

Abstract
Nitric oxide (NO) production by mammalian NO synthase (NOS) is believed to be regulated by the docking of the FMN domain in one subunit of the dimer onto the heme domain of the adjacent subunit. Glu546, a conserved charged surface residue of the FMN domain in human inducible NOS (iNOS), is proposed to participate in the interdomain FMN/heme interactions [Sempombe et al. Inorg. Chem. 2011, 50, 6869–6861]. In the present work, we further investigated the role of the E546 residue in the FMN–heme interdomain electron transfer (IET), a catalytically essential step in the NOS enzymes. Laser flash photolysis was employed to directly measure the FMN–heme IET kinetics for the E546N mutant of human iNOS oxygenase/FMN (oxyFMN) construct. The temperature dependence of the IET kinetics was also measured over the temperature range of 283 – 304 K to determine changes in the IET activation parameters. The E546N mutation was found to retard the IET by significantly raising the activation entropic barrier. Moreover, pulsed electron paramagnetic resonance data showed that the geometry of the docked FMN/heme complex in the mutant is basically the same as in the wild type construct, whereas the probability of formation of such a complex is about twice lower. These results indicate that the retarded IET in the E546N mutant is not caused by an altered conformation of the docked FMN/heme complex, but by a lower population of the IET-active conformation. In addition, the negative activation entropy of the mutant is substantially lower than that of the holoenzyme. This supports a mechanism by which the FMN domain can modify the IET, through altering probability of the docked state formation.
Introduction
Nitric oxide synthases (NOSs) are the enzymes responsible for biosynthesis of nitric oxide (NO) in mammals. NOS catalysis is a two-step process: the substrate, l-arginine (l-Arg), is first converted to N-hydroxy-l-arginine (NOHA), which in turn is converted to NO and citrulline.1, 2 There are three NOS isoforms: endothelial, neuronal and inducible NOS (eNOS, nNOS and iNOS, respectively). Mammalian NOS is a homodimeric flavo-hemoprotein. Each subunit contains a C-terminal electron-supplying reductase domain with binding sites for NADPH (the electron source), FAD and FMN, and an N-terminal catalytic heme-containing oxygenase domain. The substrate, l-Arg, and cofactor 6R-5,6,7,8-tetrahydrobiopterin (H4B), bind near the heme center in the oxygenase domain.3 The oxygenase and reductase domains are connected by a calmodulin(CaM)-binding linker, occupation of which is required for the electron transfer from the reductase to the oxygenase domain. In nNOS and eNOS the CaM binding is Ca2+-dependent.4 In contrast, in the Ca2+-independent iNOS isoform, the linker is tightly bound to CaM. Although crystal structures are available for truncated NOS constructs,3, 5–9 the structures of the complete NOS holoenzyme still remain elusive.
The interdomain electron transfer (IET) process couples the reactions in the NOS domains, and thus represents a key step in NOS catalysis.1, 4, 10 Tight coupling is important for physiological function and prevention of disease, because partial or complete uncoupling in NOS results in formation of reactive oxygen species, such as superoxide and peroxynitrite.11–13 In particular, the CaM-controlled intersubunit FMN–heme IET is essential for coupling the electron transfer in the reductase domain with the NO synthesis in the heme domain by delivery of the electrons required for O2 activation at the catalytic heme site.14 It is thus of current interest to study mechanisms of the CaM-dependent FMN–heme IET in the NOS isoforms.10, 15–17 A new laser flash photolysis approach18 has been developed which allows direct determination of the FMN–heme IET rate between catalytically significant redox couples of the two centers (see a recent review for details17). Kinetics results for NOS proteins indicate that the FMN–heme IET is gated by a large conformational change (moving the FMN and Fe centers into close proximity, which results in efficient FMN–heme IET; Figure 1).19, 20 The interdomain FMN/heme docking is proposed to be guided by electrostatic interactions between the charged surface residues in the two domains (red and blue regions in Figure 1 represent negatively and positively charged regions in each domain).21, 22 It is important and timely to study the role of the FMN domain motion in the NOS isoform function.23–25 Bi-domain NOS oxyFMN constructs contain the FMN and heme domains, as well as a CaM binding region, but lack the NADPH and FAD binding domains. The truncated oxyFMN construct is a minimal electron transfer complex, and is designed to favor the interactions between the FMN and heme domains.26 The oxyFMN construct has been expressed and purified as a homogeneous dimeric protein with the activity comparable to corresponding oxygenase constructs.26 This construct is a valid model of the electron-donating (output) state of the FMN domain.18, 26–28
Figure 1.
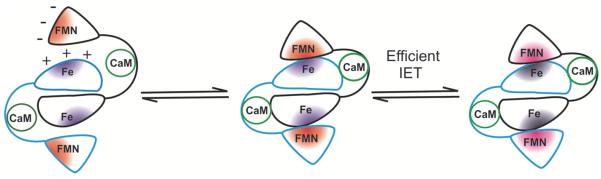
Proposed mechanism of the intersubunit IET between the FMN and heme domains in iNOS. A large conformational change of the FMN domain moves the FMN and Fe centers into close proximity, which results in efficient FMN–heme IET in the electron-donating (output) state of the FMN domain. The interdomain docking is proposed to be guided by electrostatic interactions between the charged surface residues (red and blue regions) in the FMN and heme domains. The human iNOS Glu546 residue at the FMN domain surface is proposed to participate in the interdomain FMN/heme interactions. Emerging evidence indicates that a series of free FMN domain conformations are involved in the conformational equilibrium. The two NOS subunits are colored in black and blue. CaM protein (green) binds to iNOS tightly. The NADPH and FAD binding domains are omitted for clarity. Adapted from ref. 19.
