

Abstract
Ca2+ is thought to be essential for the exocytosis and endocytosis of synaptic vesicles. However, the manner in which Ca2+ coordinates these processes remains unclear, particularly at mature synapses. Using membrane capacitance measurements from calyx of Held nerve terminals in rats, we found that vesicle endocytosis is initiated primarily in Ca2+ nanodomains around Ca2+ channels, where exocytosis is triggered. Bulk Ca2+ outside of the domain could also be involved in endocytosis at immature synapses, although only after extensive exocytosis at more mature synapses. This bulk Ca2+-dependent endocytosis required calmodulin and calcineurin activation at immature synapses, but not at more mature synapses. Similarly, GTP-independent endocytosis, which occurred after extensive exocytosis at immature synapses, became negligible after maturation. We propose that nanodomain Ca2+ simultaneously triggers exocytosis and endocytosis of synaptic vesicles and that the molecular mechanisms underlying Ca2+-dependent endocytosis undergo major developmental changes at this fast central synapse.
On arrival of an action potential at a nerve terminal, Ca2+ enters through voltage-gated Ca2+ channels and forms a high intracellular Ca2+ concentration ([Ca2+]i) domain of tens (nanodomain) to hundreds (microdomain) of nanometers in diameter. Ca2+ in this domain triggers the fusion of synaptic vesicles with the plasma membrane for exocytic neurotransmitter release. Fused vesicle membrane is subsequently re-internalized by endocytosis and recycled1,2 into the vesicle pool to be reused for synaptic transmission. Although the critical role of intracellular Ca2+ in exocytosis is well established, its role in endocytosis remains less certain. In secretory cells3 and at synapses4-6, intracellular Ca2+ is involved in fast vesicle endocytosis having a time constant of subseconds to seconds. At the auditory calyx of Held synapse in immature rats, slow endocytosis, having a time constant of tens of seconds, depends on intracellular Ca2+, with the endocytic rate being faster at higher [Ca2+]i7,8. However, at more mature calyces9 or at cultured hippocampal synapses10,11, endocytic rate is relatively constant for various magnitudes of Ca2+ influx and exocytosis.
The Ca2+ chelators EGTA and BAPTA have similar Ca2+-binding affinities, but BAPTA has a more than hundred-fold faster Ca2+-binding rate than EGTA12. These Ca2+ buffers are often used for assessing the distance from Ca2+ source to its sensor13. At immature calyces of Held, before hearing onset (postnatal day 10–12, P10–12)14, EGTA loaded into the terminal strongly reduces transmitter release15. After rodents start to hear sound, however, the inhibitory effect of EGTA becomes much weaker, whereas that of BAPTA is unchanged16, suggesting that the domains, where Ca2+-secretion coupling occurs, undergo developmental reduction in diameter from microdomain to nanodomain. At immature calyces, similar to vesicle exocytosis, endocytosis is also attenuated by EGTA when loaded into the terminal7,8, suggesting the involvement of bulk [Ca2+]i elevation that extends to outside of the nanodomain in vesicle endocytosis. However, the question as to whether [Ca2+]i in the nanodomain affects vesicle endocytosis remains open, especially at more mature synapses.
Another question of vesicle endocytosis is what is the molecular mechanism downstream of Ca2+. In chromaffin cells17 and at immature calyces of Held8, calmodulin (CaM) is thought to mediate Ca2+-dependent vesicle endocytosis. Furthermore, in chromaffin cells18,19 and brain synaptosomes20, the Ca2+/CaM-dependent protein phosphatase calcineurin (CaN) is thought to underlie endocytosis via dephoshorylation of a group of endocytic proteins21, which promotes their assemblies22. Vesicle endocytosis generally depends on GTP hydrolysis23,24, but GTP-independent endocytosis has also been reported at retinal bipolar terminals25 and immature calyces of Held26. We asked whether these endocytic mechanisms operate at the calyx of Held after hearing onset. Our results indicate that nanodomain Ca2+ is involved in the fast and slow modes of vesicle endocytosis, both of which are independent of CaM/CaN, but entirely dependent on GTP hydrolysis at the calyx of Held after hearing onset.
RESULTS
Effects of Ca2+ buffers on vesicle endocytosis
At the calyx of Held presynaptic terminal of rats before (P7–9, pre-hearing) and after (P13–14, post-hearing) hearing onset, we elicited exocytosis using Ca2+ currents induced by a 20-ms square depolarizing pulse (from −80 to +10 mV) and recorded the exocytic increase in membrane capacitance (Cm) followed by a slow endocytic Cm decrease6-9,23. The endocytic Cm decay time course decreased as the rats aged9 (time constant, τ = 30.3 ± 4.2 s (n = 8) at P7–9 and 17.8 ± 1.2 s (n = 9) at P13–14, P < 0.05; Fig. 1). To examine whether intracellular Ca2+ is involved in such slow endocytosis, we first loaded the slow Ca2+ chelator EGTA at 10 mM into calyceal terminals via whole-cell patch pipettes. EGTA slowed the endocytic Cm decay at P7–9 calyces (Fig. 1a,b), as previously reported7,8, but had no effect at P13–14 (Fig. 1c,d). The fast Ca2+ chelator BAPTA loaded at 1 mM also slowed endocytosis to a greater extent than 10 mM EGTA at P7–9 calyces7. However, 1 mM BAPTA had no effect at P13–14 (Fig. 1d). At pre-hearing calyces, the rate of endocytosis becomes slower as the amount of exocytosis becomes larger6,7,9,23, whereas at P13–14, the endocytic τ was constant when evoked by a short (1–20 ms) depolarizing pulse (Supplementary Fig. 1)9. Thus, the lack of effect of 10 mM EGTA and 1 mM BAPTA on endocytic rate at post-hearing calyces would not be secondary to their inhibitory effects on exocytosis.
Figure 1.
