

Abstract
Free radical co-oxidation of polyunsaturated lipids with tyrosine or phenolic analogs of tyrosine gave rise to lipid peroxide-tyrosine (phenol) adducts in both aqueous micellar and organic solutions. The novel adducts were isolated and characterized by 1D and 2D NMR as well as by mass spectrometry. The spectral data suggest that the polyunsaturated lipid peroxyl radicals give stable peroxide coupling products exclusively at the para position of the tyrosyl (phenoxy) radicals. These adducts have characteristic 13C chemical shifts at 185 ppm due to the cross-conjugated carbonyl of the phenol-derived cyclohexadienone. The primary peroxide adducts subsequently undergo intramolecular Diels-Alder (IMDA) cyclization, affording a number of diastereomeric tricyclic adducts that have characteristic carbonyl 13C chemical shifts at ~198 ppm. All NMR HMBC and HSQC correlations support the structure assignment of the primary and Diels-Alder adducts, as does MS collision induced dissociation. Kinetic rate constants and activation parameters for the IMDA reaction were determined and the primary adducts were reduced with cuprous ion giving a phenol-derived 4-hydroxycyclohexa-2,5-dienone. No products from adduction of peroxyls at the phenolic ortho position were found either in the primary or the cuprous reduction product mixtures. These studies provide a framework for understanding the nature of lipid-protein adducts formed by peroxyl-tyrosyl radical-radical termination processes. Coupling of lipid peroxyl radicals with tyrosyl radicals leads to cyclohexenone and cyclohexadienone adducts which are of interest in and of themselves since, as electrophiles, they are likely targets for protein nucleophiles. One consequence of lipid peroxyl reactions with tyrosyls may therefore be protein-protein crosslinks via interprotein Michael adducts.
Keywords: Tyrosine oxidation, lipid peroxidation, para-hydroxy tyrosine, lipid protein adduct, Intramolecular Diels-Alder cyclization
Introduction
Protein-derived tyrosine radicals can act as cofactors in some enzymatic reactions, including those of class I ribonucleotide reductase, prostaglandin H synthase, photosystem II1,2 and cytochrome C oxidase.3 On the other hand, protein-derived tyrosyl radicals can also be formed in vivo as a result of oxidative stress, by mechanisms including both free radical4–7 and hemeperoxidase-dependent oxidations.8 Tyrosine-nitration, for example, is thought to occur by a two-step reaction involving the initial formation of a tyrosyl radical that subsequently reacts with nitrogen dioxide (Scheme 1).4,9–11 The formation of 3-nitro-tyrosine is an important protein post-translational modification that can alter structure and function.12 It is associated with acute and chronic disease states and it can be a predictor of disease risk and progression.11,13 The extent of protein tyrosine nitration depends on the particular protein structure and the environment and location of the individual tyrosine residue.14
Scheme 1.

Free radical mediated tyrosine oxidation reactions.
Tyrosyl radicals also couple to form 3,3′-dityrosine,15–17 see Scheme 1, and the formation of tyrosine dimers is frequently cited as evidence for the formation of tyrosyl radical species in vivo. Dimerization has been associated with protein crosslinking and the formation of high molecular weight species.16,17 A recent report, for example, shows that dimerization of a SLC30A family of zinc transporters depends on redox-regulated covalent tyrosine dimerization and suggests that dityrosine-dependent membrane protein oligomerization may regulate the function of diverse membrane proteins in normal and disease states.18
Most of the mechanistic studies of tyrosine nitration and dimerization have been carried out under aqueous conditions but the same chemistry is observed in tyrosine-containing proteins that interact with membrane lipids or are located in hydrophobic environments.14,19 It is important to note that tyrosines are frequently observed in membrane-binding regions of proteins and a recent analysis suggests that placement of tyrosine residues in protein membrane locations minimizes free energy relative to their placement in solvent exposed or transitional sites.20 The co-localization of tyrosine protein residues and membrane lipids suggests the possibility of reactions of tyrosyl and lipid-derived radicals9,10 since membranes, particularly those containing polyunsaturated fatty acid (PUFA) esters,21–23 are subject to peroxidation via chain-carrying peroxyl free radicals. Membrane lipid peroxidation has been linked to a number of human diseases including atherosclerosis 24,25 and neurodegenerative disorders. 26–30
Oxidation of tyrosine residues in ApoA1, a principal constituent of high-density lipoprotein (HDL), has been of particular interest since HDL plays an important role in the transport of cholesterol from peripheral tissues to the liver. Recent studies have provided evidence that oxidative modification of ApoA1 leads to loss of HDL function.31,32 Modification of HDL tyrosines by nitration or chlorination has been the principal focus of most studies, but HDL tyrosines are prime targets for peroxidative modification since HDL are lipid and protein rich but are poorly protected from peroxyl free radicals by Natures antioxidant, α-tocopherol.33,34
We report here studies of lipid peroxyl coupling with alkyl phenoxy radicals that serve as models for tyrosyls, see Scheme 2. The product mixture formed in these reactions is complex and includes dialkyl peroxide coupling compounds as well as novel Diels-Alder adducts. It seems likely that products like the ones identified here will be major products in the free radical peroxidation of tyrosine-containing membranes since cross-termination has been established as a major pathway in radical reactions involving both peroxyl and phenoxy species. 35
Scheme 2.
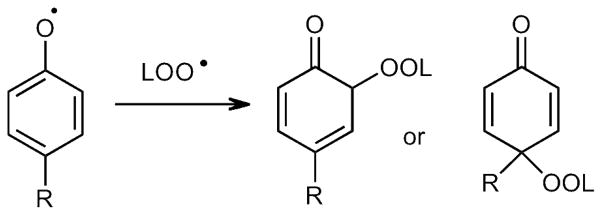
Primary coupling products of tyrosyl and lipid peroxyl radicals.
Results
Co-oxidation of hydrophobic tyrosine (1) with 1-palmitoyl-13(S)-hydroperoxyoctadecadienoyl-glycerylphosphatidyl choline (2)
A hydrophobic tyrosine analog10,36 (1, N-t-BOC-L-tyrosine tert-Butyl Ester, BTBE, 2 mM) was mixed with enantiopure PLPC-13(S)-hydroperoxide (2, 6.8 mM) generated by reaction of the 2-linoleoyl glycerophospholipid with soybean lipoxygenase. The mixture was sonicated at room temperature to form micelles in the presence of the azo free radical initiator MeOAMVN (1 mM, 2,2′-azobis(4-methoxy-2,4-dimethylvaleronitrile). The micellar solution was let stand in a 37 °C sand bath for 4h and then analyzed by HPLC-MS. A typical chromatogram is presented in Figure 1.
Figure 1.
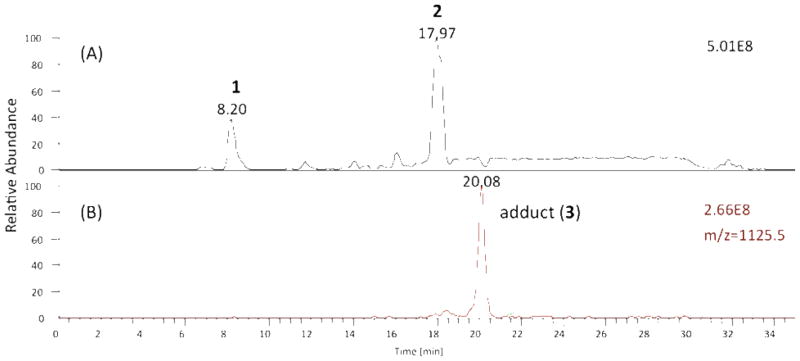
HPLC-MS analysis of reaction mixture BTBE and PLPC-13(S)-OOH. (A) Total ion current of reaction mixture, the early eluting peak at 8.2 min is BTBE (1) and later eluting peak at 18.0 min is PLPC-13(S)-OOH (2) using reverse-phase HPLC on C-18 with solvents (A) 10 mM NH4OAc in H2O and (B) 10 mM NH4OAc 95 % CH3OH/H2O with the gradient starting at 70 % B to 100% B for 10 min and then 100 % B for 15 min (B) Extracted ion current detecting adduct (3), m/z=1125.5.
MS analysis indicated the formation of adduct(s) having a parent ion of m/z = 1125.5, the expected mass of adduct 3. Attempts to isolate and further characterize the adduct(s) proved futile, however, since the initial products were only moderately stable and appeared to undergo further transformations upon attempted purification. Furthermore, the yield of the m/z=1125 adduct(s) formed under the micellar conditions was low and the products were not well-behaved on silica gel. For these reasons we sought to simplify both the tyrosine (phenol) and membrane lipid reactants so that any lipid-tyrosine adducts formed could be isolated on a scale that would permit characterization of products by the normal tools of organic chemistry, including 2D NMR spectroscopy.
Examination of reactions of several tyrosine derivatives with the simplest oxidizable fatty acids (or esters) still gave complex mixtures of products that were difficult to purify. The final simplification of the tyrosine model used was to reduce the complexity to phenol 4 [(7-methoxy-2-oxo-2H-chromen-4-yl)methyl 3-(4-hydroxyphenyl)propanoate], a compound that has no stereogenic center but a built-in chromophore unrelated to the oxidizable tyrosine moiety. The compound was prepared in one step in 87 % yield providing a model substrate having a λmax at 330 nm that could be used as a tag for products derived from the phenol. The fatty acid component was also simplified in order to minimize the number of isomeric products generated during the oxidation reaction. This reduction of complexity of the system studied led to a series of reactions of the azo initiated co-oxidation of phenol 4 with the symmetrical diene 5.