Glu546, located at the edge of the FMN domain surface of human iNOS (Figure 2), is a conserved charged residue, and is proposed to participate in the interdomain FMN/heme interactions.29, 30 Extensive studies were also conducted on the analogous residue in nNOS.21, 31 These two earlier papers showed that neutralization at the equivalent residue of rat nNOS (E762) induced the largest effect on heme reduction (decreasing the heme reduction rate by about 60 %),21 and the slower electron transfer may be caused by a more positive FMN sq/hq midpoint potential.31 Their findings were used primarily to understand how the mutational effect increased the activity of the nNOS enzyme. However, the underlying mechanism for the slower electron transfer in the nNOS E762N mutant remained unclear. In the present study we investigated the mechanistic role of the equivalent human iNOS E546 residue in modulating the FMN–heme IET rate, which in turn may provide insights about the interdomain FMN/heme alignment. Moreover, because the distance between redox centers is a key factor in controlling the electron transfer in proteins,32 pulsed electron paramagnetic resonance (EPR) was employed to determine the FMN…Fe distance in the mutant. To investigate if the mutation at E546 affects the structural properties of the heme center, resonance Raman (rR) spectra of ferric and ferrous-CO derivatives of wild type (wt) and E546N mutant proteins were also obtained.
Figure 2.
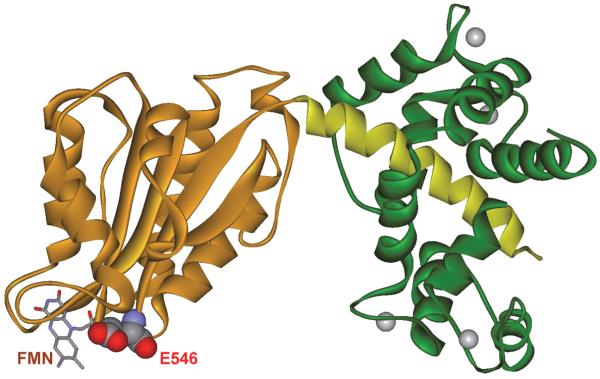
Structure of a CaM bound human iNOS FMN domain (PDB 3HR4). The iNOS FMN domain is gold, and the CaM-binding linker is yellow. The conserved E546 residue (shown in CPK) is at the edge of the FMN domain. The CaM protein is green, and the bound Ca2+ ions are grey spheres. The missing NOS heme domain in the same monomer is at the other end of the CaM-binding linker. The heme domain of the other monomer interacts with the FMN-binding domain for efficient intersubunit FMN–heme IET.
Experimental Details
Preparation of Wild Type and E546N Mutant Proteins of Human iNOS oxyFMN
The iNOS oxyFMN plasmid was co-transfected with a CaM expression vector (p209) into E. coli BL21 (DE3) cells by electroporation (MicroPulser, Bio-Rad). The iNOS protein must be co-expressed with CaM in vitro because of its tendency to aggregate when residues of the highly hydrophobic CaM-binding region are exposed to an aqueous environment. Wild type and E546N human iNOS oxyFMN constructs were expressed and purified as previously described.30 Fractions with A280/A400 < 2.2 were pooled and concentrated. The pooled protein was then dialyzed into the storage buffer (40 mM Tris-Cl, 200 mM NaCl, 10 % glycerol, 1 mM DTT, 4 μM H4B, pH 7.6); concentrated protein (~ 200 μM) was aliquoted and stored at −80 °C. The protein molar concentration was determined based on the heme content via difference spectra of the NOS ferrous-CO adduct.33
Laser Flash Photolysis
CO photolysis experiments were conducted on an Edinburgh LP920 laser flash photolysis spectrometer, in combination with a Q-switched Continuum Surelite I-10 Nd:YAG laser and a Continuum Surelite OPO. A 446 nm laser pulse (out of the OPO module) was focused onto a sample cell to trigger the IET reactions. A 50 W halogen lamp was used as the light source for measuring the kinetics on the ms – s time scales. A LVF-HL filter (Ocean Optics, FL) with passband peaking at 465 nm was placed before the partially reduced protein sample to protect it from photo-bleaching and further photo-reduction by the white monitor beam.17 The sample temperature was controlled by using a TLC 50 cuvette holder coupled with a TC 125 temperature-controller (Quantum Northwest, WA). Dry nitrogen was passed over the cuvette surface to avoid moisture buildup at lower temperature.
Briefly, a typical experiment was set up as follows. A solution of ~ 350 μL containing 20 μM 5-deazariboflavin (dRF) and 5 mM fresh semicarbazide in pH 7.6 buffer (40 mM Bis-Tris propane, 400 mM NaCl, 2 mM l-Arg, 20 μM H4B, 1 mM Ca2+ and 10 % glycerol) was degassed in a laser photolysis cuvette by a mixture of CO and Ar (with a volume ratio of ~ 1:3) for 90 min. L-Arg was present to keep the oxidized heme in the catalytically relevant high spin state.1 Aliquots of the concentrated NOS protein were subsequently injected through a septum to achieve the desired concentration, and the protein solution was kept in ice and further purged by passing the CO/Ar mixture over the solution surface for 60 min (to remove minor oxygen contamination before being subjected to illumination). The protein solution was then illuminated for an appropriate period to obtain a partially reduced form of NOS [Fe(II)–CO][FMNH•]. The sample was subsequently flashed with a 446 nm laser excitation to trigger the FMN–heme IET, which was followed by monitoring the loss of absorbance of Fe(II) at 465 nm.34 All experiments were conducted at least twice. Transient absorbance changes were averaged and analyzed using OriginPro 9.0 (OriginLab Corporation, MA).
