

Abstract
Tubulin is the proposed target for drugs against cancer and helminths and is also a validated target in kinetoplastid parasites. With the aim of identifying new lead compounds against Leishmania sp., tubulin isolated from L. tarentolae was used to screen a 10 000 compound library. One compound, Chembridge No. 7992831 (5), displayed an IC50 of 13 μm against Leishmania tubulin in an in vitro assembly assay and showed a greater than threefold selectivity over mammalian tubulin. Another compound, Chembridge No. 9067250 (8), exhibited good activity against mammalian tubulin (IC50 = 5.0 μm). This compound was also toxic to several cancer cell lines with IC50 values in the region of 1 μM. Subsequent testing of analogues of 8 contained within the library identified two compounds with greater potency against mammalian tubulin (IC50 values of 1.1 and 2.8 μM). The more potent antitubulin agent also showed promising activity against cancer cell lines in vitro, with IC50 values ranging from 0.18 to 0.73 μM.
Keywords: Tubulin, screen, library, Leishmania
Tubulin is an accepted target for treatments against cancer and helminths (1–3). This protein exists as a heterodimer consisting of α and β subunits which polymerize to form microtubules. These microtubules have a number of functions within eukaryotic organisms including chromosome segregation, motility and the maintenance of cellular morphology (4–6). Given these vital roles, tubulin is essential to all eukaryotes. The assembly–disassembly process is critical for the proper functioning of microtubules within the cell. Taxol, one of the more well-known compounds which act against tubulin, does so by stabilizing the protofilaments and thereby prevents disassembly (7). Other compounds such as colchicine and vinblastine inhibit the assembly of tubulin (8–10). Therefore, compounds which affect this assembly–disassembly process could serve as good lead compounds in drug discovery efforts targeting pathogenic eukaryotic cells.
Tubulin is a validated target in kinetoplastid parasites (11,12) and therefore offers an excellent target against which to develop drug treatments against these organisms. Leishmania sp. is responsible for the disease leishmaniasis, which in the case of visceral leishmaniasis can be fatal. An estimated 12 million people currently suffer from leishmaniasisa. The current treatments are far from ideal and there is a clear need to identify new lead compounds.
Previous work from our laboratory has attempted to optimize the lead compound oryzalin (1) (13) for inhibition of Leishmania tubulin assembly. A series of oryzalin analogues have been synthesized with the most promising leads, GB-II-5 (2), GB-II-150 (3) and 4, showing low micromolar activity against Leishmania donovani axenic amastigotes and mid-nanomolar activity against Trypanosoma brucei bloodstream forms (14–17) (Figure 1). However, these compounds suffer from metabolic instability, which limits their activity in vivo (16,18). While we continue to optimize these compounds for antikinetoplastid activity and improved metabolic stability, we are also seeking to identify new lead compounds that selectively interfere with parasite microtubules. Given the selectivity observed with these compounds in vitro and the selectivity observed with tubulin targeting anthelminths in vivo (19,20) there is clearly the potential to develop antikinetoplastid compounds with selectivity in vivo. This potential is perhaps greater for antikinetoplastids in comparison with anthelminths as the homology of Leishmania sp. and helminth α- and β-tubulin with porcine tubulin are in the regions of >80 and >90% respectively (21,22).
Figure 1.
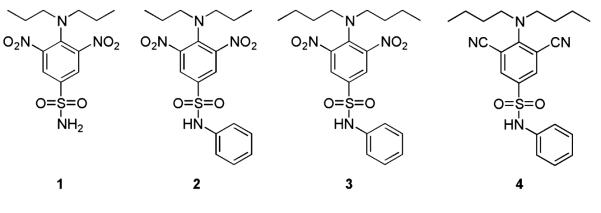
Structures of oryzalin (1) and analogues with known antitubulin activity.
We have isolated tubulin from Leishmania tarentolae in our laboratory and have shown that this protein is a suitable alternative to tubulin purified from Leishmania amazonensis for use in antiparasitic drug discovery efforts (22). Leishmania tarentolae tubulin is nearly identical in amino acid sequence to tubulins from other Leishmania sp. (>98%), and dinitroaniline compounds display indistinguishable activity against and binding affinity for L. tarentolae and L. amazonensis tubulin. In addition to the obvious safety advantages of purifying tubulin from a non-pathogenic species, L. tarentolae is also more readily and inexpensively cultured than Leishmania species that infect humans. This means that the large scale production of protein required for a high throughput screen becomes more feasible using L. tarentolae tubulin. The use of high throughput screens to identify novel lead compounds has increased because of the greater accessibility of the required technology. This is reflected in the number of screens against protozoan parasites reported over the last few years (23–28). Given the need for new lead compounds against kinetoplastid parasites and the greater accessibility of Leishmania tubulin, a 10 000 compound library was screened against this protein purified from L. tarentolae. Secondary assays were used to determine the selectivity of these compounds for leishmanial tubulin over mammalian tubulin. The cellular effects on Leishmania of the most potent hit were examined. In addition, three compounds were identified that displayed activity against mammalian tubulin and cancer cell lines.