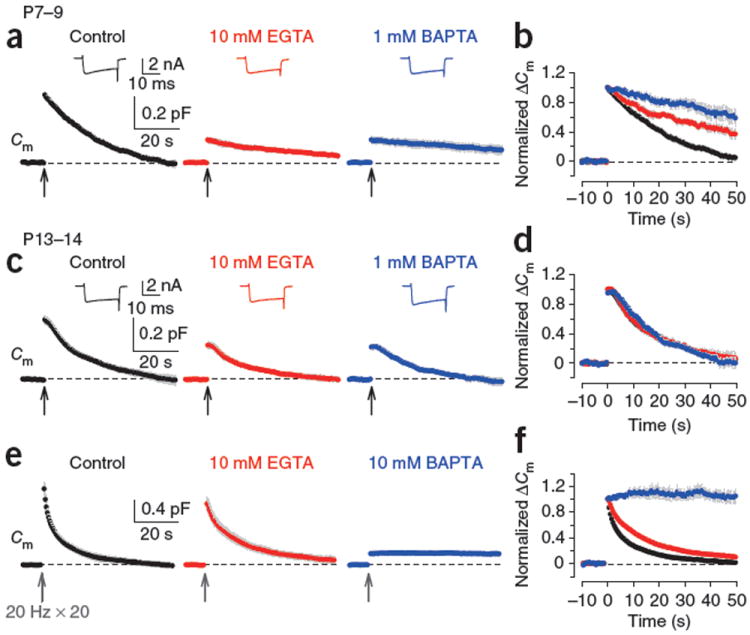
Different effects of EGTA and BAPTA on vesicle endocytosis at the developing calyces of Held. (a) At P7–9 calyceal terminals, presynaptic Ca2+ currents (ICa, upper insets) and Cm changes were evoked by a 20-ms depolarizing command pulse (from −80 to +10 mV, onset is indicated by an arrow) in the presence of 0.5 mM EGTA (control, black), 10 mM EGTA (red) or 1 mM BAPTA (blue) in the whole-cell patch pipette. Average Cm traces from six to eight presynaptic terminals and representative ICa traces are shown. Gray shading indicates s.e.m. (b) Cm traces in a normalized at the peak. (c) The stimulation protocol described in a was applied to P13–14 calyces. Averaged Cm traces were obtained from 5–9 calyceal terminals. (d) Cm traces in c normalized at the peak. (e) Cm changes induced by a train of 20 depolarizing pulses (20-ms duration) at 20 Hz in the presence of 0.5 mM EGTA (control, black, n = 9), 10 mM EGTA (red, n = 8) or 10 mM BAPTA (blue, n = 5) loaded into P13–14 calyces. EGTA (10 mM) had no significant effect on exocytic ΔCm (1.07 ± 0.10 pF, P = 0.098). (f) Cm traces in e normalized at the peak.
Might endocytosis at mature synapses be independent of Ca2+ or dependent exclusively on a transient increase of [Ca2+]i in Ca2+ nanodomains, which cannot be attenuated by 10 mM EGTA or 1 mM BAPTA12,13? To investigate this issue, we loaded P13–14 calyces with 10 mM BAPTA. Exocytosis evoked by single 20-ms depolarizing pulse was abolished by 10 mM BAPTA (data not shown), so we evoked a larger exocytosis using a short (1 s) train of repetitive depolarizing pulses (20 times at 20 Hz; Fig. 1e). This train induced a larger exocytic Cm change (ΔCm, 1.33 ± 0.11 pF, n = 9) followed by a bi-exponential endocytic Cm decay (τfast = 1.5 ± 0.3 s, 39 ± 5%, τslow = 14.9 ± 2.8 s, τmean = 9.9 ± 1.9 s, n = 9), which was slightly slowed by 10 mM EGTA (τmean = 17.7 ± 3.1 s, n = 8, P = 0.031; Fig. 1e,f). After loading with 10 mM BAPTA, exocytic ΔCm was strongly reduced, but a small exocytic fraction remained (0.20 ± 0.02 pF, n = 5). Notably, this exocytosis was not followed by an endocytic Cm decay (Fig. 1e,f). Thus, 10 mM BAPTA completely abolished endocytosis at more mature calyces. These results suggest that vesicle endocytosis after exocytosis is triggered primarily by nanodomain Ca2+ at post-hearing nerve terminals.
Ca2+-dependent fast and slow endocytosis
We next investigated the mechanisms underlying fast and slow endocytosis induced by repetitive stimulation6 (Fig. 2). When the calyx of Held terminal in P7–9 rats was repetitively stimulated by 20-ms depolarizing pulses at 1 Hz, Cm increases summed, and concomitantly the endocytic Cm decay after each pulse (initial rate, 41 ± 7 fF s−1, n = 16) became gradually faster and reached a maximal rate (ratemax) at around the eighth pulse (Fig. 2a) of 160 ± 11 fF s−1 (mean value of 10th–20th events; Fig. 2b). In P13–14 calyces, the 1-Hz train also induced endocytic acceleration (Fig. 2d) and reached a ratemax (207 ± 32 fF s−1, n = 8; Fig. 2a) similar to that at P7–9 (P = 0.2). After the 1-Hz train, Cm slowly recovered to the baseline, with a τmean of 38.8 ± 5.6 s (n = 7) at P7–9 (Fig. 2a,c) and 21.6 ± 0.9 s (n = 7) at P13–14 (Fig. 2d,f). Thus, the endocytic rate after repetitive stimulation became faster as the rats aged (P = 0.021).
Figure 2.
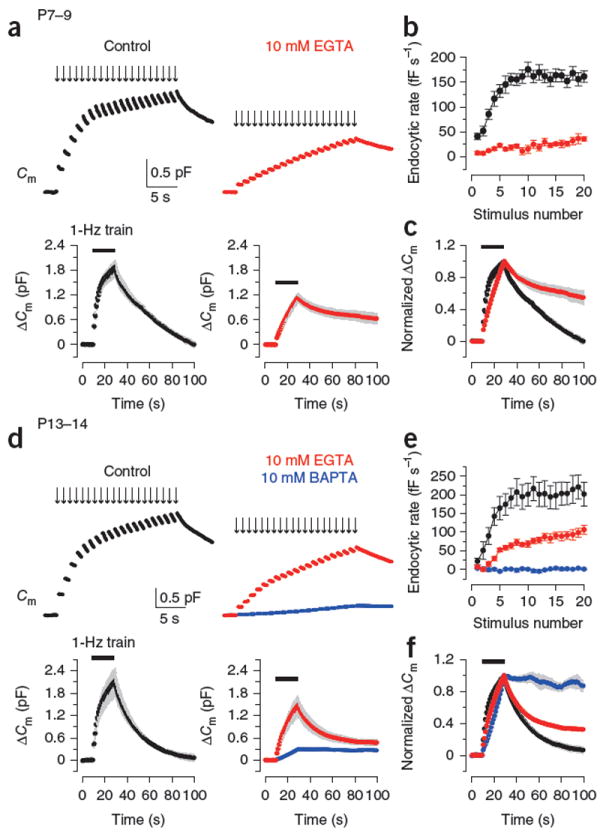
Ca2+ dependence of fast and slow endocytosis elicited by repetitive stimulation at the calyx of Held. (a) Top, Cm changes induced by 20-ms depolarizing pulses (arrows indicate onsets) repeated at 1 Hz for 20 s at P7–9 calyceal terminals preloaded with EGTA at 0.5 mM (black, control) or 10 mM (red). Bottom, averaged Cm records, shown at a longer time scale, from P7–9 calyceal terminals with 0.5 mM (n = 7) or 10 mM EGTA (n = 5). Gray shading indicates s.e.m. Black bars indicate the period of stimulation. (b) Endocytic rate during the stimulus train (fF s−1, ordinate), estimated from linear regression line fit to the Cm decay 0.45–1 s after each pulse versus stimulus number (abscissa) in P7–9 calyces with 0.5 mM (black, n = 16) or 10 mM EGTA (red, n = 6). (c) Cm traces from a normalized at the peak (superimposed). (d–f) Data are presented as in a–c for P13–14 calyces (n = 5–9). Data from terminals loaded with 10 mM BAPTA (blue) are also shown. Error bars represent s.e.m.