Pulsed EPR Spectroscopy
The [Fe(III)][FMNH•] form of the E546N mutant was prepared as previously described.15 The pulsed EPR experiments were performed on a homebuilt Ka-band (26–40 GHz) pulsed EPR spectrometer35 at the microwave frequency of 29.454 GHz. The refocused stimulated electron spin echo (ESE) pulse sequence used for relaxation-induced dipolar modulation enhancement (RIDME) measurements was previously described.15 The measurement temperatures were 15 K and 25 K.
Fluorescence Spectra of NOS
The NOS flavin fluorescence spectra were measured with a Cary Eclipse Fluorescence Spectrophotometer (Agilent Technologies) at room temperature. 1 – 7 μM of the iNOS protein in degassed buffer (40 mM Bis-Tris propane, 400 mM NaCl, 2 mM l-Arg, 10 % glycerol, pH 7.6) was filtered using a 0.2 μm membrane filter. All samples were excited at 446 nm, and the fluorescence spectra were recorded from 480 to 700 nm. The flavin fluorescence intensities at 525 nm were plotted versus protein concentrations. Linear regression analysis of the data yielded the NOS flavin fluorescence per μM as the slope of the line for each sample.
Resonance Raman Spectra of NOS
Resonance Raman spectra of the wt and E546N mutant of human iNOS oxyFMN proteins were collected at room temperature using procedures described in the literature.36
Results
Kinetics of Electron Transfer between the Heme and FMN Domains in Partially Reduced iNOS oxyFMN Proteins
The primary technique used to determine the IET kinetics in this work is a laser flash photolysis approach34 in which the iNOS heme and FMN centers are first photo-reduced to the [Fe(II)–CO][FMNH•] form in the presence of dRF and CO. A 446 nm laser pulse then serves to dissociate the pre-bound CO from the reduced heme, which triggers an IET from the free Fe(II) to the FMNH•, followed by a much slower phase in which CO re-binds the ferrous heme.18, 34, 37
As expected, CO dissociation by laser photolysis of the partially reduced [Fe(II)–CO][FMNH•] form results in a spectral “transition” (i.e., a change in direction of absorption changes over time) in the 465 nm traces of the human iNOS oxyFMN E546N mutant. Figure 3 shows the typical absorbance changes at 465 nm that follow irradiation of the E546N mutant with a 446 nm laser pulse at 21 °C. The initial rapid positive deflection in A465 tracks the laser-initiated dissociation of CO. This is followed by a rapid decay (with a rate constant k1 of 139 ± 3 s−1) to below the pre-flash baseline of A465, which is attributable to the IET process shown in eq 1. On a much longer timescale (k2 = 1.30 ± 0.03 s−1), A465 returns to the baseline as CO re-binds to the heme Fe(II) (Figure S1 of Supporting Information).
| eq 1 |
In eq 1, FMNhq stands for the FMN hydroquinone species generated by the IET reaction. As expected for an intramolecular process, the value of k1 shows no dependence on concentration of the reduced protein, which can be estimated from the value of the associated ΔA465 (data not shown).
Figure 3.
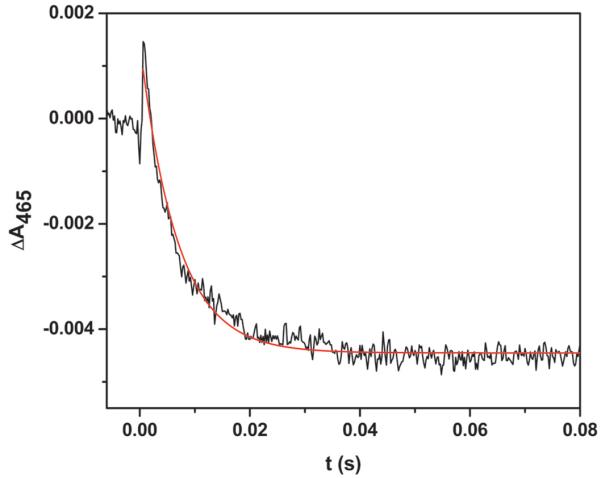
Transient trace at 465 nm from 0 to 0.1 s obtained for the [Fe(II)-CO][FMNH•] form of the human iNOS oxyFMN E546N mutant flashed by a 446 nm laser excitation. The sample temperature was set at 21 °C. The anaerobic solution contained ~ 10 μM NOS protein, ~ 20 μM dRF and 5 mM fresh semicarbazide in pH 7.6 buffer (40 mM Bis-Tris propane, 400 mM NaCl, 2 mM l-Arg, 20 μM H4B, 1 mM Ca2+ and 10 % glycerol). The solid line corresponds to the best single-exponential fit to the data.
The FMN–heme IET Kinetics as a Function of Temperature
To determine the activation parameters of the FMN–heme IET process, the kinetic parameters of the E546N mutant were determined over the temperature range of 283–304 K, and compared to those previously obtained for the wild type (wt) oxyFMN construct.20 The numerical values are tabulated in Table S1 of Supporting Information. An appreciable increase in the IET rate constant was observed concomitant with an increase in the temperature. Importantly, the obtained rate constant of the rapid decay (Figure 3) is independent of the ΔA465 signal amplitude in the studied temperature range (data not shown), again confirming an intra-protein process.