Experimental
Compounds and other reagents
The CNS-Set™ of 10 000 drug-like compounds was purchased from ChemBridge Corporation (Suite G, San Diego, CA, USA). These compounds were supplied as 10 mm stock solutions in dimethyl sulphoxide (DMSO) in 96-well plates. Hit compounds were repurchased from ChemBridge as 5 mg of solid. Unless otherwise noted, all other reagents were from Sigma-Aldrich (St Louis, MO, USA).
Tubulin purification
Tubulin from L. tarentolae was isolated using a larger scale version of the previously reported protocol (22). Leishmania tarentolae were grown in brain heart infusion medium supplemented with Hemin (10 mg/mL). The parasites were cultured in 2 L flasks to a maximum cell density of approximately 2 × 108 cells/mL. In a typical purification, 20 × 1011 cells were used as the starting material and an average of 50 mg of protein were isolated at concentrations ranging from 10 to 30 mg/mL. This quantity of tubulin was sufficient to screen approximately 1600 compounds in the primary assay under the conditions described below. Tubulin from pig brain was isolated as outlined earlier (15).
Compound screening
Primary assay
Compounds were diluted in DMSO to 2 mm, then diluted to 1 mm with H2O, giving a 50:50 DMSO:H2O solution. Dimethyl sulphoxide and 3 (at 40 μM) were used as controls. The compounds were screened using a modification of the previously reported assay (22,29). Primary assays were carried out in 384-well plates at Leishmania tubulin concentrations of 1.2 mg/mL. Compounds were preincubated with tubulin for 10 min at 4 °C followed by 5 min at RT. Addition of 4 μL of assembly solution (25% DMSO, 5 mm GTP) followed, giving a final assay volume of 20 μL. The plates were read using a SpectraMax Plus microplate reader (Molecular Devices, Sunnyvale, CA, USA) at 30 °C for 15 min with intervals of 15 seconds between readings. Successful assembly was considered an increase in optical density of >0.02 absorbance units at 351 nm.
IC50 determinations
IC50 values against Leishmania and mammalian tubulin were determined in half area 96-well plates at tubulin concentrations of 1.5 mg/mL using previously published assay conditions (15,22).
Susceptibility testing of L. donovani, Vero cells and T. b. brucei
The susceptibility of L. donovani axenic amastigote-like parasites and Vero cells (African green monkey kidney epithelial cells) to growth inhibition by compounds of interest was assayed as described previously (14,30). The susceptibility of T. b. brucei to 5 was assayed as described previously (14).
Growth inhibition assay against cancer cell lines
Viable cells were quantified using the sulphorhodamine B (SRB) assay in several solid tumor cell lines, including four prostate cancer cell lines (LNCaP, PC-3, DU145, PPC-1), a bladder cancer cell line (TSU-Pr1), a colon cancer cell line (HT-29) and a breast cancer cell line (MCF-7). Cells were plated in 96-well plates at a density of 800–5000 cells/well (depending on the cell line) for 1 day prior to the addition of different compounds at a range of concentrations (0–100 μm). After 96 h incubation at 37 °C, cells were fixed by the addition of 50 μL of 50% cold trichloroacetic acid and incubated at 4 °C for 3 h. The plate was washed three times with tap water and was allowed to air dry. The cells were then stained with 0.4% SRB dissolved in 1% acetic acid for 30 min at room temperature. Unbound SRB was washed away with five washes of 1% acetic acid. The plate was again allowed to air dry and the bound SRB stain, representing surviving cells, was dissolved in 200 μL of Tris base (10 mm). The optical density was determined at 540 nm using a microplate reader (Dynex Technologies, Chantilly, VA, USA). Mean values were obtained from at least four wells per treatment condition. Plots of per cent inhibition of cell growth versus drug concentration were constructed, and the concentration that inhibited cell growth by 50% relative to the untreated control (IC50) was determined by nonlinear least squares regression using winnonlin software (version 5.2, Pharsight Corp., Mountain View, CA, USA). winnonlin was provided by a Pharsight Academic License to The Ohio State University.