To examine the Ca2+ dependence of endocytosis induced by this stimulation protocol, we loaded EGTA (10 mM) into the calyceal terminals. In P7–9 calyces, EGTA potently inhibited the endocytic acceleration during the stimulus train as previously reported6, with its ratemax being reduced to 27.1 ± 4.8 fF s−1 (n = 6, P < 0.0001; Fig. 2a,b). In P13–14 calyces, EGTA also attenuated the endocytic acceleration (Fig. 2d,e), but to a lesser extent than in P7–9 calyces (ratemax, 87.2 ± 9.6 fF s−1, n = 9). Essentially the same results were obtained for the percentage of Cm recovery during the 1-Hz pulse (Supplementary Fig. 2). In both age groups, the slow Cm recovery after the stimulus train was inhibited by 10 mM EGTA, with its effect being weaker in post-hearing calyces (Fig. 2). Thus, bulk [Ca2+]i elevation contributed to both fast and slow types of endocytosis and its contribution decreased as the rats matured. In P13–14 calyces, 10 mM BAPTA nearly abolished the EGTA-resistant endocytosis during and after the repetitive stimulation (Fig. 2d–f), suggesting that nanodomain Ca2+ is primarily involved in endocytosis with different kinetics at post-hearing calyceal terminals.
CaM/CaN-dependent endocytosis at immature terminals
These results suggest that both the fast and slow modes of endocytosis can be triggered by intracellular Ca2+ at both pre-hearing and post-hearing calyces of Held. At pre-hearing calyces, it has recently been reported that CaM initiates all forms of endocytosis8. We re-examined this issue in pre-hearing and post-hearing calyces with intraterminal loadings of the CaM-specific inhibitors, myosin light-chain kinase (MLCK) peptide or CaM-binding domain (CBD) peptide. At pre-hearing calyces, MLCK peptide (0.2 μM) significantly attenuated endocytic acceleration during the 1-Hz stimulation (Fig. 3a,b) and reduced the ratemax (81.3 ± 13 fF s−1, n = 6) compared with the control peptide (0.2 μM), which cannot bind to CaM27 (194.6 ± 20.8 fF s−1, P < 0.01). MLCK peptide at a higher concentration (30 μM) had a similar inhibitory effect (ratemax = 83.8 ± 14 fF s−1, n = 7), suggesting that its maximal effect was attained at 0.2 μM. CBD peptide (20 μM) also reduced the ratemax (Fig. 3a,b) to 70.4 ± 9.1 fF s−1 (n = 5). In addition to its effect on fast endocytosis, MLCK peptide (0.2 μM) significantly prolonged the slow endocytosis after the 1-Hz train, with its half decay time (τ0.5, 58.1 ± 8.0 s, n = 5) being longer than that observed after control peptide loading (21.1 ± 1.1 s, n = 5, P < 0.01; Fig. 3a,c). CBD peptide (20 μM) showed a similar inhibitory effect on the slow endocytosis (τ0.5 = 55.5 ± 8.7 s, n = 5; Fig. 3a,c). These results confirm that CaM activity is involved in Ca2+-dependent fast and slow endocytosis at pre-hearing calyceal synapses8.
Figure 3.
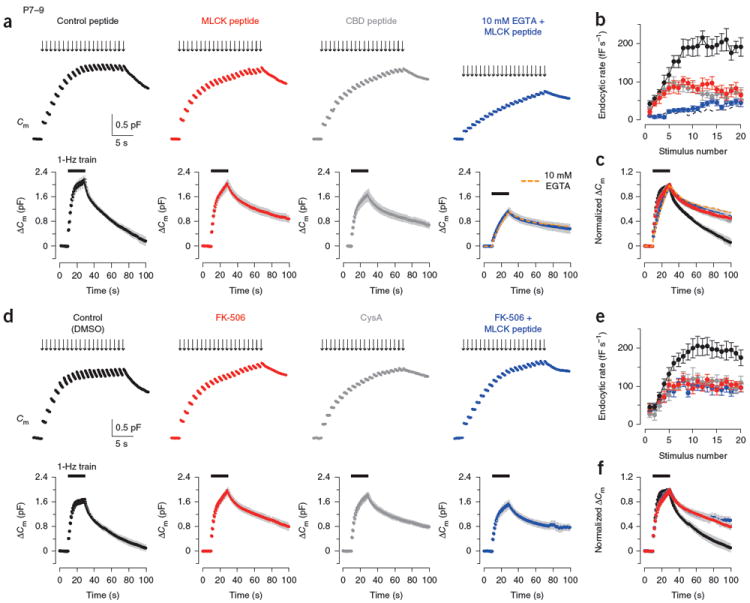
Effects of inhibitors for CaM and CaN in fast and slow endocytosis at P7–9 calyces. (a) Top, Cm changes induced by a stimulus train (20 pulses of 20-ms depolarization at 1 Hz) in the terminal preloaded with control peptide (0.2 μM, black, P8), MLCK peptide (0.2 μM, red, P8), CBD peptide (20 μM, dark gray, P8) or MLCK peptide (0.2 μM) and 10 mM EGTA (blue, P9). Bottom, averaged Cm records, shown at a slow time scale, from P7–9 calyceal terminals (n = 5 for each). Data from terminals preloaded with 10 mM EGTA (Fig. 2a) were superimposed (dashed orange line) on data from terminals preloaded with 10 mM EGTA and MLCK peptide (blue) in the bottom right panel. Gray shading indicates s.e.m. Black bars indicate the period of stimulation. (b) Summary data for the Cm decay rate (ordinate) measured after each pulse during the 1-Hz train versus stimulus number (abscissa) in the presence of control peptide (black, n = 6), MLCK peptide (red, n = 6), CBD peptide (dark gray, n = 5) or MLCK peptide and 10 mM EGTA (blue, n = 6). Dashed line indicates data from 10 mM EGTA-loaded terminals (Fig. 2a). (c) Averaged Cm traces shown in a normalized at the peak (superimposed). (d–f) Data are presented as in a–c for terminals loaded with 0.1% DMSO (black) or calyceal terminals loaded with FK-506 (0.1 μM, with 0.1% DMSO, red), cyclosporin A (CysA, 1 μM, with 0.1% DMSO, dark gray) or FK-506 and MLCK peptide (0.2 μM, blue) in P7–9 rats. Sample records (top panels of d) were derived from different calyceal terminals in P8 rats. Averaged Cm records (bottom panels of d) were obtained from 6–11 terminals. Error bars represent s.e.m.
To determine whether CaM-dependent endocytosis depends on nanodomain Ca2+, we loaded calyces with 10 mM EGTA, together with MLCK peptide (0.2 μM). In the presence of 10 mM EGTA, MLCK peptide had no further inhibitory effect on endocytosis evoked by the 1-Hz train (Fig. 3a,c). Thus, elevation of bulk [Ca2+]i is required for activation of CaM mediating fast and slow endocytosis.