Our previous viscosity study indicated that the NOS FMN–heme IET process is conformationally gated.19 The temperature dependence of the IET rate constant ket was thus analyzed by transition state theory using the Eyring equation,
| eq 2 |
where ΔH‡ is the activation enthalpy, ΔS‡ is the activation entropy, h is the Planck's constant, kB is the Boltzmann constant, and R is the gas constant. Temperature dependence profiles of gated electron transfer processes in other proteins have been analyzed similarly.38, 39 The fit to the E546N data using eq 2 led to ΔH‡ = 45.7 ± 3.3 kJ/mol and ΔS‡ = −47.0 ± 3.4 J/mol/K (Figure 4); the parameters are listed in Table 1, along with those of other iNOS proteins. The obtained activation values are comparable to those of inter-flavin electron transfer in human cytochrome P450 reductase,39 which is homologous to the NOS reductase domain. For the wt oxyFMN construct, the activation enthalpy value (52.5 ± 3.3 kJ/mol) is comparable with that of E546N, while the activation entropy value (−17.1 ± 1.2 J/mol/K) is only about one-third of that of the E546N mutant (Table 1).20 This indicates that the E546N mutation retards the IET process by raising the activation entropic barrier (by a factor of three).
Figure 4.
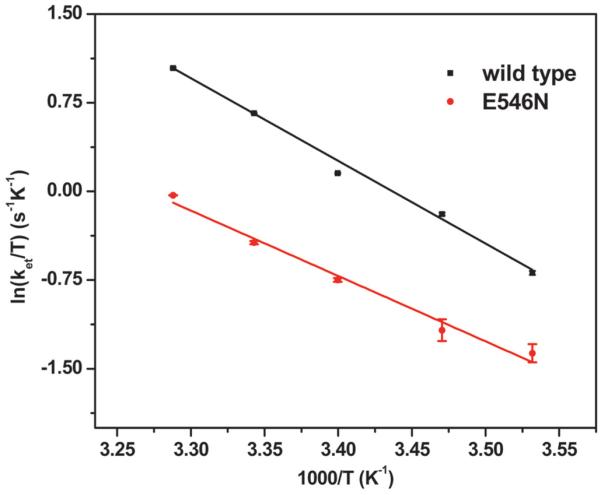
Eyring plots showing the temperature dependence of ket measured for wild type (square) and E546N (circle) human iNOS oxyFMN constructs. The obtained ΔH‡ and ΔS‡ values are listed in Table 1.
Table 1.
Eyring parameters from temperature dependence analysis of observed rate constant for the FMN–heme IET in wild type and E546N human iNOS oxyFMN proteins, along with those of wild type iNOS holoenzyme.
| ΔH‡ (kJ/mol) | ΔS‡ (J/mol/K) | |
|---|---|---|
| wild type human iNOS oxyFMN a | 52.6 ± 3.7 | −17.1 ± 1.2 |
| E546N human iNOS oxyFMN | 45.7 ± 3.3 | −47.0 ± 3.4 |
| wild type human iNOS holoenzyme a | 36.9 ± 1.6 | −89.7 ± 4.5 |
Data taken from ref. 20.
Pulsed EPR Studies of the E546N Mutant
The RIDME technique40 was used to determine the distance between the FMN and heme centers in the [Fe(III)][FMNH•] form of the E546N mutant. These experiments were identical to those previously used by us to study the wt oxyFMN construct.15 The solid line in Figure 5 shows the spectrum of the magnetic dipolar interactions between FMNH• and the low spin ferric heme center in the mutant. This spectrum is very similar to that obtained for the wt iNOS oxyFMN15 (dashed line in Figure 5) in that it shows a prominent line at the frequency of 8.3 MHz. The amplitude of this line in the spectrum of E546N iNOS is, however, only about half that observed for the wt protein, which is a significant observation because both spectra were normalized by the corresponding ESE signal amplitudes.
Figure 5.

Cosine Fourier transformation of the dipolar electron spin echo envelope modulation (ESEEM) spectrum shown in quotient RIDME trace of human iNOS E546N oxyFMN construct. Experimental conditions were the same as those used for the wt oxyFMN construct.15 The total protein concentration was 312 μM, and 42% ethylene glycol was added as a glassing agent; buffer: 100 mM bis-Tris-propane, 200 mM NaCl, 3 mM imidazole, pH 7.6.
The position of the maximum in the RIDME spectrum of FMNH• (Figure 5) is largely determined by the distance between Fe(III) and FMNH•. In addition, because of the relatively strong g-anisotropy of the ferric heme center, the maximum position depends on the orientation of the Fe(III) – FMNH• radius vector, R, with respect to the heme g-frame.15 It is therefore theoretically possible that the same maximum position in the RIDME spectra of E546N and wt iNOS could result from somewhat different geometries of the FMN domain docking, with the spectral changes due to the altered distance being somehow compensated for by the altered orientation of R. However, we consider a simultaneous change in the two geometrical parameters resulting in zero total spectroscopic effect to be quite unlikely, especially taking into account that only one surface residue was mutated, while the specific docking geometry is probably determined by significantly more than a single residue (see Discussion). We conclude therefore that the docking geometry of the FMN domain in E546N iNOS is basically the same as in wt iNOS, with the Fe(III)⋯FMNH• distance of about 19 Å.15
Since both the wt and E546N iNOS samples were prepared using identical procedures, the difference in normalized amplitudes of the RIDME spectra (Figure 5) should be caused by different probabilities of forming a well-defined Fe(III) – FMNH• structural pair, or in other words, different docking probabilities. Thus, the RIDME spectroscopic results suggest that the slower IET rate in the mutant iNOS is most likely not caused by a disturbed geometrical alignment of the FMN and heme domains in the docked complex, but rather by a lower probability of forming such a complex (see Discussion).