Flow cytometry
Leishmania donovani promastigotes were incubated with either 1% DMSO, 3 (2 μM), or 5 (10 or 20 μm) for 48 h at 37 °C and were processed for cell cycle analysis as described previously (14).
Binding site reaction
Interaction of 8 and 5 with the colchicine site was measured by competition with a known colchicine site agent which becomes fluorescent when bound (31). Binding reaction consisted of 5 μm mammalian tubulin and 5 μm 2-methoxy-5-(2′,3′,4′-trimethoxyphenyl)tropone (MTPT) in tubulin buffer (0.1 m Pipes, 0.5 mm MgCl2, pH 7.0). 2-Methoxy-5-(2′,3′,4′-trimethoxyphenyl)tropone is a ****wellestablished probe of the colchicine site on tubulin. Fluorescence was excited at 350 nm and emission measured from 380 to 480 nm, using an ISS PC1 spectrofluoromter (ISS Inc., Champaign, IL, USA). The decrease in peak emission intensity was measured following addition of the known competitor dihydrocombretastatin A4 or the test compounds 8 and 5. Decrease in fluorescence was taken as a direct measure of displacement of MTPT.
LCMS analysis of compounds
The DMSO stock solutions of 5, 8, 23 and 25 used in the assays were analysed by LCMS to confirm the identity and purity of the compounds. Compounds were evaluated for purity by separation and detection via LC/UV/MS on an Agilent 1100 liquid chromatography (LC) system linked to a ThermoFinnigan TSQ Quantum Discovery Max triple quadrupole mass spectrometer (MS). Initial MS tuning was completed to identify parent and primary fragment ion masses for each compound. All four compounds were detectable with negative mode electrospray (ESI, compounds 8, 23 and 25) or atmospheric pressure chemical ionization (compound 5). Each compound yielded a parent ion peak with a mass corresponding to the loss of a hydrogen atom, [M–H]−. For evaluation of compound purity, a gradient from 95% water to 95% methanol over 20 min was used to separate injected samples through a Thermo Aquasil C18 column (3 μm, 100 × 2.1 mm). The LC variable wavelength detector was set to 260 nm and MS filters were established for both full scan (100–1500 m/z) and single reaction monitoring (SRM) modes (SRM filters were established to monitor parent >fragment transitions). The DMSO stock compound solutions were diluted to 10 and 50 μM in 50% methanol for LC/MS injection (50 μL). For all sample separations, overlapping primary peaks were observed in the UV and MS filters. All observed peaks were integrated and the area of the primary analyte peak was divided by the total area of all peaks in each chromatogram. The analyte signal represented greater than 99% of the total peak area in each chromatogram. Compound purity was therefore determined to be greater than 99% for each compound evaluated.
Results and Discussion
Tubulin purified from L. tarentolae was used to screen a 10 000 member library of drug-like molecules. The library selected was the CNS set. While leishmaniasis is not a disease of the central nervous system, this library was selected because of the physiochemical properties of its compounds. This library contains compounds that have a high probability of being orally bioavailable, a property that is especially important when developing drugs against neglected diseases where alternative methods of drug administration are often not viable. The assay used was a modification of the established assembly assay which monitors increases in absorbance at 351 nm as an indication of tubulin polymerization (22). The established assay involves testing in a half-area 96-well plate (final assay volume 50 μL) with tubulin at a concentration of 1.5 mg/mL. The use of L. tarentolae, which grows to higher cell density in a less expensive growth medium compared with other Leishmania species, as a source of tubulin means lower collection volumes are required for tubulin isolation (22). We have previously shown that the use of diethylaminoethyl–Sepharose chromatography together with one cycle of assembly–disassembly allows for the ready isolation of assembly competent L. tarentolae tubulin of excellent purity as assessed by SDS-PAGE (22). However, isolating sufficient tubulin to screen 10 000 compounds at the volumes and concentrations reported in the previous 96-well plate assay was undesirable. Selection of a 384-well plate to carry out the primary screen facilitated the use of lower assay volumes, and preliminary experiments showed that the volume could reasonably be reduced to 20 μL (data not shown). In addition, tubulin concentrations of 1.2 mg/mL produced reproducible results (data not shown). However, the rate of Leishmania tubulin assembly still presented an obstacle to screening. Leishmania tubulin assembles rapidly, with complete assembly observed within 3 min (see Figure 2). This makes the simultaneous measurement of tubulin-containing samples in a full 384-well plate difficult, given the time required to both add the assembly solution across the entire plate (see Figure 2) and read the first time-point. Attempts to slow the rate of assembly by modifying the assay conditions proved unsuccessful (data not shown). The final assay conditions used were the result of a series of investigations into optimizing throughput and reproducibility. These included the variation of the selected buffer, magnesium concentrations, pH, DMSO concentration, and the concentration and use of chelating agents. We determined that three columns of the 384-well plate could be tested successfully in each experiment, enough to evaluate 16 compounds with appropriate controls per assay. The use of only three columns not only decreased the time required to add the assembly solution, thereby allowing for an earlier reading of the initial time-point, but also allowed smaller time intervals between data points.
