitt B, Remme W, Zannad F, Neaton J, Martinez F, et al. Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med. 2003;348:1309–21. doi: 10.1056/NEJMoa030207. [DOI] [PubMed] [Google Scholar]
- 91.Pitt B, Zannad F, Remme WJ, Cody R, Castaigne A, et al. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized aldactone evaluation study investigators. N Engl J Med. 1999;341:709–17. doi: 10.1056/NEJM199909023411001. [DOI] [PubMed] [Google Scholar]
- 92.Munck A, Mendel DB, Smith LI, Orti E. Glucocorticoid receptors and actions. Am Rev Respir Dis. 1990;141:S2–10. [PubMed] [Google Scholar]
- 93.Munck A, Guyre PM. Glucocorticoid physiology, pharmacology and stress. Adv Exp Med Biol. 1986;196:81–96. doi: 10.1007/978-1-4684-5101-6_6. [DOI] [PubMed] [Google Scholar]
- 94.Sapolsky RM, Romero LM, Munck AU. How do glucocorticoids influence stress responses? Integrating permissive, suppressive, stimulatory, and preparative actions. Endocr Rev. 2000;21:55–89. doi: 10.1210/edrv.21.1.0389. [DOI] [PubMed] [Google Scholar]
- 95.Schacke H, Docke WD, Asadullah K. Mechanisms involved in the side effects of glucocorticoids. Pharmacol Ther. 2002;96:23–43. doi: 10.1016/s0163-7258(02)00297-8. [DOI] [PubMed] [Google Scholar]
- 96.Rosen J, Miner JN. The search for safer glucocorticoid receptor ligands. Endocr Rev. 2005;26:452–64. doi: 10.1210/er.2005-0002. [DOI] [PubMed] [Google Scholar]
- 97.Bruna A, Nicolas M, Munoz A, Kyriakis JM, Caelles C. Glucocorticoid receptor-JNK interaction mediates inhibition of the JNK pathway by glucocorticoids. EMBO J. 2003;22:6035–44. doi: 10.1093/emboj/cdg590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yang-Yen HF, Chambard JC, Sun YL, Smeal T, Schmidt TJ, et al. Transcriptional interference between c-Jun and the glucocorticoid receptor: mutual inhibition of DNA binding due to direct protein-protein interaction. Cell. 1990;62:1205–15. doi: 10.1016/0092-8674(90)90396-v. [DOI] [PubMed] [Google Scholar]
- 99.Schule R, Rangarajan P, Kliewer S, Ransone LJ, Bolado J, et al. Functional antagonism between oncoprotein c-Jun and the glucocorticoid receptor. Cell. 1990;62:1217–26. doi: 10.1016/0092-8674(90)90397-w. [DOI] [PubMed] [Google Scholar]
- 100.Vayssiere BM, Dupont S, Choquart A, Petit F, Garcia T, et al. Synthetic glucocorticoids that dissociate transactivation and AP-1 transrepression exhibit antiinflammatory activity in vivo. Mol Endocrinol. 1997;11:1245–55. doi: 10.1210/mend.11.9.9979. [DOI] [PubMed] [Google Scholar]
- 101.Schacke H, Hennekes H, Schottelius A, Jaroch S, Lehmann M, et al. SEGRAs: a novel class of anti-inflammatory compounds. Ernst Schering Res Found Workshop. 2002:357–71. doi: 10.1007/978-3-662-04660-9_20. [DOI] [PubMed] [Google Scholar]
- 102.Shah N, Scanlan TS. Design and evaluation of novel nonsteroidal dissociating glucocorticoid receptor ligands. Bioorg Med Chem Lett. 2004;14:5199–203. doi: 10.1016/j.bmcl.2004.07.052. [DOI] [PubMed] [Google Scholar]