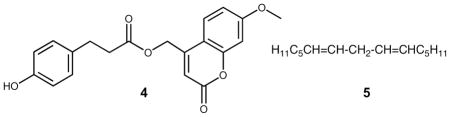
Peroxide Adducts of Peroxyl-phenoxy Coupling
The UV-active tyrosine analogue (4) was co-oxidized with E,E-6,9-pentadecadiene (5) 37 in benzene for 9 h at 37 ºC using MeOAMVN as initiator. Reaction progress was monitored at 330nm by normal phase (NP) HPLC-UV using 25% ethyl acetate in hexanes as the mobile phase (Figure 2A). In the early stages of oxidation, a peak at 16 min appeared (fraction IV) while additional peaks were detected at longer reaction times. The peak at 16 min (Figure 2A, Fraction IV) was collected, evaporated to dryness and re-suspended in 1:1 H2O:acetonitrile. Reverse phase (RP) HPLC of Fraction IV (Figure 2B) showed that it was homogeneous and the purified compound was dissolved in CDCl3 and 1H, COSY, NOESY, HSQC, HMBC, and 13C spectra were obtained at room temperature. The spectral data acquired for IV led to assignment of structure 6 to this major product of the co-oxidation reaction. Compound 6 has a characteristic 13C chemical shift for the carbonyl carbon (C4) at 185 ppm and in the HMBC experiment, C4 exhibited only one three-bond cross peak to H2. Extraction of coupling constants suggests that the conjugated diene has E,E geometry and the integration and coupling pattern is consistent with the para peroxyl-phenoxy coupling product proposed.
Figure 2.

HPLC-UV (330 nm) analysis of the co-oxidation reaction of 4 and 5. (A) Normal phase separation of the reaction mixture into fractions I–IV, 4 elutes at 23 min. (B, C, and D) reverse phase analysis of separated fractions I, II and IV.
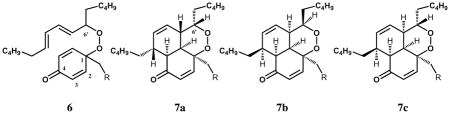
The adduct 6 was found to be unstable and it formed a mixture of stereoisomeric products with structures assigned as the intramolecular Diels-Alder (IMDA) cyclization products 7a, 7b and 7c. The same normal phase-reverse phase protocol used to purify 6 was also used to isolate the stereoisomeric IMDA products. Thus, the reaction mixture was purified first with normal phase to collect acyclic and cyclic adduct fractions, the peaks were collected as Fractions I–IV and then analyzed for purity with reversed phase HPLC. Fraction I contains only one product (7a) eluting at 14.3 min on reverse phase (Figure 2D) and Fraction II separates into two peaks eluting at 14.1 (7b) and 14.9 (7c) min with reversed phase HPLC (Figure 2C). It is noteworthy that all of the IMDA products 7a–c, were formed from the single peroxide precursor 6, i.e. Fraction IV.
Fraction III cannot be separated completely from Fraction IV and it appears to be a product closely related to 6. Thus, NMR analysis of Fraction III (Figure S5) shows it to be a mixture of compounds that are isomeric with 6 at the conjugated diene center adjacent to C6′. The structures suggested have one of the double bonds of the conjugated structure in the Z configuration. MS and UV analysis of Fraction III are consistent with the structures of the proposed constituents.
Distinctive differences between para coupling and IMDA adducts are not only the 13C chemical shifts but also the HMBC correlations. As noted, the 13C chemical shift for carbonyl carbon (C4) of 6 was observed at 185 ppm while that of the IMDA products were generally seen at 199 ppm. In the HMBC experiment, C4 of 6 exhibited only one three-bond cross peak to H2 whereas the cyclic adduct gave rise to two additional cross peaks to the ring junction protons, as shown in Figure 3. The stereochemistry of each IMDA adduct was assigned based on NOESY correlations between protons that are within the allowed spatial vicinity.
Figure 3.
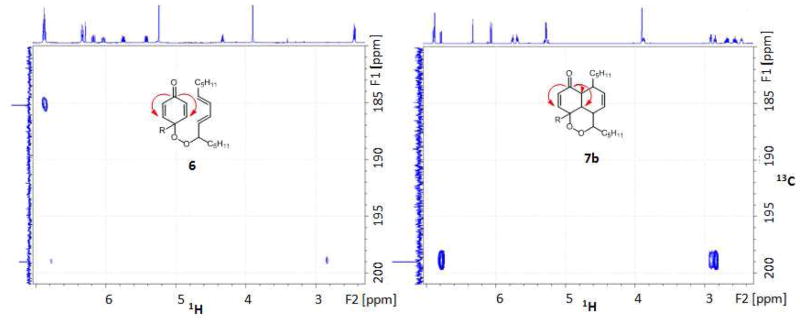
Representative HMBC correlations of adducts 6 and 7b: (A) adduct 6, C4 (185 ppm) to H2 (6.8 ppm) and (B) adduct 7b, C4 (199 ppm) to H2 and H6 (6.8, 2.9, 2.8 respectively). There is a weak two-bond correlation to H5 with this diastereomer.
HSQC spectra provide one bond correlation between carbon and protons along with the number of attached protons. The 13C signals of the peroxide bearing carbons of 6 and the IMDA adducts showed only one HSQC cross peak corresponding to C6′ at 86.0 ppm for 6 and 82.2 ppm for the 7 diasteomeric adducts. Any ortho coupling adducts such as 8, or potential derivative IMDA products such as 9 and 10, 38–42 would exhibit two HSQC cross peaks for both C5 and C6′ in each adduct. We therefore conclude that significant amounts of stable ortho coupling products are not formed. Both HSQC and HMBC spectra therefore support the notion that para phenoxy radical coupling with peroxyl occurs to form adducts 6,7a, 7b, and 7c as the major stable adducts. A more detailed discussion of the NMR analyses is presented in Supporting Information.
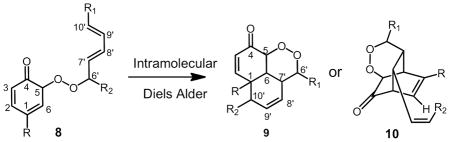
Silver coordinated (LC-Ag+)-CID-MS analysis of acyclic and cyclic adducts
While molecular ion MS information of the adducts was collected by ESI or APCI methods, good collision induced dissociation spectra could not be obtained by these conventional approaches. Silver ion forms stable complexes with unsaturated compounds and silver ion coordination MS proved to be a useful approach to provide additional support for the assigned structures of 6 and 7. Precursor mass m/z of all the isolated adducts were observed at 699 and 701 [M+Ag] due to the two isotopes 107Ag and 109Ag (Figure 4A). The silver ion complex of the primary adduct 6 displayed a major fragment ion of 476 [M-223+Ag] due to the cleavage of the peroxide bond (Figure 4B). On the other hand, the silver ion complex of the cyclic adduct exhibited multiple fragmentations as assigned in Figure 4C.
Figure 4.
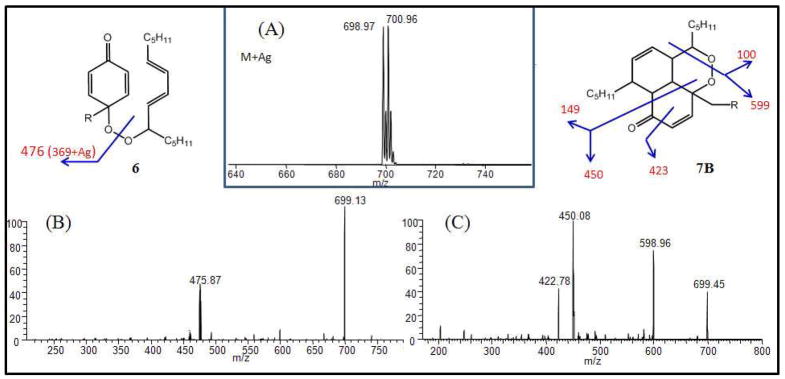
Representative MS spectra primary and IMDA adducts in 1 μM AgNO3 in MeOH sprayed directly into the MS source (collision pressure, 1.40 mTorr; collision dissociation energy, 25 eV) (A) precursor mass of the primary and IMDA adducts [M+Ag] at 699 and 701 m/z and (B) Collision induced dissociation (CID) of the silver ion complex at 699 m/z of the primary adduct 6 and (C) CID of adduct 7b.
Kinetics of Intramolecular Diels-Alder reaction of Adduct 6
Rate constants were determined for the intramolecular Diels-Alder (IMDA) reaction under several conditions. The transformation was monitored at 231 nm, the λmax of the conjugated diene, E,E-7′,9′-pentadecadiene. The decrease in absorbance at 231 nm was due to loss of the conjugated diene as the reaction proceeded, as shown in Figure 5. The data fit a single exponential decay, indicating a first order process with a rate constant; k = (9 ± 1) × 10−5 s−1 in ethanol at 37°C. The rate constant is lower in aprotic solvents such as acetonitrile (k = (6.8 ± 0.8) × 10−5 s−1) and hexanes (k = (1.3 ± 0.3) × 10−5 s−1) at 37 ºC, also having an apparent correlation with polarity. An Eyring analysis gives ΔH‡ of 68 kJ mol−1 and ΔS‡ of −98 J K−1mol−1 in ethanol in the 25–50 ºC range.