Figure 2.

Assays used to evaluate the 10 000 compound library for activity against Leishmania tubulin. (A) Schematic of the primary assay used for screening. (B) Examples of positive and negative control data obtained in the primary assay. The data shown are taken from validation studies using DMSO as the negative control and GB-II-150 as the positive control. (C) Schematic for the screening process, including secondary assays.
To validate the assay, plates with known assembly inhibitors and DMSO controls were tested (see Figure 2B). The identification of these known inhibitors of Leishmania tubulin assembly indicated that false negatives would not be a concern. While a few false positives were observed in the validation assays, the protocol was not changed because such false positives would be eliminated in a secondary screen where all hits would be retested.
The 10 000 member compound library was then screened at concentrations of 100 μM with one determination per compound. To eliminate false positives, hits were retested at least twice to verify the results. Through this process, 14 compounds were identified as inhibitors of Leishmania tubulin polymerization.
Having identified compounds which were active at 100 μM, the next step was to quantify the activity of the compounds by generating IC50 values against Leishmania tubulin assembly. To provide a direct comparison with antileishmanial tubulin compounds already in the literature, the IC50 determinations were made using the original assay format carried out in half-area 96-well plates and employing tubulin concentrations of 1.5 mg/mL. Most of the hits possess IC50 values in the region 50–90 μm. However, one compound, Chembridge No. 7992831 (5), has an IC50 of 13 μm, making 5 the most interesting hit identified from the antileishmanial screen. Some hits belong to similar structural classes, indicating that modifications to these scaffolds could generate compounds with increased potency. One compound was insoluble at the concentrations required for testing. Given the potential problems generated by these solubility issues, this compound was not studied further. The structures of the 13 hits are shown in Figure 3 and their biological activities are given in Table 1.
Figure 3.

Structures of the 13 hits.
Table 1.
In vitro activity of the 13 hits identified from the screen (μm)
| Compound | IC50 versus Leishmania tarentolae tubulin assembly |
IC50 versus porcine brain tubulin assembly |
IC50 versus L. donovani axenic amastigotes |
IC50 versus Vero Cells |
|---|---|---|---|---|
| 1 | >40 (14) | >40 (14) | 72 ± 10 (14) | |
| 3 | 6.4 ± 1.0 | >40 (16) | 2.3 ± 0.5 (15) | 9.7 ± 1.1 (16) |
| 4 | 6.6 ± 0.7 (17) | >50a (17) | 4.4 ± 0.09 (17) | 16 ± 1 (17) |
| 5 | 13 ± 1 | 38 ± 4 | 11 ± 5 | 29 ± 15 |
| 6 | 81 ± 18 | 120 ± 40 | >50 | >100 |
| 7 | 78 ± 14 | 140 ± 30 | >50 | >100 |
| 8 | 55 ± 5 | 5.2 ± 0.7 | >100 | >50 |
| 9 | 58 ± 7 | 67 ± 11 | >100 | 31 ± 5 |
| 10 | 64 ± 4 | 68 ± 10 | >100 | 37 ± 17 |
| 11 | 68 ± 6 | 63 ± 13 | >100 | >100 |
| 12 | 66 ± 3 | 61 ± 50 | >50 | >100 |
| 13 | 60 ± 11 | 63 ± 30 | >100 | >100 |
| 14 | 89 ± 4 | 111 ± 33 | >100 | >50 |
| 15 | 107 ± 12 | 72 ± 13 | >100 | >100 |
| 16 | 95 ± 11 | 71 ± 13 | >100 | >100 |
| 17 | 70 ± 7 | 70 ± 6 | >100 | >100 |
| Podophyllotoxin | NT | 1.4 ± 0.6 | NT | 0.05 ± 0.03 |
| Pentamidine | NT | NT | 3.2 ± 1.5 | NT |
IC50 values represent the mean ± standard deviation of at least three independent experiments.
NT, not tested.
Compound not soluble at higher concentrations.