- 103.Schacke H, Schottelius A, Docke WD, Strehlke P, Jaroch S, et al. Dissociation of transactivation from transrepression by a selective glucocorticoid receptor agonist leads to separation of therapeutic effects from side effects. Proc Natl Acad Sci USA. 2004;101:227–32. doi: 10.1073/pnas.0300372101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ali A, Thompson CF, Balkovec JM, Graham DW, Hammond ML, et al. Novel N-arylpyrazolo[3,2-c]-based ligands for the glucocorticoid receptor: receptor binding and in vivo activity. J Med Chem. 2004;47:2441–52. doi: 10.1021/jm030585i. [DOI] [PubMed] [Google Scholar]
- 105.Bledsoe RK, Montana VG, Stanley TB, Delves CJ, Apolito CJ, et al. Crystal structure of the gluco-corticoid receptor ligand binding domain reveals a novel mode of receptor dimerization and coactivator recognition. Cell. 2002;110:93–105. doi: 10.1016/s0092-8674(02)00817-6. [DOI] [PubMed] [Google Scholar]
- 106.Kauppi B, Jakob C, Farnegardh M, Yang J, Ahola H, et al. The three-dimensional structures of antagonistic and agonistic forms of the glucocorticoid receptor ligand-binding domain: RU-486 induces a transconformation that leads to active antagonism. J Biol Chem. 2003;278:22748–54. doi: 10.1074/jbc.M212711200. [DOI] [PubMed] [Google Scholar]
- 107.Biggadike K, Bledsoe RK, Hassell AM, Kirk BE, McLay IM, et al. X-ray crystal structure of the novel enhanced-affinity glucocorticoid agonist fluticasone furoate in the glucocorticoid receptor-ligand binding domain. J Med Chem. 2008;51:3349–52. doi: 10.1021/jm800279t. [DOI] [PubMed] [Google Scholar]
- 108.Madauss KP, Bledsoe RK, McLay I, Stewart EL, Uings IJ, et al. The first X-ray crystal structure of the glucocorticoid receptor bound to a non-steroidal agonist. Bioorg Med Chem Lett. 2008;18:6097–99. doi: 10.1016/j.bmcl.2008.10.021. [DOI] [PubMed] [Google Scholar]
- 109.Wagner RL, Apriletti JW, McGrath ME, West BL, Baxter JD, Fletterick RJ. A structural role for hormone in the thyroid hormone receptor. Nature. 1995;378:690–97. doi: 10.1038/378690a0. [DOI] [PubMed] [Google Scholar]
- 110.Baxter JD, Webb P. Thyroid hormone mimetics: potential applications in atherosclerosis, obesity and type 2 diabetes. Nat Rev Drug Discov. 2009;8:308–20. doi: 10.1038/nrd2830. [DOI] [PubMed] [Google Scholar]
- 111.Bleicher L, Aparicio R, Nunes FM, Martinez L, Gomes Dias SM, et al. Structural basis of GC-1 selectivity for thyroid hormone receptor isoforms. BMC Struct Biol. 2008;8:8. doi: 10.1186/1472-6807-8-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Brenta G, Danzi S, Klein I. Potential therapeutic applications of thyroid hormone analogs. Nat Clin Pract Endocrinol Metab. 2007;3:632–40. doi: 10.1038/ncpendmet0590. [DOI] [PubMed] [Google Scholar]
- 113.Wagner RL, Huber BR, Shiau AK, Kelly A, Cunha Lima ST, et al. Hormone selectivity in thyroid hormone receptors. Mol Endocrinol. 2001;15:398–410. doi: 10.1210/mend.15.3.0608. [DOI] [PubMed] [Google Scholar]
- 114.Baxter JD, Goede P, Apriletti JW, West BL, Feng W, et al. Structure-based design and synthesis of a thyroid hormone receptor (TR) antagonist. Endocrinology. 2002;143:517–24. doi: 10.1210/endo.143.2.8617. [DOI] [PubMed] [Google Scholar]
- 115.Togashi M, Borngraeber S, Sandler B, Fletterick RJ, Webb P, Baxter JD. Conformational adaptation of nuclear receptor ligand binding domains to agonists: potential for novel approaches to ligand design. J Steroid Biochem Mol Biol. 2005;93:127–37. doi: 10.1016/j.jsbmb.2005.01.004. [DOI] [PubMed] [Google Scholar]