Figure 5.
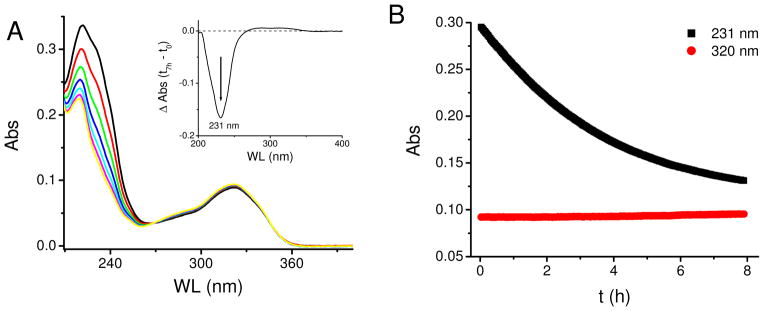
Kinetics of the intramolecular Diels-Alder reaction of adduct 6 in ethanol at 37 ºC. (A) Spectra collected every 1 hour, the inset shows the differential spectrum between 7 h and initial time. (B). The decay at 231 nm fit as a single exponential decay, indicating a first order reaction, with a rate constant k = (9 ± 1) × 10−5 s−1 (n = 3). Absorbance at 320 nm remained constant (coumarin moiety), and was used as a normalizing element.
Reduction of Adduct 6
While a rich chemistry is expected for 6, we have limited the scope of our current studies to an exploration of reduction reactions that lead to stable end products. Thus, reaction of 6 with cuprous ion or thiourea gave the p-hydroxy cyclohexadienone 11 in modest yield. We found no evidence for formation of the ortho hydroquinone 12 in the crude product mixture formed from co-oxidation of 4 and 5, also suggesting that coupling of the peroxyl radical is exclusively para. Cuprous ion was found to be the most effective way to reduce 6, while ferrous salts, thiourea and glutathione generated some compound 11, albeit in low yield. The p-OH product 11 was independently synthesized by reaction of the phenol precursor with PhI(OAc)2,43,44 the isomeric hydroquinone 12 was also independently synthesized so that authentic standards of both products were available for comparison.
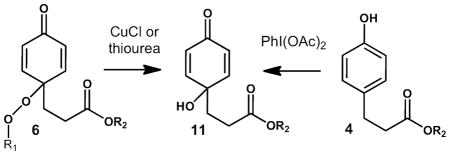
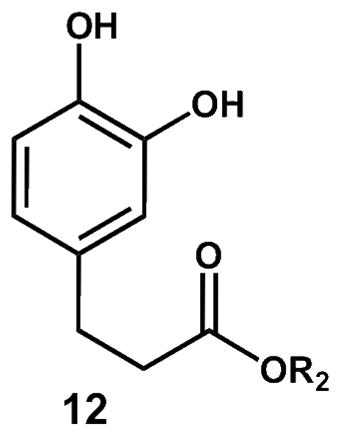
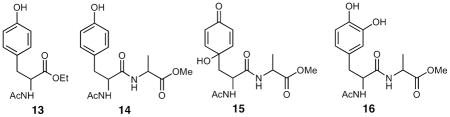
Co-oxidation of tyrosine derivatives with methyl linoleate
The formation and isolation of phenol-peroxyl radical cross-termination products in the simple model system served as a template for the reaction of more complex systems including N-acetyltyrosine ethyl ester 13 and the tyrosine-alanine dipeptide, 14. Reaction of both substrates with methyl linoleate gave rise to adducts that had the appropriate MS for the tyrosyl-linoleate peroxyl radical cross-termination product and the dipeptide system was subjected to a more detailed analysis. The primary product of co-oxidation of methyl linoleate and the dipeptide 14 had a UV λmax at 235 nm, indicating the presence of conjugated diene, the molecular ion was observed at m/z = 633.4 daltons (Figure S13) and NMR analysis indicates the presence of a major product or mixture of major products having a 13C chemical shift for a carbonyl carbon at ~185 ppm and 2D spectra consistent with formation of a product or products having a structure analogous to 6 (Figure S15). It should be noted that four linoleate hydroperoxide stereoisomers are formed in the peroxidation reaction and four coupling products with structures analogous to 6 are thus likely to be formed. This accounts for the complexity of the product mixture as analyzed by NMR and HPLC. Products were also found in the reaction mixture of 14 and methyl linoleate that had 13C chemical shift for a carbonyl carbon at ~196–8 ppm and 2D spectra consistent with formation of a product or products having structures analogous to 7a–c.
Reaction of the linoleate-peroxyl adduct mixture of 14 with CuCl gave rise to the 4-hydroxycyclohexa-2,5-dione 15, (Figure S18) a product that was synthesized independently from 14 with PhI(OAc)2.43,44 The chromatogram shown in Figure 6 illustrates the fact that the para coupling product is the only adduct formed. Chromatograms D and E in the Figure show an analysis of the reaction product formed from dipeptide 14 and methyl linoleate scanned for the M+18 adduct before (D) and after (E) reaction with CuCl. Chromatogram B shows a synthetic standard of the ortho hydroquinone 16 and the synthetic para hydroxy tyrosyl dipeptide 15 is shown in chromatogram C. Clearly, there is no evidence for formation of the hydroquinone that would form from ortho coupling. In summary, all of the data gathered from the reaction of 14 and methyl linoleate are consistent with formation of para cross-termination primary products in this co-oxidation.
Figure 6.
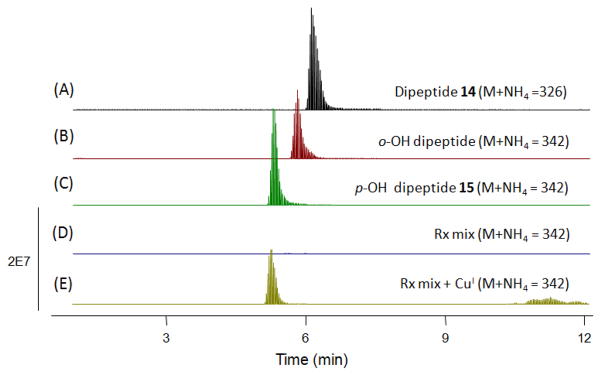
Extracted Ion Chromatograms of CuCl reduction mixture of 14-linoleate adducts. (A) 14 (B) synthetic 16 (C) synthetic 15 (D) crude co-oxidation mixture before reduction (E) crude co-oxidation mixture followed by CuCl reduction.
Discussion
Phenolic antioxidants trap peroxyl radicals in a two-step sequence, the first is an H-atom transfer from phenol to the peroxyl radical, Eq. 1, the second is reaction of the intermediate phenoxy radical with
| (1) |
| (2) |
another peroxyl radical, Eq. 2, to give non-radical products. Nature’s phenolic antioxidant, α-tocopherol, reacts with peroxyl radicals with a rate constant kinh of over 2 • 106 M−1s−1 and the second step, a radical-radical coupling occurs with a rate close or equal to diffusion control. The rate constant for inhibition, kinh, depends dramatically on the nature of substituents on the phenol. Tyrosine, a para alkyl substituted phenol, has a bond dissociation enthalpy (85.2 kcal/mol) 45 nearly 9 kcal/mol higher than that of tocopherol and as expected, the inhibition rate constant recently reported for tyrosine is substantially lower than that of tocopherol.9,46 Nevertheless, as a p-alkyl phenol, tyrosine is expected to be a modestly active antioxidant and trap peroxyl radicals to give non-radical termination products.
The phenolic radical termination step leading to non-radical products has been studied in some detail for α-tocopherol as well as for commercial products like BHT, BHA and simple phenols.35 The termination products observed are usually the result of peroxyl coupling at the para and ortho positions of the aryloxy ring. Simple phenols such as p-cresol give cross-coupling products with t-Bu peroxyl radicals bonded exclusively at the para position, see Scheme 4. Products from aryloxy radical dimerization are also formed but the cross coupling product 16 is the major product formed under most reaction conditions.35 Given this precedent, it is notable that cross termination products of tyrosine, itself a p-alkyl phenol, and lipid peroxyl radicals have not been previously detected in extracts from cells, tissues and biological fluids. One suspects that the primary peroxide coupling products may not survive biological reducing agents present in fluids and tissues and for this reason, we chose to examine tyrosyl-peroxyl radical coupling at the fundamental chemical level with the idea that the knowledge gained from this study could be used in studies of relevant biological systems.
Scheme 4.
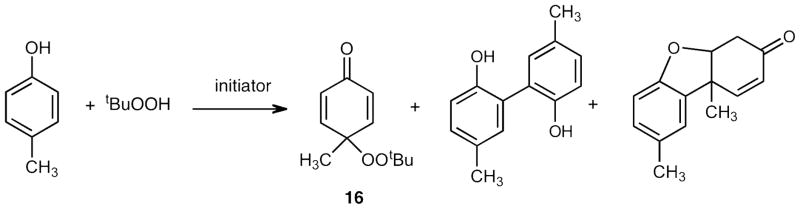
Radical termination products of para-cresol and t-butyl hydroperoxide.