Compound selectivity
The selectivity of these hit compounds for Leishmania tubulin compared with the corresponding mammalian protein is important if the hits are to be considered further for antiparasitic drug discovery. Therefore, the IC50 values of the 13 compounds against porcine brain tubulin polymerization were also determined. Compound 5, the most promising compound from the Leishmania tubulin screen, inhibited porcine brain tubulin assembly with an IC50 of 40 μm. Although 5 displays only a threefold selectivity for Leishmania tubulin over mammalian tubulin, greater selectivity may be obtainable following synthetic optimization. Aside from 5, only Chembridge No. 9044486 (6) and Chembridge No. 9056942 (7) displayed any selectivity for Leishmania tubulin (less than twofold). One compound Chembridge No. 9067250 (8) showed significant activity against mammalian tubulin, displaying an IC50 of 5.2 μm. This compound is 10-fold more active against mammalian tubulin than the parasite protein. Although compound 8 is not suitable as an antileishmanial candidate, it may be of interest as an anticancer lead (see below).
The activity of the 13 compounds against L. donovani axenic amastigotes and the cytotoxicity of these agents against Vero cells were also determined. Compound 5 is the most active of the hits against the parasite, exhibiting an IC50 value of 11 μm, similar to its IC50 against Leishmania tubulin. The remaining compounds possessed IC50 values against axenic amastigotes >50 μm. While the majority of the compounds exhibited no toxicity to Vero cells at concentrations up to 100 μm, a few were toxic at lower concentrations. Interestingly, compound 8 was not toxic to Vero cells up to 50 μm concentrations, which is surprising given the effect of this molecule on mammalian tubulin and its activity against mammalian cancer cell lines (see later sections). Compound 5 displayed an IC50 value against Vero cells of 29 μm. Two compounds, Chembridge No. 7946770 (9) and Chembridge No. 7960631 (10), also gave IC50 values in the region of 30 μm. Given that both compounds 9 and 10 have IC50 values in the region of 70 μm against mammalian tubulin, their activity against Vero cells may be due to another target besides tubulin. However, other factors, for example cell permeability, may be the cause of the observed differences in IC50 values between isolated tubulin and Vero cells.
Structural classes within the hits
Among the hits, there are clearly four sets of compounds that are structurally related: (i) 8, 9, 10 and Chembridge No. 9063043 (17); (ii) Chembridge No. 9041709 (11), Chembridge No. 9035650 (12), Chembridge No. 9035165 (13) and Chembridge No. 7944936 (16); (iii) Chembridge No. 9026860 (14) and 7; (iv) Chembridge No. 9049411 (15) and 6. Class (i) contains N-substituted benzamides. Two of the compounds in this class, 9 and 10, show IC50 values in the region of 60 μm against both Leishmania and mammalian tubulin. One definite trend is that these two compounds both have IC50 values against Vero cells in the region of 30 μm, suggesting that these two compounds may possesses activity against mammalian cells through mechanism(s) other than or in addition to inhibiting tubulin assembly. However, it should also be considered that other effects, such as drug accumulation, could be the cause of these differences in the IC50 values. Class (ii) contains substituted aryl 4-piperazin-1-yl compounds. Three of these compounds, 11, 12 and 13, contain a 2-phenyl-1,3-oxazole-4-carbonitrile functional group, all these agents have IC50 values against both Leishmania and mammamlian tubulin of approximately 60 μm. This is in contrast to 16 which gives much lower IC50 values, indicating the significance of the 2-phenyl-1,3-oxazole-4-carbonitrile group. Compound 12 showed slightly increased activity against L. donovani axenic amastigotes, displaying toxicity to the parasites at 50 μm. Class (iii) compounds contain a 1,2,4-oxadiazol-5-yl ring system. These compounds have lower activity against Leishmania tubulin with IC50s around 80 μm. They are some of the least active against mammalian tubulin with IC50s of over 100 μm. Interestingly, compound 7 shows slightly improved activity against L. donovani axenic amastigotes, displaying toxicity to the parasites at 50 μm, whereas compound 14 has slightly better activity against Vero cells. The agents in class (iv) contain a thioacetamide unit but differ in the ring system attached to the sulphur atom. These compounds possess low activity in both the tubulin and cell-based assays.
Compound 5, the most potent against Leishmania tubulin assembly
Compound 5 is the most promising antileishmanial compound of the hits identified, with its IC50 of 13 μm against Leishmania tubulin and threefold selectivity compared with mammalian tubulin (see Figure 4 for concentration-dependent inhibition of Leishmania tubulin assembly by 5).
Figure 4.