- 116.Borngraeber S, Budny MJ, Chiellini G, Cunha-Lima ST, Togashi M, et al. Ligand selectivity by seeking hydrophobicity in thyroid hormone receptor. Proc Natl Acad Sci USA. 2003;100:15358–63. doi: 10.1073/pnas.2136689100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Suckling K. Selective thyromimetics for atherosclerosis and dyslipidaemia: another old target making progress. Expert Opin Investig Drugs. 2008;17:615–8. doi: 10.1517/13543784.17.5.615. [DOI] [PubMed] [Google Scholar]
- 118.Svensson S, Ostberg T, Jacobsson M, Norstrom C, Stefansson K, et al. Crystal structure of the heterodimeric complex of LXRα and RXRβ ligand-binding domains in a fully agonistic conformation. EMBO J. 2003;22:4625–33. doi: 10.1093/emboj/cdg456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Quinet EM, Savio DA, Halpern AR, Chen L, Schuster GU, et al. Liver X receptor (LXR)-β regulation in LXRα-deficient mice: implications for therapeutic targeting. Mol Pharmacol. 2006;70:1340–49. doi: 10.1124/mol.106.022608. [DOI] [PubMed] [Google Scholar]
- 120.Joseph SB, McKilligin E, Pei L, Watson MA, Collins AR, et al. Synthetic LXR ligand inhibits the development of atherosclerosis in mice. Proc Natl Acad Sci USA. 2002;99:7604–9. doi: 10.1073/pnas.112059299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Hu B, Collini M, Unwalla R, Miller C, Singhaus R, et al. Discovery of phenyl acetic acid substituted quinolines as novel liver X receptor agonists for the treatment of atherosclerosis. J Med Chem. 2006;49:6151–54. doi: 10.1021/jm0609566. [DOI] [PubMed] [Google Scholar]
- 122.Kratzer A, Buchebner M, Pfeifer T, Becker TM, Uray G, et al. Synthetic LXR agonist attenuates plaque formation in apoE−/− mice without inducing liver steatosis and hypertriglyceridemia. J Lipid Res. 2009;50:312–26. doi: 10.1194/jlr.M800376-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Terasaka N, Hiroshima A, Koieyama T, Ubukata N, Morikawa Y, et al. T-0901317, a synthetic liver X receptor ligand, inhibits development of atherosclerosis in LDL receptor-deficient mice. FEBS Lett. 2003;536:6–11. doi: 10.1016/s0014-5793(02)03578-0. [DOI] [PubMed] [Google Scholar]
- 124.Peng D, Hiipakka RA, Dai Q, Guo J, Reardon CA, et al. Antiatherosclerotic effects of a novel synthetic tissue-selective steroidal liver X receptor agonist in low-density lipoprotein receptor-deficient mice. J Pharmacol Exp Ther. 2008;327:332–42. doi: 10.1124/jpet.108.142687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Schultz JR, Tu H, Luk A, Repa JJ, Medina JC, et al. Role of LXRs in control of lipogenesis. Genes Dev. 2000;14:2831–38. doi: 10.1101/gad.850400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Collins JL, Fivush AM, Watson MA, Galardi CM, Lewis MC, et al. Identification of a nonsteroidal liver X receptor agonist through parallel array synthesis of tertiary amines. J Med Chem. 2002;45:1963–66. doi: 10.1021/jm0255116. [DOI] [PubMed] [Google Scholar]