Fluorescence Spectra of the NOS Flavin Cofactor
Flavins bound to a protein can exhibit fluorescence that is both specific and characteristic of the particular protein. The characteristic fluorescence maximum of the NOS flavin at 525 nm is a sensitive probe for the local environment of the FMN cofactor (including solvent exposure)41, 42 and proximity to the heme (due to paramagnetic quenching).37 To examine the FMN domain conformation, we measured the fluorescence spectra of the wt and E546N mutant of human iNOS oxyFMN proteins under identical conditions, i.e., same protein concentration, buffer, and fluorometer settings (Figure S2 in Supporting Information). Additionally, the fluorescence intensities (per μM) of the oxidized proteins were determined (inset of Figure S2). The E546N mutant exhibits similar fluorescence per μM as that of the wt protein.
Resonance Raman Spectra of the Heme Center
Resonance Raman spectra of the substrate-free, l-Arg-bound and NOHA-bound complexes of the ferric and ferrous CO-derivatives of the E546N mutant, with respect to those of the wt oxyFMN construct and oxygenase domain, are shown in Figures S3 and S4 in Supporting Information. The data show that the wt oxyFMN construct exhibits spectral features similar to those of the oxygenase domain alone. In addition, the E546N mutation basically does not perturb the spectra. These results indicate that the docking of the FMN domain to the heme domain does not notably affect electronic and structural properties of the ground state of the heme active center. In addition, the data support the concept above that the mutation does not affect the docked structure, but only the population of the active conformation. It is noteworthy that the rR spectra of the oxyFMN construct are also similar to those the NOS holoenzyme obtained under similar conditions,43 confirming proper folding of the protein samples examined.
Discussion
The FMN domain motion is proposed to be important for efficient intersubunit FMN#x2013;heme IET (Figure 1), a catalytically essential step during NOS catalysis.19,20 E546 is a conserved charged surface residue located at the edge of the human iNOS FMN domain (Figure 2), and is not in close contact with either heme center of the dimeric NOS protein, based on a computationally docked model.29 The E546 residue has been recently proposed to participate in the iNOS interdomain FMN/heme interactions.30 In the present work we directly investigated the mechanistic role of the equivalent iNOS residue in the key FMN#x2013;heme IET process by examining the kinetics of the discrete step and spectroscopic characteristics of IET-relevant NOS forms. Specifically, we compared the FMN–heme IET kinetics and pulsed EPR properties of the wild type and E546N human iNOS oxyFMN constructs, which provided a detailed probe of the role that E546 has in formation of the interdomain FMN/heme complex.
The FMN#x2013;heme IET rate constant of the E546N mutant at 21 °C is decreased to one third of the rate in the wt construct (139 s−1 in the mutant versus 343 s−1 in the wt20, 34). Moreover, the activation entropy of E546N is about three times that of the wt construct (Table 1). Importantly, pulsed EPR data (Figure 5) showed that the geometry of the docked FMN/heme complex in the mutant is basically the same as in the wt construct, whereas the probability of formation of such a complex is about twice lower. These results together strongly indicate that the retarded IET in the E546N mutant is not directly caused by a disturbed conformation of the docked FMN/heme complex, but by a smaller probability of formation of such a complex. The larger negative activation entropy of the E546N mutant is as a result of the FMN domain must search for the optimum IET-active conformation among a larger number of conformations that are not IET-competent (due to less optimized guiding forces) than in the wt protein. The weaker interaction between the docked domains will probably also result in shorter lifetime of the docked complex. The increase of the entropy of the undocked state is concomitant with the decrease in the entropy of the docked state (since the lower probability of formation is equivalent to a smaller number of microscopic states), resulting in the observed increase in the activation entropy.
Next we compare the IET data for the oxyFMN constructs with those obtained for the holoenzyme. The negative entropy of activation is significantly increased in the mutant compared to the wild type oxyFMN construct, but is still substantially lower than that of the holoenzyme (Table 1). During NOS catalysis, the FMN domain cycles between the interaction with the NADPH/FAD domain (to receive electrons) and the interaction with the heme-containing oxygenase domain (to deliver electrons to the heme).44 It has been shown that the NADPH/FAD domain has a much greater capability to interact with the FMN domain than does the oxygenase domain.28 In the NOS holoenzyme, even in the presence of bound CaM, its dominant conformational state is thus in the docked FAD/FMN state (because of the stronger intra-domain FAD/FMN interactions),45 which must undergo a transformation to an IET-active docked FMN/heme conformation before the FMN#x2013;heme IET takes place. On the other hand, the oxyFMN construct does not contain the NADPH/FAD domain, and is thus better poised for the FMN#x2013;heme IET (while in the E546N oxyFMN mutant, the docking process is perturbed). The substantially larger negative activation entropy of the holoenzyme is due to the fact that the FMN domain in the holoenzyme must “test” more conformations than in the oxyFMN constructs prior to the IET. The entropy of the “free” state of the holoenzyme should be larger because there are a larger number of undocked microscopic states (due to the larger size of the holoenzyme).