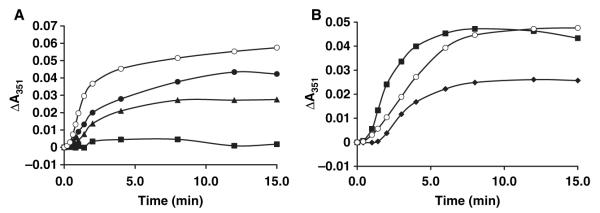
Inhibition of the assembly of Leishmania tubulin (A) and mammalian tubulin (B) by compound 5. The assembly of 1.5 mg/mL purified Leishmania and mammalian tubulin was assessed as described previously (15,22,29) in the absence (open circles) or presence of compound 5 at 5 (closed circles), 10 (triangles), 20 (squares) or 40 μm (diamonds). Data shown are from a representative experiment performed on three separate occasions.
We suspected that the observed antileishmanial properties of this compound were related to its ability to inhibit tubulin polymerization. To test this hypothesis, compound 5 was incubated with L. donovani promastigotes at 10 and 20 μm for 48 h, then cell cycle analysis was performed to determine if 5 arrested cells in mitosis (see Figure 5). Compound 5 appears to have little effect on the cell cycle of L. donovani when compared with the DMSO control. This is even more evident when compared with 3, a known Leishmania tubulin inhibitor, tested at 2 μm under the same experimental conditions, which clearly shows a dramatic increase in the percentage of cells in the G2/M phase compared with G1 and a number of cells containing four times the amount of DNA observed in G1. The results observed for compound 3 in this experiment were consistent with data previously observed for this class of compounds (15). Thus, although compound 5 clearly inhibits Leishmania tubulin polymerization in vitro, its inhibition of parasite growth could be due to an off target effect. Additionally, compound 5 was assayed against T. b. brucei, a related parasite, and surprisingly showed no activity at 100 μm.
Figure 5.
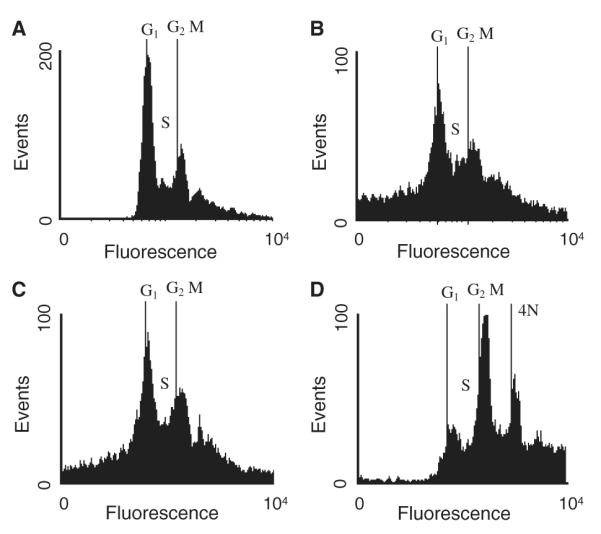
Cell cycle analysis of Leishmania donovani promastigotes treated with compounds 3 and 5. After 48 h incubation in the presence of 1% DMSO (A) 5 at 20 μm (B), 5 at 10 μm (C) or 3 at 2 μm (D), parasites were fixed, stained with propidium iodide, and analysed by flow cytometry as described in the Experimental section.
SAR information around compound 5 from other compounds in the library
The compound library that was screened contains more than one compound per structural class. Given the knowledge that any compound not identified as a hit is inactive at 100 μm, some preliminary SAR information can be obtained by searching the library for compounds with structures related to the hits. To acquire some information regarding the SAR for Leishmania tubulin assembly inhibition around compound 5, the library was initially searched for compounds containing substructure X shown in Figure 6. There are 40 compounds in the library with this substructure, including 19 compounds with a halogen at the 5th position. The most noticeable trend is that only one compound other than the hit contains the propyn-1-yoloxy group, Chembridge No. 7905398 (18). Given that the structure of this compound differs significantly in two of the other functional groups, it is difficult to make any conclusions from this compound with regards the requirement of the propyn-1-yoloxy group. However, two compounds Chembridge No. 7983651 (19) and Chembridge No. 7961265 (20) are also present in the library; the inactivity of these compounds where the only structural difference is in the propyn-1-yoloxy group implies that this moiety may be required for activity. There are seven compounds where only the functional group of the 4th position oxygen is different and none of these displayed any activity. This supports the hypothesis that the propyn-1-yoloxy group is required for activity. The inactivity of 18 would suggest that the propyn-1-yoloxy group is not the only requirement for activity and that the halogen and/or the cyano group are also required for activity. Further investigations into the SAR of compound 5 should examine the activity of compounds where individual groups are held constant and the other groups are systematically modified.