- 127.Fievet C, Staels B. Liver X receptor modulators: effects on lipid metabolism and potential use in the treatment of atherosclerosis. Biochem Pharmacol. 2009;77:1316–27. doi: 10.1016/j.bcp.2008.11.026. [DOI] [PubMed] [Google Scholar]
- 128.Pellicciari R, Fiorucci S, Camaioni E, Clerici C, Costantino G, et al. 6α-Ethyl-chenodeoxycholic acid (6-ECDCA), a potent and selective FXR agonist endowed with anticholestatic activity. J Med Chem. 2002;45:3569–72. doi: 10.1021/jm025529g. [DOI] [PubMed] [Google Scholar]
- 129.Maloney PR, Parks DJ, Haffner CD, Fivush AM, Chandra G, et al. Identification of a chemical tool for the orphan nuclear receptor FXR. J Med Chem. 2000;43:2971–74. doi: 10.1021/jm0002127. [DOI] [PubMed] [Google Scholar]
- 130.Nicolaou KC, Evans RM, Roecker AJ, Hughes R, Downes M, Pfefferkorn JA. Discovery and optimization of non-steroidal FXR agonists from natural product-like libraries. Org Biomol Chem. 2003;1:908–20. [PubMed] [Google Scholar]
- 131.Goodwin B, Jones SA, Price RR, Watson MA, McKee DD, et al. A regulatory cascade of the nuclear receptors FXR, SHP-1, and LRH-1 represses bile acid biosynthesis. Mol Cell. 2000;6:517–26. doi: 10.1016/s1097-2765(00)00051-4. [DOI] [PubMed] [Google Scholar]
- 132.Lu TT, Makishima M, Repa JJ, Schoonjans K, Kerr TA, et al. Molecular basis for feedback regulation of bile acid synthesis by nuclear receptors. Mol Cell. 2000;6:507–15. doi: 10.1016/s1097-2765(00)00050-2. [DOI] [PubMed] [Google Scholar]
- 133.Sinal CJ, Tohkin M, Miyata M, Ward JM, Lambert G, Gonzalez FJ. Targeted disruption of the nuclear receptor FXR/BAR impairs bile acid and lipid homeostasis. Cell. 2000;102:731–44. doi: 10.1016/s0092-8674(00)00062-3. [DOI] [PubMed] [Google Scholar]
- 134.Duran-Sandoval D, Mautino G, Martin G, Percevault F, Barbier O, et al. Glucose regulates the expression of the farnesoid X receptor in liver. Diabetes. 2004;53:890–98. doi: 10.2337/diabetes.53.4.890. [DOI] [PubMed] [Google Scholar]
- 135.Stayrook KR, Bramlett KS, Savkur RS, Ficorilli J, Cook T, et al. Regulation of carbohydrate metabolism by the farnesoid X receptor. Endocrinology. 2005;146:984–91. doi: 10.1210/en.2004-0965. [DOI] [PubMed] [Google Scholar]
- 136.Duran-Sandoval D, Cariou B, Fruchart JC, Staels B. Potential regulatory role of the farnesoid X receptor in the metabolic syndrome. Biochimie. 2005;87:93–98. doi: 10.1016/j.biochi.2004.11.018. [DOI] [PubMed] [Google Scholar]
- 137.Cariou B, Duran-Sandoval D, Kuipers F, Staels B. Farnesoid X receptor: a new player in glucose metabolism? Endocrinology. 2005;146:981–83. doi: 10.1210/en.2004-1595. [DOI] [PubMed] [Google Scholar]
- 138.Lee YK, Moore DD. Liver receptor homolog-1, an emerging metabolic modulator. Front Biosci. 2008;13:5950–58. doi: 10.2741/3128. [DOI] [PubMed] [Google Scholar]
- 139.Desvergne B, Wahli W. Peroxisome proliferator-activated receptors: nuclear control of metabolism. Endocr Rev. 1999;20:649–88. doi: 10.1210/edrv.20.5.0380. [DOI] [PubMed] [Google Scholar]
- 140.Robinson E, Grieve DJ. Significance of peroxisome proliferator-activated receptors in the cardiovascular system in health and disease. Pharmacol Ther. 2009;122:246–63. doi: 10.1016/j.pharmthera.2009.03.003. [DOI] [PubMed] [Google Scholar]