The free radical initiated reaction of the hydrophobic tyrosine, BTBE, and the phospholipid hydroperoxide, PLPC-13(S)-OOH, did indeed give a product fraction having the correct m/z in the precursor ion mass spectrum as well as an MS2 fragmentation pattern consistent with an adduct of tyrosine and peroxide. The isolation of homogeneous products from BTBE proved difficult but co-oxidation of the simple phenol (4) and symmetrical diene (5) did give sufficient quantities of adducts for a comprehensive NMR study. The major product identified at early stages of reaction was the para peroxide coupling product 6, with no evidence for the formation of the phenol dimer or ortho coupling products. The peroxide 6 slowly undergoes an intramolecular Diels-Alder reaction to give at least three tricyclic products, one of the endo stereoisomers 7b being the major IMDA product formed. While the half-life for IMDA cyclization at 37°C is on the order of 2 hrs, we note that the rate is solvent dependent and likely subject to acid catalysis. Of particular note is the fact that the IMDA reaction leads to new C-C bonds between the lipid and phenol (or tyrosine). Thus, while the peroxide bond of a lipid-protein adduct like 6 should be readily reduced and the lipid-protein bond severed, any IMDA lipid-protein coupling products formed are more likely to be of a permanent nature.
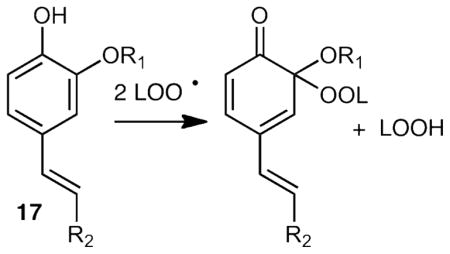
Peroxyl radical coupling with phenoxy radicals followed by IMDA cycloaddition has been reported in the reactions of curcumin 38,39 as well as caffeic 40 and ferulic acid 41,42 esters. Caffeic acid, curcumin and ferulic acid have phenol ring substitution as in 17 and the products isolated appear to be the result of ortho coupling to the phenolic OH. The preference for ortho coupling in 17 is likely due to the fact that para coupling in 17 disrupts olefin conjugation while that is not the case for tyrosine, which has para alkyl substitution.
The system studied in detail, 4 + 5 → 6, does serve as a model for the reactions of systems more relevant to proteins and fatty acids. Thus, data accumulated from the reactions of 13 or 14 with methyl linoleate gave evidence of a linoleate-tyrosine para adduct. While the product mixture was complex and adducts could not be isolated in pure form, characteristic MS, MS2 and NMR data were acquired and indicated that the chemistry observed in the model system translates to the more complex peptide-lipid reactions. Evidence for formation of para adducts and reduction of the primary products to the 4-hydroxycyclohexa-2,5-dienone was obtained.
We note that both the lipid peroxide tyrosine adducts, any derived 4-hydroxycyclohexadienones and the IMDA cyclo-adducts are electrophiles and should be subject to attack by nucleophiles from proteins or nucleic acids. Cross-conjugated cyclohexadienones are prone to undergo Michael addition and cysteines, lysines and histidines are potential protein-derived nucleophiles. A Michael addition of a cysteine on one protein and a tyrosine-derived electrophile on another would lead to a protein-lipid-protein adduct and could account for the formation of high molecular weight protein species as shown in Scheme 5 for systems under conditions of oxidative stress.
Scheme 5.
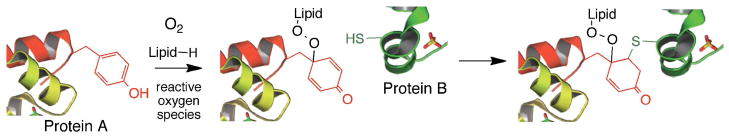
Tyrosine Lipid-Mediated Protein Crosslink
High molecular weight protein-protein adducts are frequently found under conditions of oxidative stress and tyrosyl-tyrosyl radical coupling has been cited as one mechanism for formation of these adducts.8,18,19,24,31,32,47–50 We suggest that lipid peroxide tyrosine adducts such as those reported here are also likely sources of such high molecular weight protein species under conditions of oxidative stress. Oxidizable lipid is likely to be present in great excess compared to tyrosine residues in biological membranes, which are known targets of reactive oxygen species. The formation of lipid-derived peroxyl radicals is, in fact, one of the cornerstone reactions of oxidative stress, and these lipid peroxyls are relatively mobile in a membrane compared to a tyrosine protein residue.51
Tyrosine radical-peroxyl radical chemistry will likely be dependent on the local protein-lipid environment including the presence or absence of antioxidant inhibitors. Tyrosine gives up its phenolic hydrogen some three orders of magnitude slower than does α-tocopherol so it seems likely that tyrosine radical chemistry will be suppressed if tocopherol is present in a lipid membrane or compartment. But circumstances exist where tocopherol intervention in tyrosine-lipid free radical chemistry seems unlikely, one such instance being in high-density lipoprotein (HDL) biology. HDL plays an important role in reverse-cholesterol transport and oxidative modification of HDL particles has been suggested to play a role in the development of cardiovascular disease. 19,31,33,47,48,50,52,53 An average HDL particle has a molecular weight of 270 kD, of which 50% is protein and 28% phospholipid. HDL contains, on average, approximately 0.67 molecules of α-tocopherol per particle.52 This means that 1 in 3 HDL particles would have no α-tocopherol while each will contain approximately 98 molecules of phospholipid and 30 to 40 tyrosine residues (see the Supporting Information for calculations that lead to this estimate).33,52 In those particles that contain no α-tocopherol, unsaturated lipids and tyrosine protein residues are among the most likely targets of peroxyl radical attack.
Once formed, the fate of membrane or lipoprotein tyrosyl radicals will be determined by whether the radical first encounters another tyrosyl radical and undergoes self-termination to give a tyrosine-tyrosine dimer or instead finds a lipid peroxyl radical to give a cross-termination product. Our studies in homogeneous solution find significant amounts of cross termination products under conditions where the oxidizable lipid is present in excess compared to the phenol. It seems reasonable to suggest that many biological membranes and lipid particles will provide local environments in which tyrosyl-tyrosyl radical coupling is limited by the specific sites and rate of diffusion of these protein radicals. Lipid peroxyl radicals, on the other hand, would likely be substantially more mobile than protein tyrosine radicals, perhaps by two or three orders of magnitude, and this should promote the cross-termination reaction.51,54–57
Finally, we note that the focus of a host of studies on protein tyrosine modification has been mainly on ortho- substitution, i.e. chlorination, hydroxylation and nitration. The stable lipid peroxide-tyrosine adducts identified here have been exclusively the result of para-addition to the tyrosine moiety and this suggests that para- substituted products may also be formed in other radical transformations. Such para addition compounds would be active electrophiles and subject to reaction with protein and nucleic acid nucleophiles. Numerous reports have suggested that oxidation and adduction of tyrosines might play an important role in numerous diseases including atherosclerosis 24,25,32,58,59 and neurodegenerative disorders such as Parkinson’s and Alzheimer’s disease.26–30,60–74 We suggest that lipid and lipid peroxyl radicals75 may play an important role in this chemistry.
Experimental Section
General methods and materials
BTBE was prepared by Dr. Joy Joseph from the Medical College of Wisconsin. The initiator, MeOAMVN (2,23-Azobis(4-methoxy-2,4 dimethylvaleronitrile) was purchased from Wako Chemicals USA, Inc. (Richmond, VA, USA) and dried under high vacuum for 2 h and kept at −80 ºC until use. Methyl linoleate and linoleic acids were purchased from NuChek Prep (Elysian, MN, USA). Symmetric E,E-6,9-pentadecadiene was synthesized following the method previously reported 37. Benzene (HPLC grade) was passed through a column of neutral alumina and stored over molecular sieves. Hexanes, ethyl acetate, methylene chloride, THF and methanol (all 99.9%) were purchased from Thermo Fisher Scientific Inc. Lipids were obtained from Avanti Biochemicals, 3-(4-Hydroxyphenyl)propionic acid and 4-Bromomethyl-7-methoxycoumarin were purchased from TCI America. 4-Dihydroxyhydrocinnamic and L-alanine methyl ester hydrochloride were purchased from Sigma Aldrich. N-acetyl-L-tyrosine was purchased from Fluka. PhI(OAc)2 (Iodobenzene diacetate) was purchased from Acros Organics. Triethylamine and N,N-Dimethylformamide (DriSolv®) were purchased from EMD® and used without further purification. All glassware was oven-dried prior to use.
HPLC Analysis Conditions
Normal phase NP-HPLC analyses were performed using a Beckman Ultrasphere 5 μm silica column (4.6 mm × 25 cm) using different methods: condition A: isocratic method using 20 % ethyl acetate in hexanes at 1.5 mL/min flow rate; condition B: 10% isopropanol in hexanes increasing to 15% isopropanol in 15 minutes for 5 minutes; condition C: 8% isopropanol in hexanes at 4 mL/min, using a semipreparative NP column (Beckman Ultrasphere Silica 5 μm, 10 mm × 25 cm). Reverse phase HPLC analyses were performed using a Luna® C-18 column (4.6 mm × 25 cm) from Phenomenex (Torrance, CA). The mobile phases used in reverse phase HPLC are: Condition 1 (A) 10 mM ammonium formate and (B) acetonitrile with an initial gradient of 80 % B then increased to 95 % B immediately followed by 99% B for 10 min.; Condition 2: (A) 10 mM NH4OAc in H2O (B) 10 mM NH4OAc in 95 % CH3OH/H2O, the gradient started with 70 % B to 100% B over 10 min and maintained at 100 % B for 15 min.; Condition 3: (A) 10 mM ammonium formate and (B) acetonitrile, the gradient started with 25% B maintained for 5 minutes then increased to 50% B for 5 minutes and then to 75% at for 5 minutes followed immediately 99% B maintained for 5 min.; Condition 4: starting with 70 % acetonitrile:10 mM ammonium formate, increased to 83% acetonitrile over 20 min., then back to 70% acetonitrile.