Figure 6.
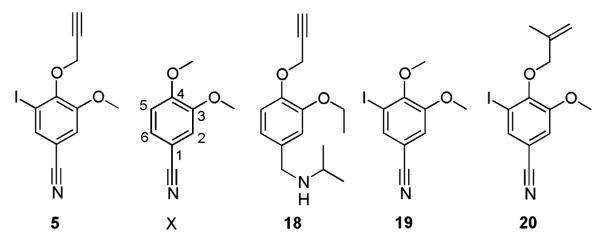
Selected analogues of compound 5 present in the compound library.
Compound 8 has potent activity against mammalian tubulin
Compound 8 was identified as a potent inhibitor of mammalian tubulin assembly. While identifying inhibitors of mammalian tubulin was not the primary goal of this work, the potency of compound 8 against mammalian tubulin assembly (see Figure 7) prompted us to investigate the activity of this compound against a panel of cancer cell lines. The IC50 values against most of these cell lines were approximately 1 μm (see Table 2). These results indicate that compound 8 is worthy of further consideration as an anticancer lead compound and encouraged us to examine other related compounds from the library for their effects on mammalian tubulin.
Figure 7.
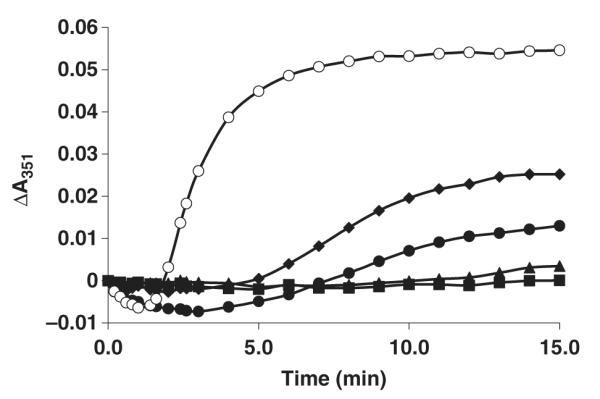
Inhibition of mammalian tubulin assembly by compound 8. The assembly of 1.5 mg/mL purified mammalian tubulin was assessed as described previously (13) in the absence (open circles) or presence of compound 8 at 2.5 (diamonds), 5 (closed circle), 10 (triangles) or 20 μm (squares). Data shown is from a representative experiment performed on three separate occasions.
Table 2.
The activity of compounds 8, 23 and 25 against several mammalian cancer cell lines
| Cancer cell line IC50 (μm) |
||||||
|---|---|---|---|---|---|---|
| Compound | HT29 | LNCaP | PPC1 | PC3 | MCF7 | TSU-Pr1 |
| 8 | 1.9 ± 0.2 | 1.6 ± 0.4 | 1.2 ± 0.3 | 3.5 ± 1.1 | 1.3 ± 0.2 | 1.0 ± 0.6 |
| 23 | 0.71 ± 0.03 | 0.20 ± 0.02 | 0.37 ± 0.04 | 0.79 ± 0.07 | 0.18 ± 0.00 | 0.33 ± 0.02 |
| 25 | 5.0 ± 0.1 | 0.88 ± 0.10 | 1.5 ± 0.2 | 5.8 ± 0.5 | 0.74 ± 0.03 | 1.2 ± 0.2 |
| Vinblastine (nm) | 2.4 ± 0.2 | 3.4 ± 0.9 | 1.0 ± 0.4 | 1.4 ± 0.3 | 1.3 ± 0.4 | 1.6 ± 0.08 |
SAR information around compound 8 from other compounds in the library
The library was searched for analogues of compound 8 which could also be tested against mammalian tubulin to gain some information regarding the SAR. Aromatic sulphonamides are a common structural motif in the library as evident by the identification of 179 compounds with the substructure Y (see Figure 8). This search was refined to identify compounds with substituents and substitution patterns on the aromatic ring systems similar to those of compound 8.
Figure 8.

Structures of analogues of compound 8 present in the library.