- 141.Rosenson RS. Fenofibrate: treatment of hyperlipidemia and beyond. Expert Rev Cardiovasc Ther. 2008;6:1319–30. doi: 10.1586/14779072.6.10.1319. [DOI] [PubMed] [Google Scholar]
- 142.Sarraf P, Mueller E, Smith WM, Wright HM, Kum JB, et al. Loss-of-function mutations in PPARγ associated with human colon cancer. Mol Cell. 1999;3:799–804. doi: 10.1016/s1097-2765(01)80012-5. [DOI] [PubMed] [Google Scholar]
- 143.Thompson EA. PPARγ physiology and pathology in gastrointestinal epithelial cells. Mol Cells. 2007;24:167–76. [PubMed] [Google Scholar]
- 144.Ondrey F. Peroxisome proliferator-activated receptor γ pathway targeting in carcinogenesis: implications for chemoprevention. Clin Cancer Res. 2009;15:2–8. doi: 10.1158/1078-0432.CCR-08-0326. [DOI] [PubMed] [Google Scholar]
- 145.Mansure JJ, Nassim R, Kassouf W. Peroxisome proliferator-activated receptor γ in bladder cancer: a promising therapeutic target. Cancer Biol Ther. 2009;8:6–15. doi: 10.4161/cbt.8.7.7853. [DOI] [PubMed] [Google Scholar]
- 146.Landreth G, Jiang Q, Mandrekar S, Heneka M. PPARγ agonists as therapeutics for the treatment of Alzheimer’s disease. Neurotherapeutics. 2008;5:481–89. doi: 10.1016/j.nurt.2008.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Jiang Q, Heneka M, Landreth GE. The role of peroxisome proliferator-activated receptor-γ (PPARγ) in Alzheimer’s disease: therapeutic implications. CNS Drugs. 2008;22:1–14. doi: 10.2165/00023210-200822010-00001. [DOI] [PubMed] [Google Scholar]
- 148.Woo EJ, Jeong DG, Lim MY, Jun Kim S, Kim KJ, et al. Structural insight into the constitutive repression function of the nuclear receptor Rev-erbβ. J Mol Biol. 2007;373:735–44. doi: 10.1016/j.jmb.2007.08.037. [DOI] [PubMed] [Google Scholar]
- 149.Wang Z, Benoit G, Liu J, Prasad S, Aarnisalo P, et al. Structure and function of Nurr1 identifies a class of ligand-independent nuclear receptors. Nature. 2003;423:555–60. doi: 10.1038/nature01645. [DOI] [PubMed] [Google Scholar]
- 150.Kruse SW, Suino-Powell K, Zhou XE, Kretschman JE, Reynolds R, et al. Identification of COUP-TFII orphan nuclear receptor as a retinoic acid-activated receptor. PLoS Biol. 2008;6:e227. doi: 10.1371/journal.pbio.0060227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Clarke N, Germain P, Altucci L, Gronemeyer H. Retinoids: potential in cancer prevention and therapy. Expert Rev Mol Med. 2004;6:1–23. doi: 10.1017/S1462399404008488. [DOI] [PubMed] [Google Scholar]
- 152.He B, Wilson EM. The NH2-terminal and carboxyl-terminal interaction in the human androgen receptor. Mol Genet Metab. 2002;75:293–98. doi: 10.1016/S1096-7192(02)00009-4. [DOI] [PubMed] [Google Scholar]
- 153.Rogerson FM, Fuller PJ. Interdomain interactions in the mineralocorticoid receptor. Mol Cell Endocrinol. 2003;200:45–55. doi: 10.1016/s0303-7207(02)00413-6. [DOI] [PubMed] [Google Scholar]
- 154.Kumar R, Baskakov IV, Srinivasan G, Bolen DW, Lee JC, Thompson EB. Interdomain signaling in a two-domain fragment of the human glucocorticoid receptor. J Biol Chem. 1999;274:24737–41. doi: 10.1074/jbc.274.35.24737. [DOI] [PubMed] [Google Scholar]