We next examined whether CaN might mediate the CaM-dependent endocytic components by loading either the CaN inhibitor FK-506 (0.1 μM) or cyclosporin A (CysA, 1 μM) into P7–9 rat calyces. Similar to the CaM inhibitors, these CaN inhibitors attenuated the endocytic acceleration, reducing the ratemax attained during the stimulus train (control, 194.5 ± 20.4 fF s−1, n = 11; FK-506, 103.0 ± 9.1 fF s−1, n = 11, P < 0.01; CysA, 110.8 ± 10.2 fF s−1, n = 8, P < 0.01; Fig. 3d,e). The CaN inhibitors also slowed endocytic Cm decay after the 1-Hz train with increased τ0.5 (control, 19.7 ± 2.4 s, n = 7; FK-506, 55.8 ± 9.0 s, n = 6, P < 0.01; CysA, 51.0 ± 5.0 s, n = 6, P < 0.01; Fig. 3d,f). Loading FK-506 or CysA at a higher concentration (10 μM) had no additional effect (data not shown), suggesting that the maximal effect was attained by FK-506 at 0.1 μM and CysA at 1 μM. When we loaded both FK-506 (0.1 μM) and MLCK peptide (0.2 μM), their combined effects on fast and slow endocytosis were not stronger than their individual effects (Fig. 3d–f), suggesting that CaN exclusively mediates the downstream effect of CaM on vesicle endocytosis. Thus, the Ca2+-CaM-CaN cascade mediates a large fraction of fast and slow endocytosis during and after repetitive stimulation at pre-hearing calyces of Held.
Developmental decline of the CaM/CaN-dependent endocytosis
We next examined whether the Ca2+-CaM-CaN–dependent endocytic mechanism persists in the post-hearing calyces of Held by loading either MLCK peptide (30 μM) or CBD peptide (20 μM) into P13–14 calyces. In contrast with pre-hearing calyces, these CaM inhibitor peptides no longer affected endocytosis during or after the 1-Hz train at P13–14 (Fig. 4a–c and Supplementary Fig. 3). Similarly, the CaN inhibitors FK-506 (10 μM) and CysA (10 μM) no longer affected endocytosis evoked by the stimulus train at P13–14 calyces (Fig. 4d–f). These results suggest that neither fast nor slow endocytosis is mediated by the Ca2+-CaM-CaN cascade at the calyx of Held after hearing onset.
Figure 4.
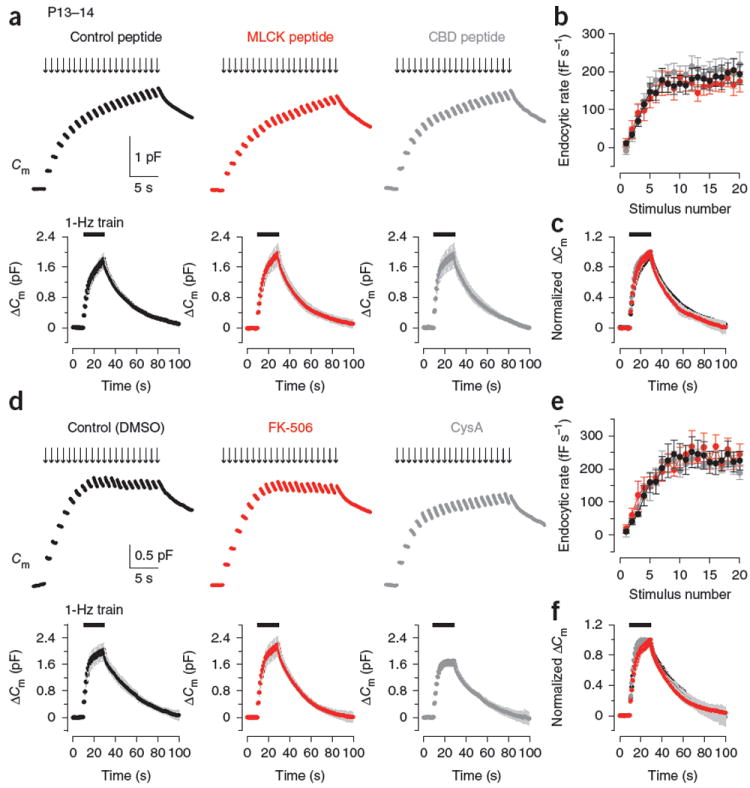
No effect of inhibitors for CaM and CaN in fast and slow endocytosis at P13–14 calyces. (a) Top, sample Cm changes induced by a 1-Hz train of 20 depolarizing pulses with the pipette solution containing control peptide (30 μM, black), MLCK peptide (30 μM, red) or CBD peptide (20 μM, dark gray) at P13–14 calyces. Bottom, averaged Cm records from P13–14 calyces loaded with control peptide (black, n = 6), MLCK peptide (red, n = 8) or CBD peptide (dark gray, n = 5). Gray shading indicates s.e.m. Black bars indicate the period of stimulation. (b) Summary data for the Cm decay rate after each pulse during the 1-Hz train from presynaptic terminals loaded with control peptide (black, n = 6), MLCK peptide (red, n = 8) or CBD peptide (dark gray, n = 6). (c) Averaged Cm traces from a normalized at the peak (superimposed). (d–f) Data are presented as in a–c for calyces loaded with 0.1% DMSO (control, black), FK-506 (10 μM, with 0.1% DMSO, red) or CysA (10 μM, with 0.1% DMSO, dark gray) in P13–14 rats (n = 5–6 terminals). Error bars represent s.e.m.
What developmental change causes the loss of functions of CaM and CaN in endocytosis? During the second postnatal week, CaM expression at calyceal terminals remains unchanged28. We therefore examined the expression of CaN in pre-hearing and post-hearing calyces. Immuno-staining with antibody to CaN (Fig. 5a) revealed that CaN was universally expressed at calyceal terminals in P7–8 rats, which were identified by synaptophysin immunoreactivity. At calyces in P14–15 rats, however, CaN immunoreactivity was much weaker (Fig. 5a). In densitometric quantification after subtracting the background in the presence of exogenous CaN, the intensity of CaN immunoreactivity at P14–15 calyces was 61% of that at P7–8 (Fig. 5b). Consistently, western blot analysis using tissue samples from the medial nucleus of trapezoid body (MNTB) regions also revealed a significant developmental decrease (to 66%, from P7 to P14, P = 0.04) in CaN immunoreactivity (Supplementary Fig. 4). Thus, CaN expression at the calyceal terminals was apparently downregulated during development, which may contribute, at least in part, to the developmental decline of the involvement of the Ca2+-CaM-CaN cascade in vesicle endocytosis.
Figure 5.
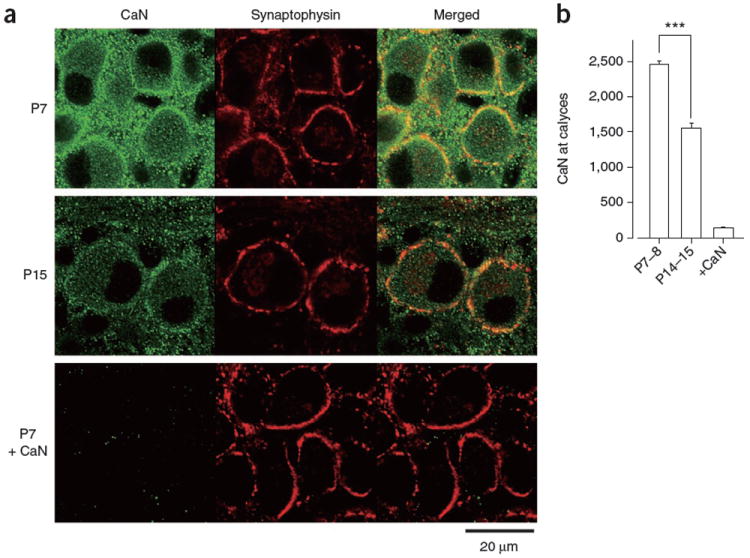
Developmental decline of CaN expression at the calyx of Held presynaptic terminals. (a) CaN immunoreactivity (left, green), synaptophysin immunoreactivity (middle, red) and their overlap (right). Bottom left (P7 + CaN), background after absorbing the primary antibodies with CaN protein. (b) Densitometric measurements of CaN immunofluorescence intensity in the regions that overlapped with synaptophysin immunofluorescence signals in P7–8 and P14–15 preparations (***P < 0.001, Scheffe test) and the background intensity measured after antibody absorption in P7–P15 preparations (+CaN). Error bars represent s.e.m.