Interestingly, the order of the IET rate constant (wt oxyFMN > E546N oxyFMN > holoenzyme) is in agreement with the order of the magnitude of the activation entropy. This can be explained by a conformational sampling model, in which sampling of a continuum of conformational states gives a range of transient donor-acceptor complexes, only a subset of which are IET-competent.24, 28, 46 This presents an important mechanism for regulation in NOS enzyme, which avoids the necessity for tight binding of the FMN domain to achieve efficient IET. The IET complexes must form, pass electrons, and then split apart. Interdomain FMN/heme interactions in the NOS enzymes are rather weak, and the docked IET complexes are short-lived.19, 28 The observed rate of heme reduction for a population of NOS conformations is thus limited by the relatively infrequent formation of the docked IET-competent complexes. More conformations to sample (as in the holoenzyme and E546N mutant) give higher magnitude of activation entropy, resulting in less frequent formation of the FMN/heme complex and slower subsequent IET. This supports a mechanism by which the FMN domain can modify the IET through altering probability of the docked state formation. Interestingly, Hoffman and coworkers have presented elegant studies of electron transfer (ET) in weakly bound protein-protein complexes that have multiple conformations, only a limited subset of which are ET-active.47, 48
The fact that the IET in the E546N mutant is not completely suppressed suggests that other residues may also participate in facilitating the interdomain FMN/heme alignment; also note that the mutation is charge neutralization, not charge reversal. What might be specific in the NOS FMN-heme interactions is complementary electrostatic surfaces but not specific ion pairing. Indeed, recent work on rat nNOS mutants showed that heme reduction and NO synthesis in nNOS is enabled by electrostatic interactions involving Lys423, Lys620, and Lys660, which form a triad of positive charges on the NOS heme domain surface.22
The flavin fluorescence intensity of the E546N mutant is not dramatically different from that of the wild type protein under comparable conditions. However, this is not necessarily unexpected. The shielding residues on either side of FMN are apparently not perturbed in the mutant and the simple swinging of the FMN domain to contact the heme domain does not necessarily perturb the quenched flavin fluorescence in this molecule. In addition, the FMN#x2013;heme IET rate constant for the E546N mutant is 139 s−1; by using the Dutton's Ruler49 with no driving force and a 0.7 eV reorganization energy, the calculated edge-to-edge distance is estimated as ~ 18 Å. This estimate is in good agreement with the direct measurement by pulsed EPR above. At such a distance, it is too far away to quench (as the Förster quenching is proportional to 1/R6).
The present work was conducted on a charge neutralization mutant. It will be interesting to look at a charge reversal mutant at this site. We are also aware that the truncated bi-domain oxyFMN construct is a model of the NOS output state for NO production.26 It is necessary to further examine the full-length enzyme mutants so that the effects of the amino acid replacements on the NOS enzyme activity and the IET kinetics can be assessed. It would be also interesting to test if the mutation might alter the thermodynamics of FMN involved in the IET, and how this could change the temperature dependence profile. Importantly, recent mutational and kinetics studies on an equivalent residue (E762) in full-length rat nNOS clearly showed that this residue is critical for nNOS NO production activity.21, 31 Their results strongly support regulatory roles of the conserved human iNOS E546 residue in NOS enzyme function.
The similar rR spectroscopic properties of the wt and E546N proteins are rather surprising. Our previous data showed notable substrate-specific effects of the mutation in the FMN domain on the magnetic circular dichroism (MCD) spectra of iNOS ferric centers in an adjacent domain.11 The apparent discrepancy between the MCD and rR results is due to the fact that the two techniques detect different structural variations at the NOS heme site: MCD probes the electronic transition from the ground to excited states, while resonance Raman provides information about the vibrational modes of the ground state. Interestingly, a recent kinetics study showed that a similar mutation at the equivalent FMN domain site of nNOS notably increases activity by enhancing ferrous heme-NO oxidation reactivity.31 It is still unclear how the mutation in the FMN domain affects MCD spectroscopic properties and reactivity of the heme site in an adjacent domain. The mutation may lead to various changes (e.g., docking pathways, conformational equilibrium etc.). At the current stage, it is difficult to envisage how the local structural change (induced by the FMN domain mutation) may propagate down to the heme site. We emphasize, however, that the resonance Raman data not only probe the structure of the heme site, but the Raman data are also useful and significant in terms of directing future studies to address how the FMN domain may influence the NOS function.
Conclusions
The primary goal of this work was to determine the mechanism responsible for the slower FMN#x2013;heme IET of the E546N human iNOS mutant. The IET kinetics and pulsed EPR data showed conclusively that the E546N mutation retards the IET process by significantly raising the activation entropic barrier, without notably disturbing the FMN/heme alignment in the docked complex. This provides an important conformational sampling mechanism by which the FMN domain can modify the FMN#x2013;heme IET, through altering the probability of the docked state.
Supplementary Material
Acknowledgement
This work was supported by grants from the National Institutes of Health (GM081811 to C.F.), the National Science Foundation (CHE-1150644 to C.F., NSF1026788 to D.L.R. and NSF0956358 to S.-R.Y.) and AHA Grant-in-Aid (12GRNT11780019 to C.F.). This project was also supported by grants from the National Center for Research Resources (5P20RR016480-12) and the National Institute of General Medical Sciences (8 P20 GM103451-12).