NMR Studies
All reported NMR spectra were acquired using a 14.1 T Bruker magnet equipped with a Bruker DRX spectrometer operating at proton Larmor frequency of 600.13 MHz. 1H spectra were acquired in 2.5 mm NMR tubes using a Bruker 5 mm cryogenically cooled NMR probe. Chemical shifts were referenced internally to chloroform (7.26 ppm), which also served as the 2H lock solvent. 1H, 2D COSY, NOESY, HSQC, and HMBC were obtained for all the adducts for structural assignment. 13C was obtained only for the major adducts. Some NMR spectra were interpreted using MestReNova NMR 6.1.1.
Silver (LC-Ag+)-CID-MS of acyclic and cyclic adducts
The adducts were purified by HPLC prior to MS analysis and they were dissolved in 0.3 mM AgNO3 solution in MeOH to obtain MS2 information.76 MS was performed using a ThermoFinnigan TSQ Quantum Ultra equipped with a Finnigan Surveyor Autosampler Plus (San Jose, CA). Nitrogen gas served both as the sheath and auxiliary gas, and argon served as the collision gas. The electrospray needle was maintained at 4.6 kV, and the heated capillary temperature was 200 °C. Collision induced dissociation (CID) for the tyrosine adducts were set either 25 eV or 35 eV under 1.4 mTorr of argon. Spectra are displayed by averaging the scans across the individual chromatography peaks. Data acquisition and analysis is performed using Xcalibur software version 2.0 SR2 (San Jose, CA).
HPLC-ESI-mass spectrometry
HPLC-MS/MS analysis was performed on a Waters Aquity UPLC system (Waters, Milford, MA) connected to a Finnigan LTQ mass spectrometer (Thermo Fisher Scientific, Waltham, MA), operating in the ESI positive ion mode. The same HPLC mobile phase and gradient described above were used except the column for the LC-MS analysis. Luna C8 with 3 μm was used with flow rate 350 μL/min. MS analysis was performed in both data dependent and SIM (selected ion monitoring) mode for the desired m/z.
Synthesis of PLPC-13(S)-OOH (2)
PLPC (5 mg) and 4 mg of soybean lipoxygenase (Sigma, St. Louise, MO) was dissolved in 5 mL of 100 mM borate and 10 mM deoxycholic acid buffer (pH = 9). The reaction mixture was stirred vigorously at room temperature for 1 h and CHCl3: CH3OH = 2:1 (3 mL) was used twice to extract PLPC and PLPC-OOH. The organic layer was collected and solvent was evaporated using a rotary evaporator. The phospholipid extracted in this way was re-suspended in CH3OH and purified with HPLC using 95 % CH3OH in H2O as solvent. The collected peak was dried and resuspended in isopropanol and used as a stock solution (2.16 mg/4mL).
Oxidation of BTBE (1, N-t-BOC-L-tyrosine tert-Butyl Ester) and PLPC-13S-hydroperoxide (2) in a liposome
BTBE (33.7 mg) was dissolved in 1 mL of CH3OH to prepare a100 mM stock solution. One mL of PLPC-13(S)-OOH (540 μg) and 2 μL of BTBE stock solutions were combined in an Eppendorf tube and 2 μL of a MeOAMVN stock solution (50 mM in benzene) was added. The solution was vortexed for 2 min and it was then dried under a nitrogen stream. NH4OAc (100 μL of 100 mM solution, pH = 7) was added to the dried mixture to make a final concentrations of PLPC-13(S)-OOH, BTBE, and MeOAMVN of 6.8 mM, 2 mM, and 1 mM respectively. The reaction mixture was sonicated for 5 min then let it stand in a 37 ºC sand bath with stirring. An aliquot (30 μL) was taken from the reaction mixture after 4 h and diluted with 100 μL of CH3OH for MS analysis.
Synthesis of tyrosine analogue (4)
In a 100 mL round bottom flask was added 3-(4-hydroxyphenyl)propionic acid (0.62 g, 3.72 mmol) and the flask was purged with argon. DMF was added (40 mL) and 4-bromomethyl-7-methoxycoumarin (1.0 g, 3.7 mmol) followed by triethylamine (0.52 mL, 3.7 mmol). The flask was wrapped with aluminum foil and stirred at room temperature under an argon-balloon for 24 h. The solvent was then removed in vacuo, yielding the crude product as a pale-brown oil. Silica flash column chromatography (60:39:1, hexanes:EtOAc:TEA) afforded the product 4 as a white solid (1.15g, 87%) upon solvent removal. 1H NMR (400 MHz, DMSO) δ 9.17 (s, 1H), 7.58 (d, J = 8.9 Hz, 1H), 7.05 – 6.98 (m, 3H), 6.94 (dd, J = 8.9, 2.5 Hz, 1H), 6.72 – 6.59 (m, 2H), 6.23 (s, 1H), 5.31 (d, J = 1.1 Hz, 2H), 3.85 (s, 3H), 2.75 (tt, J = 6.8, 3.4 Hz, 4H). 13C NMR (101 MHz, DMSO) δ 172.29, 162.90, 160.29, 156.03, 155.35, 150.72, 130.69, 129.49, 126.20, 115.48, 112.69, 110.67, 109.58, 101.38, 61.54, 56.35, 35.60, 29.78. HRMS (M+Na) calculated 377.0996, found 377.0996.
Oxidation of tyrosine analogue (4) with E,E-6,9-pentadecadiene (5)
To a solution of E,E-6,9-pentadecadiene (5, 0.5 g, 2.4 mmol) in benzene (3 mL) at 37 ºC was added a solution of the tyrosine analogue 4 (21 mg, 0.06 mmol in 2 mL of MeCN) and the radical initiator MeOAMVN (30 mg, 0.1 mmol). The reaction mixture was kept at 37 ºC under an oxygen atmosphere for 9 hours. Reaction progress was monitored by HPLC after 5 h of oxidation. The reaction was stopped at 9 h to obtain pure acyclic adduct 6 for NMR experiments (Table S1). HRMS (M+H) calculated 593.3114, found 593.3107.
Kinetics of Intramolecular Diels-Alder of adduct 6
Adduct 6 was purified by normal phase HPLC using 20 % ethyl acetate in hexanes. The peak corresponding to the acyclic adduct was collected and the purity was assessed by reverse phase HPLC and NMR. In order to assess the rate of cyclization from adduct 6 to 7 via Intramolecular Diels-Alder reaction, a small volume of the work solution was introduced into a much larger volume of solvent that has been pre-equilibrated at the desired temperature, yielding a final concentration of 50 μM of adduct 6. The conversion was followed by UV spectroscopy in an Agilent 8453 photo diode array spectrophotometer in quartz cuvettes with Teflon stoppers. The temperature was maintained @ 37 °C by a circulating bath. The absorbance of the solution was measured at 231 nm, the λmax of conjugated diene, every 2 min for 8 h. The decrease in absorbance at 231 nm was fitted to a single exponential decay.
Reduction of adduct 6
To 100 μL of a 100 μM solution of 6 in 10% isopropanol in ethyl acetate, was added with a variety of reducing reagents. Thus, the following reagents were examined in exploratory experiments: (A) 5mg of CuCl crystals; (B) 5 mg Fe(NH4)2(SO4)2; (C) 10 mM thiourea in methanol; (D) 5 mg of CuCl crystals and 8.4 M acetic acid; (E) 5 mg Fe(NH4)2(SO4)2 and 8.4 M acetic acid (F) 5 mg Zn (dust) and 8.4 M acetic acid 77 (G) 20 μM CuSO4 and 100 μM or 1 mM ascorbic acid; (H) freshly scratched Mg turnings and iodine (in methanol); (I) Dimethyl sulfide at 1 mM, 10 mM, and 100 mM; (J) 100 mM 2-mercaptoethanol; (K) glutathione 1 mM and 100 mM; (L) sodium borohydride 1 mM and 35 mM; (M) 60 mM SnCl2. Each reaction was left at room temperature for 18 h in the dark and then analyzed by reverse-phase HPLC on a C18 column using solvent Condition 3. The reaction progress was monitored at 320 nm. The reaction mixtures were compared with authentic standards, tyrosine analogue (4), p-hydroxy tyrosine analogue (11), and o-hydroxy tyrosine analogue (12).