GTP dependence of endocytosis at the calyx of Held
At pre-hearing calyces (P7–9), the nonhydrolysable GTP analog GTPγS nearly abolishes slow endocytosis induced by a single depolarizing pulse (duration, 1–20 ms) when loaded into the terminal at 0.2–0.3 mM23,26. However, a more recent study reported the presence of GTPγS-resistant endocytosis, which became apparent as the net exocytic Cm increases by more than 2 pF during intensive stimulation26. At the retinal bipolar cell terminal, GTPγS has no effect on endocytosis at 3.5 mM25, but abolishes it at 5 mM24. We therefore asked whether vesicle endocytosis can be observed in the presence of 5 mM GTPγS (tetralithium salt) at pre-hearing calyces. To minimize GTP synthesis from ATP, we removed the ATP-generating reagent phosphocreatine and reduced the ATP concentration to 2 mM in the pipette solution. In this condition, small fractions of fast and slow endocytosis were discerned during and after the 1-Hz train (Fig. 6a–c), confirming a previous report26. To determine whether this GTPγS-resistant component is CaM dependent or not, we loaded MLCK peptide (0.2 μM) and GTPγS (5 mM) into P7–9 calyces. In the presence of GTPγS, MLCK peptide had no additional effect on endocytosis during or after the 1-Hz train (Fig. 6a–c), indicating that GTPγS occluded the inhibitory effect of MLCK peptide on vesicle endocytosis. Thus, at immature calyces, the GTP-independent minor endocytic component was not mediated by CaM and the CaM-dependent fast and slow endocytosis was entirely GTP dependent.
Figure 6.

Developmental decline of GTP-independent endocytic component. (a) Top, sample Cm changes induced by a train of 20 depolarizing pulses, with low-[ATP] pipette solution containing 20 mM LiCl (control, with 1 mM GTP, black), 5 mM GTPγS (tetralithium salt, red) or 5 mM GTPγS and 0.2 μM MLCK peptide (blue) at P7–9 calyces. Each depolarizing pulse (from −80 to +10 mV for 20 ms) was preceded by a pre-pulse (stepping to +80 mV for 5 ms) to relieve Ca2+ currents from trimeric G protein–dependent inhibition by GTPγS. Bottom, averaged Cm records from the calyces loaded with LiCl (black, n = 6), GTPγS (red, n = 5) or GTPγS and MLCK peptide (blue, n = 5) at P7–9 calyces. Gray shading indicates s.e.m. Black bars indicate the period of stimulation. (b) Summary data for the Cm decay rate after each pulse during the 1-Hz train in the terminals loaded with LiCl (black, n = 7), GTPγS (red, n = 6) or GTPγS and MLCK peptide (blue, n = 6) in P7–9 rats. (c) Data in a normalized at the peaks after the train. (d–f) Data are presented as in a–c from P13–14 calyces (n = 5–8). Error bars represent s.e.m.
We next examined whether this GTP-independent endocytosis persists after hearing onset. In P13–14 calyces, GTPγS (5 mM) completely abolished endocytosis during and after the 1-Hz train, despite the fact that the exocytic ΔCm accumulated during the train (3.4 ± 0.3 pF, n = 8) was greater than that at pre-hearing calyces (P = 0.043) (Fig. 6d–f). Thus, vesicle endocytosis appears to become entirely GTP dependent during the second postnatal week at the calyx of Held.
DISCUSSION
Nanodomain Ca2+ couples endocytosis to exocytosis
To maintain transmitter release by vesicle recycling and to keep the area of presynaptic membrane constant, the amount of vesicle exocytosis must be somehow reported to the endocytic machinery. It has been suggested that Ca2+ does this in the case of massive exocytosis4-7. Our results suggest that Ca2+ acts, also for moderate exocytosis in a physiological range, by simultaneously triggering both exocytosis and endocytosis of synaptic vesicles in the Ca2+ nanodomain at relatively mature calyces of Held. This is consistent with endocytic rate not being dependent on stimulus intensity over a wide range at relatively mature synapses9-11 (Supplementary Fig. 1). In response to an action potential, [Ca2+] in the nanodomain (within 50 nm of Ca2+ channels13) rises to hundreds of micromolar and dissipates in milliseconds29, thereby triggering exocytic transmitter release16. During such a short period, Ca2+ may prime vesicles for endocytosis, which occurs after exocytosis over tens of seconds and is not Ca2+ dependent30, possibly at the peri-active zone31,32. The actual distance between the sites of Ca2+ entry and vesicle endocytosis remains unknown.
Simultaneous activation of exocytic and endocytic machineries by nanodomain Ca2+ would allow low-affinity Ca2+-binding proteins such as synaptotagmin33,34 and synaptophysin35 to be involved in endocytosis, supporting the idea that these proteins have dual roles in vesicle exocytosis and endocytosis. This is also consistent with the hypothesis that the C terminus36 or synprint site37 of the α subunit of voltage-gated Ca2+ channel is involved in vesicle endocytosis via its direct interaction with endocytic proteins. On prolonged repetitive stimulation, Ca2+ would spill out of the nanodomain. In such a condition, bulk Ca2+ would become involved in vesicle endocytosis to prevent the nerve terminal from aberrant swellings and from severe synaptic vesicle pool depletion.
Developmental changes in endocytic mechanisms
Consistent with previous studies in secretory cells3 and nerve terminals4-8, our results indicate that bulk Ca2+ that accumulates outside of the Ca2+ nanodomain contributes to fast and slow endocytosis at the pre-hearing and post-hearing calyces. The CaM-CaN–signaling mechanism was downstream of bulk Ca2+ at pre-hearing calyces, as in secretory cells18,19 and synaptosomes20. However, this mechanism no longer operated after hearing onset. Similarly, Ca2+ and CaM underlie inactivation of presynaptic Ca2+ currents28,38 at pre-hearing calyces of Held, but not at post-hearing calyces28, presumably as a result of a developmental decline in residual bulk [Ca2+]i39 below the threshold for CaM activation (Kd, 2–9 μM)40. This developmental change, together with the downregulation of CaN expression, might explain the lack of CaM- and CaN-dependent endocytosis in the post-hearing calyces. Although NCS-1 family, CaBP1 and hippocalcin have a Ca2+-binding affinity that is 1–2 orders of magnitude higher than CaM41, it remains to be seen whether these Ca2+-binding proteins might be involved in vesicle endocytosis at mature synapses with lower bulk [Ca2+]i.