Abbreviations
- NOS
nitric oxide synthase
- iNOS
inducible NOS
- nNOS
neuronal NOS
- CaM
calmodulin
- oxyFMN
bi-domain NOS construct in which only oxygenase and FMN domains along with CaM-binding region are present
- IET
intramolecular interdomain electron transfer
- dRF
5-deazariboflavin
- H4B
6R-5,6,7,8-tetrahydrobiopterin
- ket
observed rate constant of the FMN–heme IET process
- EPR
electron paramagnetic resonance
- RIDME
relaxation-induced dipolar modulation enhancement
- ESE
electron spin echo (ESE)
- rR
resonance Raman
- MCD
magnetic circular dichroism
- wt
wild type
- l-Arg
l-arginine
- NOHA
N-hydroxy-l-arginine
Footnotes
Author Contributions The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Supporting Information. Transient trace at 465 nm from 0 to 4 s, flavin fluorescence spectra, and resonance Raman spectra of iNOS heme domain, wild type and E546N mutant of human iNOS oxyFMN proteins, along with table of obtained IET rate constant over temperature range of 283304 K.
REFERENCES
- 1.Alderton WK, Cooper CE, Knowles RG. Biochem. J. 2001;357:593–615. doi: 10.1042/0264-6021:3570593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhu Y, Silverman RB. Biochemistry. 2008;47:2231–2243. doi: 10.1021/bi7023817. [DOI] [PubMed] [Google Scholar]
- 3.Crane BR, Arvai AS, Ghosh DK, Wu CQ, Getzoff ED, Stuehr DJ, Tainer JA. Science. 1998;279:2121–2126. doi: 10.1126/science.279.5359.2121. [DOI] [PubMed] [Google Scholar]
- 4.Roman LJ, Martasek P, Masters BSS. Chem. Rev. 2002;102:1179–1189. doi: 10.1021/cr000661e. [DOI] [PubMed] [Google Scholar]
- 5.Raman CS, Li HY, Martasek P, Kral V, Masters BSS, Poulos TL. Cell. 1998;95:939–950. doi: 10.1016/s0092-8674(00)81718-3. [DOI] [PubMed] [Google Scholar]
- 6.Li HY, Shimizu H, Flinspach M, Jamal J, Yang WP, Xian M, Cai TW, Wen EZ, Jia QA, Wang PG, Poulos TL. Biochemistry. 2002;41:13868–13875. doi: 10.1021/bi020417c. [DOI] [PubMed] [Google Scholar]
- 7.Zhang J, Martasek P, Paschke R, Shea T, Masters BSS, Kim JJP. J. Biol. Chem. 2001;276:37506–37513. doi: 10.1074/jbc.M105503200. [DOI] [PubMed] [Google Scholar]
- 8.Garcin ED, Bruns CM, Lloyd SJ, Hosfield DJ, Tiso M, Gachhui R, Stuehr DJ, Tainer JA, Getzoff ED. J. Biol. Chem. 2004;279:37918–37927. doi: 10.1074/jbc.M406204200. [DOI] [PubMed] [Google Scholar]
- 9.Xia C, Misra I, Iyanagi T, Kim J-JP. J. Biol. Chem. 2009;284:30708–30717. doi: 10.1074/jbc.M109.031682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Feng CJ, Tollin G. Dalton Trans. 2009:6692–6700. doi: 10.1039/b902884f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xia Y, Roman LJ, Masters BSS, Zweier JL. Circulation. 1998;98:387–388. [Google Scholar]
- 12.Vasquez-Vivar J, Kalyanaraman B, Martasek P, Hogg N, Masters BSS, Karoui H, Tordo P, Pritchard KA. Proc Natl Acad Sci U S A. 1998;95:9220–9225. doi: 10.1073/pnas.95.16.9220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen CA, Druhan LJ, Varadharaj S, Chen YR, Zweier JL. J. Biol. Chem. 2008;283:27038–27047. doi: 10.1074/jbc.M802269200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Panda K, Ghosh S, Stuehr DJ. J. Biol. Chem. 2001;276:23349–23356. doi: 10.1074/jbc.M100687200. [DOI] [PubMed] [Google Scholar]
- 15.Astashkin AV, Elmore BO, Fan W, Guillemette JG, Feng C. J. Am. Chem. Soc. 2010;132:12059–12067. doi: 10.1021/ja104461p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stuehr DJ, Tejero J, Haque MM. FEBS J. 2009;276:3959–3974. doi: 10.1111/j.1742-4658.2009.07120.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Feng C. Coord. Chem. Rev. 2012;256:393–411. doi: 10.1016/j.ccr.2011.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Feng CJ, Thomas C, Holliday MA, Tollin G, Salerno JC, Ghosh DK, Enemark JH. J. Am. Chem. Soc. 2006;128:3808–3811. doi: 10.1021/ja0578606. [DOI] [PubMed] [Google Scholar]
- 19.Li W, Fan W, Elmore BO, Feng C. FEBS Lett. 2011;585:2622–2626. doi: 10.1016/j.febslet.2011.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li W, Chen L, Fan W, Feng C. FEBS Lett. 2012;586:159–162. doi: 10.1016/j.febslet.2011.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Panda K, Haque MM, Garcin-Hosfield ED, Durra D, Getzoff ED, Stuehr DJ. J. Biol. Chem. 2006;281:36819–36827. doi: 10.1074/jbc.M606129200. [DOI] [PubMed] [Google Scholar]