- 155.Glaros S, Atanaskova N, Zhao C, Skafar DF, Reddy KB. Activation function-1 domain of estrogen receptor regulates the agonistic and antagonistic actions of tamoxifen. Mol Endocrinol. 2006;20:996–1008. doi: 10.1210/me.2005-0285. [DOI] [PubMed] [Google Scholar]
- 156.Skafar DF, Zhao C. The multifunctional estrogen receptor-α F domain. Endocrine. 2008;33:1–8. doi: 10.1007/s12020-008-9054-1. [DOI] [PubMed] [Google Scholar]
- 157.Chandra V, Huang P, Hamuro Y, Raghuram S, Wang Y, et al. Structure of the intact PPAR-γ-RXR-nuclear receptor complex on DNA. Nature. 2008;456:350–56. doi: 10.1038/nature07413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Lefstin JA, Thomas JR, Yamamoto KR. Influence of a steroid receptor DNA-binding domain on transcriptional regulatory functions. Genes Dev. 1994;8:2842–56. doi: 10.1101/gad.8.23.2842. [DOI] [PubMed] [Google Scholar]
- 159.Gronemeyer H, Bourguet W. Allosteric effects govern nuclear receptor action: DNA appears as a player. Sci Signal. 2009;2:pe34. doi: 10.1126/scisignal.273pe34. [DOI] [PubMed] [Google Scholar]
- 160.Meijsing SH, Pufall MA, So AY, Bates DL, Chen L, Yamamoto KR. DNA binding site sequence directs glucocorticoid receptor structure and activity. Science. 2009;324:407–10. doi: 10.1126/science.1164265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Rogatsky I, Hittelman AB, Pearce D, Garabedian MJ. Distinct glucocorticoid receptor transcriptional regulatory surfaces mediate the cytotoxic and cytostatic effects of glucocorticoids. Mol Cell Biol. 1999;19:5036–49. doi: 10.1128/mcb.19.7.5036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Kurokawa R, Soderstrom M, Horlein A, Halachmi S, Brown M, et al. Polarity-specific activities of retinoic acid receptors determined by a co-repressor. Nature. 1995;377:451–54. doi: 10.1038/377451a0. [DOI] [PubMed] [Google Scholar]
- 163.Kurokawa R, DiRenzo J, Boehm M, Sugarman J, Gloss B, et al. Regulation of retinoid signalling by receptor polarity and allosteric control of ligand binding. Nature. 1994;371:528–31. doi: 10.1038/371528a0. [DOI] [PubMed] [Google Scholar]
- 164.Rochel N, Wurtz JM, Mitschler A, Klaholz B, Moras D. The crystal structure of the nuclear receptor for vitamin D bound to its natural ligand. Mol Cell. 2000;5:173–79. doi: 10.1016/s1097-2765(00)80413-x. [DOI] [PubMed] [Google Scholar]
- 165.Spera D, Cabrera G, Fiaschi R, Carlson KE, Katzenellenbogen JA, Napolitano E. Estradiol derivatives bearing sulfur-containing substituents at the 11β or 7α positions: versatile reagents for the preparation of estrogen conjugates. Bioorg Med Chem. 2004;12:4393–401. doi: 10.1016/j.bmc.2004.06.017. [DOI] [PubMed] [Google Scholar]
- 166.Dayan G, Lupien M, Auger A, Anghel SI, Rocha W, et al. Tamoxifen and raloxifene differ in their functional interactions with aspartate 351 of estrogen receptor α. Mol Pharmacol. 2006;70:579–88. doi: 10.1124/mol.105.021931. [DOI] [PubMed] [Google Scholar]
- 167.Asim M, El-Salfiti M, Qian Y, Choueiri C, Salari S, et al. Deconstructing estradiol: removal of B-ring generates compounds which are potent and subtype-selective estrogen receptor agonists. Bioorg Med Chem Lett. 2009;19:1250–53. doi: 10.1016/j.bmcl.2008.12.080. [DOI] [PubMed] [Google Scholar]