Another controversial issue of vesicle endocytosis is its GTP dependence. Although we confirmed the presence of the GTP-independent endocytosis at pre-hearing calyces26, as has been seen at retinal bipolar terminals25, this mechanism no longer operated after hearing onset. Similarly, at the neuromuscular synapse in Drosophila, some forms of short-term depression do not depend on dynamin GTPase at the larval stage42, but there is an acute need for dynamin during repetitive stimulation at mature synapses43 and the synaptic vesicle pool is severely depleted in dynamin-deficient flies2. Dynamin 1 is essential for the slow mode of endocytosis after strong stimuli44, and pretreatment of synapses with dynamin inhibitors blocks synaptic transmission after prolonged repetitive stimulation23,45. Thus, a GTP- and dynamin-dependent form of endocytosis may be critical for mature synapses to properly function.
Vesicle endocytosis is typically classified into fast and slow modes46,47, whose relative prominence depends on Ca2+ buffer type and concentration4,7,8. However, our results suggest that this classification does not agree with underlying mechanisms. At post-hearing calyces, 10 mM EGTA had no effect on slow endocytosis lasting tens of seconds following a short pulse (20 ms) stimulation, whereas it attenuated endocytosis with similar kinetics following a repetitive stimulation. The remaining EGTA-resistant endocytic components were abolished by 10 mM BAPTA. These results suggest that slow endocytosis can be mediated by distinct Ca2+ sensors residing inside and outside of the Ca2+ nanodomain. At pre-hearing calyces, fast endocytosis during a repetitive stimulus and slow endocytosis after the train had similar pharmacological properties, with both being sensitive to 10 mM EGTA and inhibitors for CaM and CaN. Thus, endocytosis with different kinetics can be mediated by a common mechanism. Fast endocytosis during the repetitive stimulation had a similar kinetics throughout P7–14, but the underlying mechanism was independent of CaM and CaN after hearing onset. Thus, a classification from a more mechanistic viewpoint remains to be established for vesicle endocytosis with different kinetics.
ONLINE METHODS
Slice preparation and solutions
All experiments were performed in accordance with the guidelines of the Physiological Society of Japan. Methods for preparing auditory brainstem slices containing the MNTB from Wistar rats (P7–15) have been described previously23,28. The extracellular solution for recordings that isolates Ca2+ currents contained 115 mM NaCl, 2.5 mM KCl, 26 mM NaHCO3, 1.25 mM NaH2PO4, 2 mM CaCl2, 1 mM MgCl2, 10 mM glucose, 3 mM myoinositol, 2 mM sodium pyruvate, 0.5 mM ascorbic acid, 10 mM tetraethyl-ammonium chloride, 0.5 mM 4-aminopyridine, 0.001 mM tetrodotoxin, 0.01 mM bicuculline methiodide and 0.0005 mM strychnine hydrochloride (pH 7.4 when bubbled with 95% O2 and 5% CO2, 310–315 mOsm). Unless otherwise noted, the pipette solution contained 118 mM cesium gluconate, 30 mM CsCl, 10 mM HEPES, 0.5 mM EGTA, 1 mM MgCl2, 12 mM sodium phosphocreatine, 3 mM Mg-ATP and 0.3 mM Na-GTP (pH 7.3–7.4 adjusted with CsOH). When we used different concentrations of Ca2+ buffers, the osmolarity of the pipette solution was set to 315–320 mOsm by changing the concentration of cesium gluconate. For blocking GTP hydrolysis in the calyceal terminals, we used a modified low-[ATP] pipette solution that contained 128 mM cesium gluconate, 30 mM CsCl, 10 mM HEPES, 0.5 mM EGTA, 1 mM MgCl2, 2 mM Mg-ATP and 5 mM GTPγS (tetralithium salt) (pH 7.3–7.4 adjusted with CsOH). For control experiments using the low-[ATP] pipette solution, CsCl (20 mM) was replaced by equimolar LiCl, GTPγS was omitted and 1 mM Na-GTP was added. GTPγS was stored as powder at −20 °C and dissolved in the pipette solution less than 2 h before recordings.
Recordings and data analysis
Cm measurement from the calyx of Held nerve terminals in whole-cell configurations were made at 26–27 °C as described previously23. Calyceal terminals were voltage clamped at the holding potential of −80 mV and the sinusoidal voltage command was applied at 1 kHz with a peak-to-peak voltage of 60 mV. Patch pipettes (5–8 MΩ) had a series resistance of 7–20 MΩ, which was compensated by up to 65% for a final value of 6.7–7.0 MΩ. The pipettes were coated with dental wax to reduce stray capacitance (4–6 pF, compensated by >80%). Data were obtained within 20 min of break-in. Single pulse or a train (1 Hz) of step depolarization (to +10 mV, 20 ms) was used for stimulation, unless otherwise noted. Cm changes within 450 ms of square-pulse stimulation were excluded from analysis to avoid contaminations of conductance-dependent capacitance artifacts23. The amplitude of exocytic Cm change (ΔCm) was measured as the difference of Cm values between the baseline and at 450–500 ms after depolarization. For measuring Cm decay rate during repetitive stimulation at 1 Hz, regression lines were obtained from Cm records 0.45–1 s after each pulse. Sample Cm records are shown as average values of each 50 data point (for 50 ms) plotted every 50 ms (for shorter time scale) or every 500 ms (for longer time scale). GTPγS, LiCl, DMSO, MLCK peptide (RRKWQKTGHAVRAIGRL, Calbiochem), MLCK two-point mutant peptide27 (control peptide, RRKEQKTGHAVRAIGRE, Calbiochem), CBD peptide (CaM kinase II 290–309, Sigma), FK-506 (Sigma) and CysA (Tocris Bioscience) were included in the pipette solution and were infused into the calyceal terminals with the access resistance less than 12 MΩ and were allowed to diffuse in the terminals for at least 5 min before recordings. All values are given as mean ± s.e.m. and significant difference was evaluated by one-way ANOVA followed by Dunnett’s post hoc test or Student’s unpaired t test, unless otherwise noted.
Immunostaining and immunoblotting
Immunostaining and immunoblotting were performed as previously described48. For immunostaining, we used primary antibody to CaN β subunit (mouse monoclonal clone CN-B1, Sigma, diluted 1:2,000) and primary antibody to synaptophysin (rabbit polyclonal, Zymed Laboratories, diluted 1:100), along with goat secondary antibodies conjugated with Alexa fluor 488 and Alexa fluor 588 (Invitrogen, diluted 1:200). In the primary antibody absorbing tests for evaluating the specificity of the CaN immunoreactivity, bovine brain CaN (Sigma; final concentration, 500 units per ml) was pre-incubated with primary antibodies overnight at 4 °C. Primary antibody dilution buffer (pH 7.3) contained 40 mM HEPES, 300 mM NaCl, 0.2% bovine serum albumin (wt/vol) and 0.05% Triton X-100 (vol/vol). Confocal images were acquired using the Olympus FV300 system with an inverted microscope (IX71) attached with a 60× objective lens (PlanApo) and analyzed using WinROOF software (Mitani). For immunoblotting, the homogenate (2 μg) of tissue samples from the MNTB region of rat brainstem were separated using Tris-tricine gel (15–20%), electro-blotted to PVDF membrane, blocked with skimmed milk, incubated with antibody to CaN β subunit (diluted 1:2,000) overnight at 4 °C and then with horseradish peroxidase–conjugated antibody to mouse IgG (GE Healthcare, diluted 1:2,000) for 40 min at 20–24 °C and detected with ECL advance (GE Healthcare). Chemiluminescent images were acquired using LAS-1000 system (Fujifilm) and analyzed with MultiGauge software (Fujifilm). To quantify the relative amount of CaN in samples, we simultaneously applied 0.5–3 μg homogenate samples to the same gel and formulated a standard curve for measuring the intensity of CaN immunoreactivity.