Synthesis of o-hydroxy tyrosine analogue (12)
To a 100ml round bottom flask under argon was added 3,4-dihydroxyhydrocinnamic acid (0.71 g, 3.90 mmol) and DMF (50 mL). To this solution was added 4-bromomethyl-7-methoxycoumarin (0.91 g, 3.38 mmol) followed by triethylamine (0.57 mL, 3.72 mmol). The reaction mixture turned light-yellow upon addition of the amine. The flask was wrapped with aluminum foil and stirred at room temperature under an argon-balloon for 24h. The solvent was removed in vacuo, yielding the crude product as a brown oil. Silica flash column chromatography (60:39:1, hexanes:EtOAc:TEA) afforded the product as a white solid (1.13 g, 90%) upon solvent removal. 1H NMR (400 MHz, DMSO) δ 8.69 (s, 2H), 7.57 (d, J = 8.9 Hz, 1H), 7.02 (d, J = 2.5 Hz, 1H), 6.94 (dd, J = 8.9, 2.5 Hz, 1H), 6.60 (m, 2H), 6.45 (dd, J = 8.0, 2.1 Hz, 1H), 6.25 (s, 1H), 5.31 (d, J = 1.0 Hz, 2H), 3.85 (s, 3H), 2.71 (s, 4H). 13C NMR (101 MHz, DMSO) δ 172.31, 162.90, 160.30, 155.36, 150.71, 145.45, 143.94, 131.45, 126.23, 119.14, 116.04, 115.84, 112.70, 110.69, 109.66, 101.40, 61.59, 56.36, 35.62, 30.00. HRMS (M+Na) calculated 393.0945, found 393.0938.
Oxidation of NATEE (13, N-acetyl-L-Tyrosine Ethyl Ester) with methyl linoleate
Methyl linoleate (118 mg) and 5 mg NATEE 13 were oxidized using 6 mg MeOAMVN in 1 mL of 10% acetonitrile in benzene. The reaction mixture was fractionated using NP HPLC. The chromatograms of adducts (Figure S10) were similar to those of model tyrosine analogue (4) and E,E-6,9-pentadecadiene (5) reaction but they were much more complex due to the formation regioisomers. Fraction A was collected and dried under vacuum, it was then resuspended in MeOH (0.1 % AA) and analyzed by MS. The corresponding adducts were identified, yielding M+H= 575.93 and M+Na=598.27 and further characterized by MS2 (Figure S11)
Synthesis of dipeptide (14, N-acetyl-L-Tyrosine L-Alanine Methyl Ester)
To a flame-dried 10 mL round bottom flask was added N-acetyl-L-tyrosine (0.50 g, 2.24 mmol), L-alanine methyl ester hydrochloride (0.31 g, 2.24 mmol) and 2-chloro-4,6-dimethoxy-1,3,5-triazine (CDMT) (0.433 g, 2.46 mmol).78 The flask was dried in vacuo for 20 minutes and then purged with argon. Acetonitrile (2.5 mL), followed by 4-methylmorpholine (NMM) (0.616 mL, 5.60 mmol) were added over 10 min and the reaction was stirred at 15 °C under argon-balloon for 2 hours. The reaction was quenched with H2O (12 mL) and extracted with ethyl acetate (25 mL). The organic layer was washed with 1N HCl (2X, 6 mL), H2O (12 mL) and brine (12 mL), dried over MgSO4, filtered and concentrated in vacuo. Silica flash column chromatography (10:90, MeOH:CH2Cl2) afforded the product 14 as white solid (0.50 g, 72.4%) upon solvent removal. 1H NMR (400 MHz, DMSO) δ 9.13 (s, 1H), 8.41 (d, J = 7.0 Hz, 1H), 7.98 (d, J = 8.5 Hz, 1H), 7.02 (d, J = 8.5 Hz, 2H), 6.63 (d, J = 8.5 Hz, 2H), 4.43 (td, J = 9.8, 4.2 Hz, 1H), 4.26 (p, J = 7.2 Hz, 1H), 3.60 (s, 3H), 2.85 (dd, J = 13.9, 4.2 Hz, 1H), 2.57 (dd, J = 13.9, 10.1 Hz, 1H), 1.73 (s, 3H), 1.27 (d, J = 7.3 Hz, 3H). 13C NMR (101 MHz, DMSO) δ 173.31, 171.97, 169.42, 156.09, 130.42, 128.41, 115.18, 55.28, 54.28, 52.23, 47.91, 37.23, 22.83, 17.24. HRMS (M+Na) calculated 331.1264, found 331.1255.
Synthesis of p-hydroxy dipeptide (15, N-acetyl-L-p-OH-Tyrosine-L-Alanine Methyl Ester)
To a 10 mL round bottom flask was added iodobenzene diacetate (PhI(OAc)2, 0.31 g, 0.973 mmol) and H2O:THF (20:80, 3.0 mL) followed by dipeptide (14, 0.15 g, 0.486 mmol) in H2O:THF (20:80, 3.0 mL), over 10 min. The reaction mixture turned light-yellow upon initial dipeptide addition and then became amber in color. The reaction mixture stirred under atmosphere at room temperature. Reaction progress was monitored via TLC (10:90, MeOH:CH2Cl2) for loss of starting dipeptide. After 2 h, the reaction was deemed complete and the solvent was removed in vacuo, yielding an orange solid. Silica flash column chromatography (5:95, MeOH: CH2Cl2) afforded the product 15 as white solid (0.11 g, 70%) upon solvent removal. 1H NMR (600 MHz, CDCl3) δ 7.08 (dd, J = 10.4, 3.1 Hz, 1H), 6.97 (d, J = 6.8 Hz, 1H), 6.91 (dd, J = 10.3, 3.1 Hz, 1H), 6.51 (d, J = 7.2 Hz, 1H), 6.24 – 6.16 (m, 2H), 4.73 (dd, J = 13.3, 6.1 Hz, 1H), 4.54 (p, J = 7.2 Hz, 1H), 3.77 (s, 3H), 3.66 (s, 1H), 2.37 (dd, J = 14.8, 5.6 Hz, 1H), 2.05 (s, 3H), 1.95 (dd, J = 14.8, 6.3 Hz, 1H), 1.44 (d, J = 7.2 Hz, 3H). 13C NMR (151 MHz, CDCl3) δ 184.78, 172.91, 170.68, 170.40, 149.86, 149.41, 128.24, 127.76, 68.15, 52.64, 49.12, 48.36, 43.56, 23.20, 17.97. HRMS (M+Na) calculated 347.1214, found 347.1210.
Synthesis of o-hydroxy dipeptide (N-acetyl-L-o-OH-Tyrosine-L-Alanine Methyl Ester)
To a 25 mL round bottom flask was added 3,4-dihydroxy-L-phenylalanine (0.50 g, 2.54 mmol) and de-ionized H2O (6.0 mL). The flask was purged with argon and acetic anhydride (Ac2O) (5.0 mL, 50.71 mmol) was added over 1 h with an addition funnel. The reaction mixture changed from milky-white to colorless upon Ac2O addition and then became a slurry after 30 minutes. After 2 hours at room temperature, the solvent was removed in vacuo. Silica flash column chromatography (40:60 MeOH:CH2Cl2) afforded the N-acetyl-3,4-dihydroxy-L-phenylalanine78 as sticky-white solid (0.30 g, 49%). 1H NMR (300 MHz, D2O) δ 6.72 (d, J = 8.0 Hz, 1H), 6.66 (s, 1H), 6.56 (d, J = 8.1 Hz, 1H), 4.31 – 4.21 (m, 1H), 2.94 (dd, J = 14.1, 4.7 Hz, 1H), 2.67 (dd, J = 14.0, 8.6 Hz, 1H), 1.82 (s, 3H).
To a 10 mL round bottom flask was added N-acetyl-3,4-dihydroxy-L-phenylalanine (0.298 g, 1.25 mmol), L-alanine methyl ester hydrochloride (0.17 g, 1.25 mmol) and 2-chloro-4,6-dimethoxy-1,3,5-triazine (CDMT) (0.24 g, 1.37 mmol). The flask was dried in vacuo for 20 minutes and then directly purged with argon. CH3CN (3.0 mL) was added followed by 4-methylmorpholine (NMM) (0.34 mL, 3.11 mmol) over 10 minutes.79 The reaction mixture turned cloudy after NMM addition. The reaction stirred at 15 °C, under argon-balloon for 2 hours, it was then quenched with H2O (12 mL) and extracted with ethyl acetate (30 mL). The organic layer was washed with 1N HCl (2X, 6 mL), H2O (12 mL) and brine (12 mL), dried over MgSO4, filtered and concentrated in vacuo. Silica flash column chromatography (10:90, MeOH:CH2Cl2) afforded the product 15 as white solid (0.28 g, 70%) upon solvent removal. 1H NMR (600 MHz, D2O) δ 6.67 (d, J = 8.1 Hz, 1H), 6.59 (d, J = 1.7 Hz, 1H), 6.52 (dd, J = 8.1, 1.7 Hz, 1H), 4.29 (t, J = 7.7 Hz, 1H), 4.19 (q, J = 7.2 Hz, 1H), 3.50 (s, 3H), 2.73 (qd, J = 13.8, 7.8 Hz, 2H), 1.79 (s, 3H), 1.15 (d, J = 7.2 Hz, 3H). 13C NMR (151 MHz, D2O) δ 174.36, 173.85, 172.80, 143.78, 142.78, 128.76, 121.46, 116.79, 116.07, 55.11, 52.80, 48.45, 36.35, 21.48, 16.12. HRMS (M+Na) calculated 347.1214, found 347.1211.