- 22.Tejero J, Hannibal L, Mustovich A, Stuehr DJ. J. Biol. Chem. 2010;285:27232–27240. doi: 10.1074/jbc.M110.138842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Haque MM, Bayachou M, Fadlalla MA, Durra D, Stuehr DJ. Biochem. J. 2013;450:607–617. doi: 10.1042/BJ20121488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Salerno JC, Ray K, Poulos T, Li H, Ghosh DK. FEBS Lett. 2013;587:44–47. doi: 10.1016/j.febslet.2012.10.039. [DOI] [PubMed] [Google Scholar]
- 25.Persechini A, Tran Q-K, Black DJ, Gogol EP. FEBS Lett. 2013;587:297–301. doi: 10.1016/j.febslet.2012.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ghosh DK, Holliday MA, Thomas C, Weinberg JB, Smith SME, Salerno JC. J. Biol. Chem. 2006;281:14173–14183. doi: 10.1074/jbc.M509937200. [DOI] [PubMed] [Google Scholar]
- 27.Feng CJ, Tollin G, Holliday MA, Thomas C, Salerno JC, Enemark JH, Ghosh DK. Biochemistry. 2006;45:6354–6362. doi: 10.1021/bi060223n. [DOI] [PubMed] [Google Scholar]
- 28.Ilagan RP, Tejero JS, Aulak KS, Ray SS, Hemann C, Wang Z-Q, Gangoda M, Zweier JL, Stuehr DJ. Biochemistry. 2009;48:3864–3876. doi: 10.1021/bi8021087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sempombe J, Galinato MGI, Elmore BO, Fan W, Guillemette JG, Lehnert N, Kirk ML, Feng C. Inorg. Chem. 2011;50:6859–6861. doi: 10.1021/ic200952c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sempombe J, Elmore BO, Sun X, Dupont A, Ghosh DK, Guillemette JG, Kirk ML, Feng C. J. Am. Chem. Soc. 2009;131:6940–6941. doi: 10.1021/ja902141v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Haque MM, Fadlalla M, Wang ZQ, Ray SS, Panda K, Stuehr DJ. J. Biol. Chem. 2009;284:19237–19247. doi: 10.1074/jbc.M109.013144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Page CC, Moser CC, Chen XX, Dutton PL. Nature. 1999;402:47–52. doi: 10.1038/46972. [DOI] [PubMed] [Google Scholar]
- 33.McMillan K, Masters BSS. Biochemistry. 1993;32:9875–9880. doi: 10.1021/bi00089a001. [DOI] [PubMed] [Google Scholar]
- 34.Feng CJ, Dupont A, Nahm N, Spratt D, Hazzard JT, Weinberg J, Guillemette J, Tollin G, Ghosh DK. J. Biol. Inorg. Chem. 2009;14:133–142. doi: 10.1007/s00775-008-0431-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Astashkin AV, Enemark JH, Raitsimring A. Concepts in Magnetic Resonance Part B-Magnetic Resonance Engineering. 2006;29B:125–136. [Google Scholar]
- 36.Li D, Stuehr DJ, Yeh SR, Rousseau DL. J. Biol. Chem. 2004;279:26489–26499. doi: 10.1074/jbc.M400968200. [DOI] [PubMed] [Google Scholar]
- 37.Li W, Fan W, Chen L, Elmore B, Piazza M, Guillemette J, Feng C. J. Biol. Inorg. Chem. 2012;17:675–685. doi: 10.1007/s00775-012-0887-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Engstrom G, Xiao K, Yu C-A, Yu L, Durham B, Millett F. J. Biol. Chem. 2002;277:31072–31078. doi: 10.1074/jbc.M202594200. [DOI] [PubMed] [Google Scholar]
- 39.Hay S, Brenner S, Khara B, Quinn AM, Rigby SEJ, Scrutton NS. J. Am. Chem. Soc. 2010;132:9738–9745. doi: 10.1021/ja1016206. [DOI] [PubMed] [Google Scholar]
- 40.Kulik LV, Dzuba SA, Grigoryev IA, Tsvetkov YD. Chem. Phys. Lett. 2001;343:315–324. [Google Scholar]
- 41.Jachymova M, Martasek P, Panda S, Roman LJ, Panda M, Shea TM, Ishimura Y, Kim JJP, Masters BSS. Proc Natl Acad Sci U S A. 2005;102:15833–15838. doi: 10.1073/pnas.0506522102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ilagan RP, Tiso M, Konas DW, Hemann C, Durra D, Hille R, Stuehr DJ. J. Biol. Chem. 2008;283:19603–19615. doi: 10.1074/jbc.M802914200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang JL, Stuehr DJ, Ikedasaito M, Rousseau DL. J. Biol. Chem. 1993;268:22255–22258. [PubMed] [Google Scholar]
- 44.Ghosh DK, Salerno JC. Front Biosci. 2003;8:D193–D209. doi: 10.2741/959. [DOI] [PubMed] [Google Scholar]
- 45.Ghosh DK, Ray K, Rogers AJ, Nahm NJ, Salerno JC. FEBS J. 2012;279:1306–1317. doi: 10.1111/j.1742-4658.2012.08525.x. [DOI] [PubMed] [Google Scholar]
- 46.Leys D, Scrutton NS. Curr Opin Struct Biol. 2004;14:642–647. doi: 10.1016/j.sbi.2004.10.002. [DOI] [PubMed] [Google Scholar]
- 47.Liang ZX, Kurnikov IV, Nocek JM, Mauk AG, Beratan DN, Hoffman BM. J. Am. Chem. Soc. 2004;126:2785–2798. doi: 10.1021/ja038163l. [DOI] [PubMed] [Google Scholar]
- 48.Liang Z-X, Nocek JM, Huang K, Hayes RT, Kurnikov IV, Beratan DN, Hoffman BM. J. Am. Chem. Soc. 2002;124:6849–6859. doi: 10.1021/ja0127032. [DOI] [PubMed] [Google Scholar]
- 49.Moser CC, Dutton PL. Biochimica et Biophysica Acta (BBA) - Bioenergetics. 1992;1101:171–176. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.