Supplementary Material
Acknowledgments
We thank E. Johnson and S. Takamori for helpful comments. This work was supported by the Core Research for Evolutional Science and Technology of Japan Science and Technology Agency (T.T.), US National Institutes of Health grant EY014043 (H.v.G.) and Grant-in-Aid for Young Scientists from the Japanese Ministry of Education, Culture, Sports, Science and Technology #20700357 (T.Y.).
Footnotes
AUTHOR CONTRIBUTIONS
T.Y. and T.T. designed the experiments. H.v.G. designed the experiments on nonhydrolysable GTP analogues. T.Y., K.E. and N.S. performed the experiments and analyzed the data. T.Y., H.v.G. and T.T. wrote the manuscript. All of authors revised and approved the final manuscript.
Note: Supplementary information is available on the Nature Neuroscience website.
COMPETING FINANCIAL INTERESTS
The authors declare no competing financial interests.
References
- 1.Heuser JE, Reese TS. Evidence for recycling of synaptic vesicle membrane during transmitter release at the frog neuromuscular junction. J Cell Biol. 1973;57:315–344. doi: 10.1083/jcb.57.2.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Koenig JH, Ikeda K. Disappearance and reformation of synaptic vesicle membrane upon transmitter release observed under reversible blockage of membrane retrieval. J Neurosci. 1989;9:3844–3860. doi: 10.1523/JNEUROSCI.09-11-03844.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Artalejo CR, Henley JR, McNiven MA, Palfrey HC. Rapid endocytosis coupled to exocytosis in adrenal chromaffin cells involves Ca2+, GTP and dynamin, but not clathrin. Proc Natl Acad Sci USA. 1995;92:8328–8332. doi: 10.1073/pnas.92.18.8328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Neves G, Gomis A, Lagnado L. Calcium influx selects the fast mode of endocytosis in the synaptic terminal of retinal bipolar cells. Proc Natl Acad Sci USA. 2001;98:15282–15287. doi: 10.1073/pnas.261311698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Beutner D, Voets T, Neher E, Moser T. Calcium dependence of exocytosis and endocytosis at the cochlear inner hair cell afferent synapse. Neuron. 2001;29:681–690. doi: 10.1016/s0896-6273(01)00243-4. [DOI] [PubMed] [Google Scholar]
- 6.Wu W, Xu J, Wu X-S, Wu L-G. Activity-dependent acceleration of endocytosis at a central synapse. J Neurosci. 2005;25:11676–11683. doi: 10.1523/JNEUROSCI.2972-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hosoi N, Holt M, Sakaba T. Calcium-dependence of exo- and endocytotic coupling at a glutamatergic synapse. Neuron. 2009;63:216–229. doi: 10.1016/j.neuron.2009.06.010. [DOI] [PubMed] [Google Scholar]
- 8.Wu X-S, et al. Ca2+ and calmodulin initiates all forms of endocytosis during depolarization at a nerve terminal. Nat Neurosci. 2009;12:1003–1010. doi: 10.1038/nn.2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Renden R, von Gersdorff H. Synaptic vesicle endocytosis at a CNS nerve terminal: faster kinetics at physiological temperatures and increased endocytotic capacity during maturation. J Neurophysiol. 2007;98:3349–3359. doi: 10.1152/jn.00898.2007. [DOI] [PubMed] [Google Scholar]
- 10.Granseth B, Odermatt B, Royle SJ, Lagnado L. Clathrin-mediated endocytosis is the dominant mechanism of vesicle retrieval at hippocampal synapses. Neuron. 2006;51:773–786. doi: 10.1016/j.neuron.2006.08.029. [DOI] [PubMed] [Google Scholar]
- 11.Balaji J, Armbruster M, Ryan TA. Calcium control of endocytic capacity at a CNS synapse. J Neurosci. 2008;28:6742–6749. doi: 10.1523/JNEUROSCI.1082-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Naraghi M, Neher E. Linearized buffered Ca2+ diffusion in microdomains and its implications for calculation of [Ca2+] at the mouth of a calcium channel. J Neurosci. 1997;17:6961–6973. doi: 10.1523/JNEUROSCI.17-18-06961.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Augustine GJ, Santamaria F, Tanaka K. Local calcium signaling in neurons. Neuron. 2003;40:331–346. doi: 10.1016/s0896-6273(03)00639-1. [DOI] [PubMed] [Google Scholar]
- 14.Jewett DL, Romano MN. Neonatal development of auditory system potentials averaged from the scalp of rat and cat. Brain Res. 1972;36:101–115. doi: 10.1016/0006-8993(72)90769-x. [DOI] [PubMed] [Google Scholar]
- 15.Borst JGG, Sakmann B. Calcium influx and transmitter release in a fast CNS synapse. Nature. 1996;383:431–434. doi: 10.1038/383431a0. [DOI] [PubMed] [Google Scholar]
- 16.Fedchyshyn MJ, Wang L-Y. Developmental transformation of the release modality at the calyx of Held synapse. J Neurosci. 2005;25:4131–4140. doi: 10.1523/JNEUROSCI.0350-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Artalejo CR, Elhamdani A, Palfrey HC. Calmodulin is the divalent cation receptor for rapid endocytosis, but not exocytosis, in adrenal chromaffin cells. Neuron. 1996;16:195–205. doi: 10.1016/s0896-6273(00)80036-7. [DOI] [PubMed] [Google Scholar]
- 18.Engisch KL, Nowycky MC. Compensatory and excess retrieval: two types of endocytosis following single step depolarizations in bovine adrenal chromaffin cells. J Physiol (Lond) 1998;506:591–608. doi: 10.1111/j.1469-7793.1998.591bv.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chan S-A, Smith C. Physiological stimuli evoke two forms of endocytosis in bovine chromaffin cells. J Physiol (Lond) 2001;537:871–885. doi: 10.1111/j.1469-7793.2001.00871.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Marks B, McMahon HT. Calcium triggers calcineurin-dependent synaptic vesicle recycling in mammalian nerve terminals. Curr Biol. 1998;8:740–749. doi: 10.1016/s0960-9822(98)70297-0. [DOI] [PubMed] [Google Scholar]
- 21.Cousin MA, Robinson PJ. The dephosphins: dephosphorylation by calcineurin triggers synaptic vesicle endocytosis. Trends Neurosci. 2001;24:659–665. doi: 10.1016/s0166-2236(00)01930-5. [DOI] [PubMed] [Google Scholar]
- 22.Slepnev VI, Ochoa G-C, Butler MH, Grabs D, De Camilli P. Role of phosphorylation in regulation of the assembly of endocytic coat complexes. Science. 1998;281:821–824. doi: 10.1126/science.281.5378.821. [DOI] [PubMed] [Google Scholar]