Oxidation of dipeptide (14) with methyl linoleate
Methyl linoleate (400 mg) and 20 mg of the dipeptide (14) were oxidized using 20 mg MeOAMVN in 3.5 mL of 60% acetonitrile in benzene. The reaction mixture was fractionated using NP HPLC. The chromatograms of the adducts were similar to those of model tyrosine analogue (4) and E,E-6,9-pentadecadiene (5) reaction but they were much more complex due to the formation of regio-, stereoisomers (Figure S12). Fractions 6_1, 6_2, 7_3 and 7_4 were collected from RP-HPLC and analyzed by mass spectrometry. The corresponding adducts yielding M+H= 633.08 and M+Na=649.75 were observed and further characterized by MS2 (Figure S13). The fractions of 6_1 and 7_4 were collected and acquired 1D and 2 D NMR. A number of peroxyl-phenol coupled adducts were formed and the new downfield shift 13C in HMBC indicated the formation of para acyclic and cyclic adducts as demonstrated in the model system.
Supplementary Material
Acknowledgments
We are grateful to Dr. Joy Joseph of the Medical College of Wisconsin for a gift of BTBE. NAP acknowledges funding from the National Science Foundation and support from the Center in Molecular Toxicology, T32 ES007028 (Training Program in Environmental Toxicology). Vanderbilt University. Support is also acknowledged from the Howard Hughes Medical Institute to R.R., National Institutes of Health to B.K. and R.R. (2 R01Hl063119-05), and Agencia Nacional de Investigación e Innovación (ANII)/Fondo Clemente Estable to S.B. (FCE_362). S.B. was partially supported by a fellowship of ANII. R.R. is a Howard Hughes International Research Scholar.
ABBREVIATIONS
- NMR
nuclear magnetic resonance
- PUFA
polyunsaturated fatty acid
- MeOAMVN
2,2′-azobis(4-methoxy-2,4-dimethylvaleronitrile)
- HPLC
high performance liquid chromatography
- EtOAc
Ethyl acetate
- DMF
N,N,-Dimethylformamide
- MS
mass spectrometry
- HSQC
Heteronuclear Single Quantum Coherence
- HMBC
Heteronuclear Multiple Bond Coherence
- NOE
nuclear Overhauser effect
- NOESY
NOE spectroscopy
- ESI
electrospray ionization
- TIC
total ion current
- m/z
mass-to-charge ratio
- IMDA
intramolecular Diels-Alder
- BTBE
N-t-BOC-L-tyrosine tert-Butyl Ester
- NATEE
N-acetyl-L-tyrosine ethyl ester
- PLPC
1-palmitoyl-2-linoleoyl glycerylphosphatidylcholine
Footnotes
Supporting Information Available: Tables of 1H- and 13C-NMR chemical shifts, 1D- and 2D-NMR spectra, HPLC-MS, experimental details. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Pesavento RP, van der Donk WA. Adv Protein Chem. 2001;58:317. doi: 10.1016/s0065-3233(01)58008-0. [DOI] [PubMed] [Google Scholar]
- 2.Hoganson CW, Tommos C. Biochim Biophys Acta. 2004;1655:116. doi: 10.1016/j.bbabio.2003.10.017. [DOI] [PubMed] [Google Scholar]
- 3.Proshlyakov DA, Pressler MA, DeMaso C, Leykam JF, DeWitt DL, Babcock GT. Science. 2000;290:1588. doi: 10.1126/science.290.5496.1588. [DOI] [PubMed] [Google Scholar]
- 4.Radi R. Proc Natl Acad Sci U S A. 2004;101:4003. doi: 10.1073/pnas.0307446101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Das AB, Nagy P, Abbott HF, Winterbourn CC, Kettle AJ. Free Rad Biol Med. 2010 doi: 10.1016/j.freeradbiomed.2010.02.039. [DOI] [PubMed] [Google Scholar]
- 6.Nagy Pt, Kettle AJ, Winterbourn CC. J Biol Chem. 2009;284:14723. doi: 10.1074/jbc.M809396200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Winterbourn CC. Nat Chem Biol. 2008;4:278. doi: 10.1038/nchembio.85. [DOI] [PubMed] [Google Scholar]
- 8.Heinecke JW. Toxicology. 2002;177:11. doi: 10.1016/s0300-483x(02)00192-0. [DOI] [PubMed] [Google Scholar]
- 9.Bartesaghi S, Wenzel J, Trujillo M, Lopez M, Joseph J, Kalyanaraman B, Radi R. Chem Res Toxicol. 2010;23:821. doi: 10.1021/tx900446r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bartesaghi S, Valez V, Trujillo M, Peluffo G, Romero N, Zhang H, Kalyanaraman B, Radi R. Biochemistry. 2006;45:6813. doi: 10.1021/bi060363x. [DOI] [PubMed] [Google Scholar]
- 11.Szabo C, Ischiropoulos H, Radi R. Nat Rev Drug Discov. 2007;6:662. doi: 10.1038/nrd2222. [DOI] [PubMed] [Google Scholar]
- 12.Ischiropoulos H. Arch Biochem Biophys. 2009;484:117. doi: 10.1016/j.abb.2008.10.034. [DOI] [PubMed] [Google Scholar]
- 13.Peluffo G, Radi R. Cardiovasc Res. 2007;75:291. doi: 10.1016/j.cardiores.2007.04.024. [DOI] [PubMed] [Google Scholar]
- 14.Bartesaghi S, Ferrer-Sueta G, Peluffo G, Valez V, Zhang H, Kalyanaraman B, Radi R. Amino Acids. 2007;32:501. doi: 10.1007/s00726-006-0425-8. [DOI] [PubMed] [Google Scholar]
- 15.Dean RT, Fu S, Stocker R, Davies MJ. Biochem J. 1997;324:1. doi: 10.1042/bj3240001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Souza JM, Peluffo G, Radi R. Free Rad Biol Med. 2008;45:357. doi: 10.1016/j.freeradbiomed.2008.04.010. [DOI] [PubMed] [Google Scholar]
- 17.Giulivi C, Traaseth NJ, Davies KJ. Amino Acids. 2003;25:227. doi: 10.1007/s00726-003-0013-0. [DOI] [PubMed] [Google Scholar]
- 18.Salazar G, Falcon-Perez JM, Harrison R, Faundez V. PLoS One. 2009;4:e5896. doi: 10.1371/journal.pone.0005896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Shao B, Bergt C, Fu X, Green P, Voss JC, Oda MN, Oram JF, Heinecke JW. J Biol Chem. 2005;280:5983. doi: 10.1074/jbc.M411484200. [DOI] [PubMed] [Google Scholar]
- 20.Koehler J, Woetzel N, Staritzbichler R, Sanders CR, Meiler J. Proteins. 2009;76:13. doi: 10.1002/prot.22315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Porter NA. Acc Chem Res. 1986;19:262. [Google Scholar]
- 22.Porter NA. Meth Enzy. 1984;105:273. doi: 10.1016/s0076-6879(84)05035-7. [DOI] [PubMed] [Google Scholar]
- 23.Xu L, Davis TA, Porter NA. J Am Chem Soc. 2009;131:13037. doi: 10.1021/ja9029076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Leeuwenburgh C, Rasmussen JE, Hsu FF, Mueller DM, Pennathur S, Heinecke JW. J Biol Chem. 1997;272:3520. doi: 10.1074/jbc.272.6.3520. [DOI] [PubMed] [Google Scholar]
- 25.Yoritaka A, Hattori N, Uchida K, Tanaka M, Stadtman ER, Mizuno Y. Proc Natl Acad Sci U S A. 1996;93:2696. doi: 10.1073/pnas.93.7.2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ames BN. Mutation Res. 2001;475:7. doi: 10.1016/s0027-5107(01)00070-7. [DOI] [PubMed] [Google Scholar]
- 27.Beckman KB, Ames BN. Physiol Rev. 1998;78:547. doi: 10.1152/physrev.1998.78.2.547. [DOI] [PubMed] [Google Scholar]
- 28.Giasson BI, Duda JE, Murray IVJ, Chen Q, Souza JM, Hurtig HI, Ischiropoulos H, Trojanowski JQ, Lee VM. Science. 2000;290:985. doi: 10.1126/science.290.5493.985. [DOI] [PubMed] [Google Scholar]
- 29.Montine TJ, Huang DY, Valentine WM, Amarnath V, Saunders A, Weisgraber KH, Graham DG, Strittmatter WJ. J Neuropathol Exp Neurol. 1996;55:202. doi: 10.1097/00005072-199602000-00009. [DOI] [PubMed] [Google Scholar]
- 30.Cai H, Harrison DG. Circ Res. 2000;87:840. doi: 10.1161/01.res.87.10.840. [DOI] [PubMed] [Google Scholar]
- 31.Wu Z, Wagner MA, Zheng L, Parks JS, Shy JM, Smith JD, Gogonea V, Hazen SL. Nat Struct Mol Biol. 2007;14:861. doi: 10.1038/nsmb1284. [DOI] [PubMed] [Google Scholar]
- 32.Shao B, Oda MN, Oram JF, Heinecke JW. Chem Res Toxicol. 2010;23:447. doi: 10.1021/tx9003775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schaefer EJ, Foster DM, Jenkins LL, Lindgren FT, Berman M, Levy RI, Brewer HB. Lipids. 1979;14:511. doi: 10.1007/BF02533471. [DOI] [PubMed] [Google Scholar]
- 34.Karlsson H, Leanderson P, Tagesson C, Lindahl M. Proteomics. 2005;5:1431. doi: 10.1002/pmic.200401010. [DOI] [PubMed] [Google Scholar]
- 35.Bravo A, Bjørsvik H, Fontana F, Liguori L, Minisci F. J Org Chem. 1997;62:3849. [Google Scholar]
- 36.Zhang H, Joseph J, Feix J, Hogg N, Kalyanaraman B. Biochemistry. 2001;40:7675. doi: 10.1021/bi002958c. [DOI] [PubMed] [Google Scholar]
- 37.Tallman KA, Roschek B, Porter NA. J Am Chem Soc. 2004;126:9240. doi: 10.1021/ja049104q. [DOI] [PubMed] [Google Scholar]
- 38.Masuda T, Bando H, Maekawa T, Takeda Y, Yamaguchi H. Tet Lett. 2000;41:2157. [Google Scholar]
- 39.Masuda T, Maekawa T, Hidaka K, Bando H, Takeda Y, Yamaguchi H. J Agr Food Chem. 2001;49:2539. doi: 10.1021/jf001442x. [DOI] [PubMed] [Google Scholar]
- 40.Masuda T, Yamada K, Akiyama J, Someya T, Odaka Y, Takeda Y, Tori M, Nakashima K, Maekawa T, Sone Y. J Agr Food Chem. 2008;56:5947. doi: 10.1021/jf800781b. [DOI] [PubMed] [Google Scholar]
- 41.Masuda T, Yamada K, Maekawa T, Takeda Y, Yamaguchi H. Food Sci Tech Res. 2006;12:173. [Google Scholar]
- 42.Masuda T, Yamada K, Maekawa T, Takeda Y, Yamaguchi H. J Agr Food Chem. 2006;54:6069. doi: 10.1021/jf060676z. [DOI] [PubMed] [Google Scholar]
- 43.Wipf P, Kim Y. Tet Lett. 1992;33:5477. [Google Scholar]
- 44.Pelter A, Satchwell P, Ward RS, Blake K. J Chem Soc, Perkin Trans. 1995;1:2201. [Google Scholar]
- 45.Nara SJ, Valgimigli, Pedulli, Pratt DA. J Am Chem Soc. 2010;132:863. doi: 10.1021/ja907921w. [DOI] [PubMed] [Google Scholar]
- 46.Jha M, Pratt DA. Chem Commun. 2008;10:1252. doi: 10.1039/b800369f. [DOI] [PubMed] [Google Scholar]
- 47.Shao B, Heinecke JW. J Lipid Res. 2009;50:599. doi: 10.1194/jlr.E900001-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shao B, Tang C, Heinecke JW, Oram JF. J Lipid Res. 2010 doi: 10.1194/jlr.M004085. jlr.M004085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Chen X, Zhang W, Laird J, Hazen SL, Salomon RG. J Lipid Res. 2008;49:832. doi: 10.1194/jlr.M700598-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Peng D, Brubaker G, Wu Z, Zheng L, Willard B, Kinter M, Hazen SL, Smith JD. Arterioscl Throm Vas. 2008;28:2063. doi: 10.1161/ATVBAHA.108.173815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Melo E, Martins J. Biophys Chem. 2006;123:77. doi: 10.1016/j.bpc.2006.05.003. [DOI] [PubMed] [Google Scholar]
- 52.Perugini C, Bagnati M, Cau C, Bordone R, Paffoni P, Re R, Zoppis E, Albano E, Bellomo G. Pharmacol Res. 2000;41:67. doi: 10.1006/phrs.1999.0553. [DOI] [PubMed] [Google Scholar]
- 53.Szapacs ME, Kim H, Porter NA, Liebler DC. J Proteome Res. 2008;7:4237. doi: 10.1021/pr8001222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Sackmann E, Trauble H, Galla HJ, Overath P. Biochemistry. 1973;12:5360. doi: 10.1021/bi00750a020. [DOI] [PubMed] [Google Scholar]
- 55.Sonnen AFP, Bakirci H, Netscher T, Nau WM. J Am Chem Soc. 2005;127:15575. doi: 10.1021/ja054367l. [DOI] [PubMed] [Google Scholar]
- 56.Vanderkooi JM, Callis JB. Biochemistry. 1974;13:4000. doi: 10.1021/bi00716a028. [DOI] [PubMed] [Google Scholar]
- 57.Pali T, Horvath LI. Acta Physiol Hung. 1989;74:311. [PubMed] [Google Scholar]
- 58.Polymeropoulos MH, Lavedan C, Leroy E, Ide SE, Dehejia A, Dutra A, Pike B, Root H, Rubenstein J, Boyer R, Stenroos ES, Chandrasekharappa S, Athanassiadou A, Papapetropoulos T, Johnson WG, Lazzarini AM, Duvoisin RC, Di Iorio G, Golbe LI, Nussbaum RL. Science. 1997;276:2045. doi: 10.1126/science.276.5321.2045. [DOI] [PubMed] [Google Scholar]
- 59.Souza JM, Giasson BI, Chen Q, Lee VMY, Ischiropoulos H. J Biol Chem. 2000;275:18344. doi: 10.1074/jbc.M000206200. [DOI] [PubMed] [Google Scholar]
- 60.Benner EJ, Banerjee R, Reynolds AD, Sherman S, Pisarev VM, Tsiperson V, Nemachek C, Ciborowski P, Przedborski S, Mosley RL, Gendelman HE. PLoS One. 2008;3:e1376. doi: 10.1371/journal.pone.0001376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.AbdAlla S, Lother H, el Missiry A, Sergeev P, Langer A, el Faramawy Y, Quitterer U. J Biol Chem. 2009;284:6566. doi: 10.1074/jbc.M808277200. [DOI] [PubMed] [Google Scholar]
- 62.Ali FE, Leung A, Cherny RA, Mavros C, Barnham KJ, Separovic F, Barrow CJ. Free Rad Res. 2006;40:1. doi: 10.1080/10715760500329721. [DOI] [PubMed] [Google Scholar]
- 63.Danielson SR, Held JM, Schilling B, Oo M, Gibson BW, Andersen JK. Anal Chem. 2009;81:7823. doi: 10.1021/ac901176t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Good PF, Hsu A, Werner P, Perl DP, Olanow CW. J Neuropathol Exp Neurol. 1998;57:338. doi: 10.1097/00005072-199804000-00006. [DOI] [PubMed] [Google Scholar]
- 65.Pennathur S, Jackson-Lewis V, Przedborski S, Heinecke JW. J Biol Chem. 1999;274:34621. doi: 10.1074/jbc.274.49.34621. [DOI] [PubMed] [Google Scholar]
- 66.Schulz JB, Matthews RT, Muqit MM, Browne SE, Beal MF. J Neurochem. 1995;64:936. doi: 10.1046/j.1471-4159.1995.64020936.x. [DOI] [PubMed] [Google Scholar]
- 67.Anantharaman M, Tangpong J, Keller JN, Murphy MP, Markesbery WR, Kiningham KK, St Clair DK. Am J Pathol. 2006;168:1608. doi: 10.2353/ajpath.2006.051223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Butterfield DA, Reed TT, Perluigi M, De Marco C, Coccia R, Keller JN, Markesbery WR, Sultana R. Brain Res. 2007;1148:243. doi: 10.1016/j.brainres.2007.02.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Good PF, Werner P, Hsu A, Olanow CW, Perl DP. Am J Pathol. 1996;149:21. [PMC free article] [PubMed] [Google Scholar]
- 70.Hensley K, Maidt ML, Yu Z, Sang H, Markesbery WR, Floyd RA. J Neurosci. 1998;18:8126. doi: 10.1523/JNEUROSCI.18-20-08126.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Reynolds MR, Berry RW, Binder LI. Biochemistry. 2005;44:1690. doi: 10.1021/bi047982v. [DOI] [PubMed] [Google Scholar]
- 72.Reynolds MR, Lukas TJ, Berry RW, Binder LI. Biochemistry. 2006;45:4314. doi: 10.1021/bi052142h. [DOI] [PubMed] [Google Scholar]
- 73.Smith MA, Richey Harris PL, Sayre LM, Beckman JS, Perry G. J Neurosci. 1997;17:2653. doi: 10.1523/JNEUROSCI.17-08-02653.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tohgi H, Abe T, Yamazaki K, Murata T, Ishizaki E, Isobe C. Neurosci Lett. 1999;269:52. doi: 10.1016/s0304-3940(99)00406-1. [DOI] [PubMed] [Google Scholar]
- 75.Spiteller G. Free Rad Biol Med. 2006;41:362. doi: 10.1016/j.freeradbiomed.2006.03.013. [DOI] [PubMed] [Google Scholar]
- 76.Havrilla CM, Hachey DL, Porter NA. J Am Chem Soc. 2000;122:8042. [Google Scholar]
- 77.Dai P, Dussault P, Trulinger T. J Org Chem. 2004;69:2851. doi: 10.1021/jo035191d. [DOI] [PubMed] [Google Scholar]
- 78.Smith KC, White RL. J Nat Prod. 1995;58:1274. [Google Scholar]
- 79.Garrett CE, Jiang X, Prasad K, Repic O. Tet Lett. 2002;43:4161. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.