- 23.Yamashita T, Hige T, Takahashi T. Vesicle endocytosis requires dynamin-dependent GTP hydrolysis at a fast CNS synapse. Science. 2005;307:124–127. doi: 10.1126/science.1103631. [DOI] [PubMed] [Google Scholar]
- 24.Jockusch WJ, Praefcke GJK, McMahon HT, Lagnado L. Clathrin-dependent and clathrin-independent retrieval of synaptic vesicles in retinal bipolar cells. Neuron. 2005;46:869–878. doi: 10.1016/j.neuron.2005.05.004. [DOI] [PubMed] [Google Scholar]
- 25.Heidelberger R. ATP is required at an early step in compensatory endocytosis in synaptic terminals. J Neurosci. 2001;21:6467–6474. doi: 10.1523/JNEUROSCI.21-17-06467.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xu J, et al. GTP-independent rapid and slow endocytosis at a central synapse. Nat Neurosci. 2008;11:45–53. doi: 10.1038/nn2021. [DOI] [PubMed] [Google Scholar]
- 27.Török K, et al. Inhibition of calmodulin-activated smooth-muscle myosin light-chain kinase by calmodulin-binding peptides and fluorescent (phosphodiesterase-activating) calmodulin derivatives. Biochemistry. 1998;37:6188–6198. doi: 10.1021/bi972773e. [DOI] [PubMed] [Google Scholar]
- 28.Nakamura T, Yamashita T, Saitoh N, Takahashi T. Developmental changes in calcium/calmodulin-dependent inactivation of calcium currents at the rat calyx of Held. J Physiol (Lond) 2008;586:2253–2261. doi: 10.1113/jphysiol.2007.142521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Neher E. Vesicle pools and Ca2+ microdomains: new tools for understanding their roles in neurotransmitter release. Neuron. 1998;20:389–399. doi: 10.1016/s0896-6273(00)80983-6. [DOI] [PubMed] [Google Scholar]
- 30.Gad H, Löw P, Zotova E, Brodin L, Shupliakov O. Dissociation between Ca2+-triggered synaptic vesicle exocytosis and clathrin-mediated endocytosis at a central synapse. Neuron. 1998;21:607–616. doi: 10.1016/s0896-6273(00)80570-x. [DOI] [PubMed] [Google Scholar]
- 31.Roos J, Kelly RB. The endocytic machinery in nerve terminals surrounds sites of exocytosis. Curr Biol. 1999;9:1411–1414. doi: 10.1016/s0960-9822(00)80087-1. [DOI] [PubMed] [Google Scholar]
- 32.Teng H, Wilkinson RS. Clathrin-mediated endocytosis near active zones in snake motor boutons. J Neurosci. 2000;20:7986–7993. doi: 10.1523/JNEUROSCI.20-21-07986.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Poskanzer KE, Marek KW, Sweeney ST, Davis GW. Synaptotagmin 1 is necessary for compensatory synaptic vesicle endocytosis in vivo. Nature. 2003;426:559–563. doi: 10.1038/nature02184. [DOI] [PubMed] [Google Scholar]
- 34.Llinás RR, Sugimori M, Moran KA, Moreira JE, Fukuda M. Vesicular reuptake inhibition by a synaptotagmin I C2B domain antibody at the squid giant synapse. Proc Natl Acad Sci USA. 2004;101:17855–17860. doi: 10.1073/pnas.0408200101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Daly C, Ziff EB. Ca2+-dependent formation of a dynamin-synaptophysin complex: potential role in synaptic vesicle endocytosis. J Biol Chem. 2002;277:9010–9015. doi: 10.1074/jbc.M110815200. [DOI] [PubMed] [Google Scholar]
- 36.Chen Y, et al. Formation of an endophillin-Ca2+ channel complex is critical for clathrin-mediated synaptic vesicle endocytosis. Cell. 2003;115:37–48. doi: 10.1016/s0092-8674(03)00726-8. [DOI] [PubMed] [Google Scholar]
- 37.Watanabe H, et al. Involvement of Ca2+ channel synprint site in synaptic vesicle endocytosis. J Neurosci. 2010;30:655–660. doi: 10.1523/JNEUROSCI.3214-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Xu J, Wu L-G. The decrease in the presynaptic calcium current is a major cause of short-term depression at a calyx-type synapse. Neuron. 2005;46:633–645. doi: 10.1016/j.neuron.2005.03.024. [DOI] [PubMed] [Google Scholar]
- 39.Nakamura Y, DiGregorio D, Takahashi T. Single action potential–evoked Ca2+ transients at the calyx of Held presynaptic terminal. Neurosci Res. 2007;58:S71. [Google Scholar]
- 40.Shifman JM, Choi MH, Mihalas S, Mayo SL, Kennedy MB. Ca2+/calmodulin-dependent protein kinase II (CaMKII) is activated by calmodulin with two bound calciums. Proc Natl Acad Sci USA. 2006;103:13968–13973. doi: 10.1073/pnas.0606433103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Burgoyne RD. Neuronal calcium sensor proteins: generating diversity in neuronal Ca2+ signaling. Nat Rev Neurosci. 2007;8:182–193. doi: 10.1038/nrn2093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kawasaki F, Hazen M, Ordway RW. Fast synaptic fatigue in shibire mutants reveals a rapid requirement for dynamin in synaptic membrane trafficking. Nat Neurosci. 2000;3:859–860. doi: 10.1038/78753. [DOI] [PubMed] [Google Scholar]
- 43.Wu Y, Kawasaki F, Ordway RW. Properties of short-term synaptic depression at larval neuromuscular synapses in wild-type and temperature-sensitive paralytic mutants of Drosophila. J Neurophysiol. 2005;93:2396–2405. doi: 10.1152/jn.01108.2004. [DOI] [PubMed] [Google Scholar]
- 44.Lou X, Paradise S, Ferguson SM, De Camilli P. Selective saturation of slow endocytosis at a giant glutamatergic central synapse lacking dynamin 1. Proc Natl Acad Sci USA. 2008;105:17555–17560. doi: 10.1073/pnas.0809621105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Newton AJ, Kirchhausen T, Murthy VN. Inhibition of dynamin completely blocks compensatory synaptic vesicle endocytosis. Proc Natl Acad Sci USA. 2006;103:17955–17960. doi: 10.1073/pnas.0606212103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.von Gersdorff H, Matthews G. Inhibition of endocytosis by elevated internal calcium in a synaptic terminal. Nature. 1994;370:652–655. doi: 10.1038/370652a0. [DOI] [PubMed] [Google Scholar]
- 47.Wu L-G, Ryan TA, Lagnado L. Modes of vesicle retrieval at ribbon synapses, calyx-type synapses and small central synapses. J Neurosci. 2007;27:11793–11802. doi: 10.1523/JNEUROSCI.3471-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kimura M, Saitoh N, Takahashi T. Adenosine A1 receptor–mediated presynaptic inhibition at the calyx of Held of immature rats. J Physiol (Lond) 2003;553:415–426. doi: 10.1113/jphysiol.2003.048371. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.