Compounds 21–27, shown in Figure 8, were identified as being closely structurally related to compound 8 and were tested against mammalian tubulin at concentrations of 100 and 50 μm. Two compounds Chembridge No. 7987157 (25) and Chembridge No. 9048759 (23) showed activity at both concentrations, while Chembridge No. 7973517 (27) displayed activity at 100 μm. All other analogues of compound 8 exhibited little or no effect on the assembly of mammalian tubulin. Determination of the IC50 values for compounds 25 and 23 revealed that they have greater potency than compound 8, with values of 2.8 and 1.1 μm, respectively. These results suggest that the methyl ester is important for activity. The potent activity of compound 23 shows that the 3′ position fluorine is not required for activity. Given that both compounds 23 and 25 are more active than compound 8, having the methyl ester in the 3rd position may increase activity. However, the inactivity of Chembridge No. 7951602 (24), which is a regioisomer of compound 25, suggests that the presence of the methyl ester is not the only requirement for activity and there could be conformational and/or steric demands. The inactivity of Chembridge No. 7973060 (26) could further indicate the importance of the position of the ester group or could demonstrate the importance of the methoxy group at the 4′ position. Further investigations should involve the synthesis and evaluations of derivatives with the methyl ester in the ortho, para and meta positions to directly determine the effect of the position of this group on activity.
Other sulphonamides have been shown to possess activity against mammalian tubulin. In particular, the compounds E7010 (ABT-751) (28), T-138067 (29) and T-900607 (30) are in clinical trials (32–39). Recent publications have reported the results of phase I and phase II clinical trials with ABT-751 (28), which is orally bioavailable (37,39). Reports on T-138067 (29) in phase II clinical trials have shown that this compound displays low levels of toxicity (35,38). Structural similarities between these compounds and other sulphonamides with antitubulin activity and compound 8 are evident (see Figure 9). Interestingly, compound 29 has a methoxy group in the 4th position and a fluorine in the 3rd (compared with 4′ and 3′ for compound 8). Compound 29 has an IC50 value in the region of 2 μm against mammalian tubulin and IC50 values from 11 to 165 nm against a panel of cancer cell lines (40). The activity against mammalian tubulin is similar to that of compound 8 (5 μm); however, it shows greater potency against cancer cell lines (1 μm in the case of compound 8). Likewise, compound 28 has an IC50 against mammalian tubulin of 2.2 μm and IC50s against tumour cell lines of 0.06–0.8 μg/mL (0.16–2.2 μm) (41), similar to the values obtained for compound 23 in this study.
Figure 9.

Structures of sulphonamides with known antitubulin activity (33,42,43,45).
One of the other striking similarities with regard to the structures is the presence of a methoxy group in the para position. The importance of this functional group is particularly illustrated when comparing the activities of compounds 30–32 (42,43). Compound 32, while active against cancer cell lines, has an IC50 of >100 μm against mammalian tubulin and is proposed to interfere with the G1 phase of the cell cycle rather than G2/M. However, compound 31, the methoxy analogue of 32, has an IC50 of 9.5 μm against mammalian tubulin. Additionally, compound 30 has an IC50 of 2.1 μm against mammalian tubulin, again highlighting the importance of the methoxy group. The difference in activity between compounds 30 and 31 illustrates the importance of the other functional groups. These observations are consistent with the data obtained for compound 8 and its analogues.
Investigating the mammalian tubulin binding sites of compounds 8 and 5
Compounds 28 and 29 are known to bind to the colchicine site of mammalian tubulin (41,44). If compound 8 binds to the same site as these compounds as expected, the observed increased activity of these analogues against mammamlian tubulin compared with Leishmania tubulin would be consistent with the inactivity of classical colchicine site agents against Leishmania tubulin (41,44).
Competition studies were performed with mammalian tubulin and MTPT, an established probe for the colchicine site on tubulin which becomes fluorescent upon binding (31) (see Table 3). Incubation of MTPT with mammalian tubulin in the presence of 2, 20 or 50 μm compound 8 caused a dose-dependent inhibition in MTPT binding as measured by a decrease in observed fluorescence. Inhibition by compound 8 was less than, but comparable with, that observed with dihydrocombretastatin A4, a known colchicine site ligand. This lends support to the argument that compound 8 and its derivatives bind tubulin at the colchicine binding site, thus explaining the increased activity of compound 8 against mammalian tubulin over Leishmania tubulin. Compound 5 had no effect on the binding of MTPT indicating that the observed inhibition of polymerization (Table 1) is caused by a binding site on mammalian tubulin different from the colchicine site. Given its increased potency against Leishmanial tubulin over mammalian tubulin, this was expected.
Table 3.
Displacement of colchicine site ligand binding of mammalian tubulin by competitors
| Inhibition (%) of MTPT binding in presence of competitors |
|||
|---|---|---|---|
| 2 μm | 20 μm | 50 μm | |
| Dihydrocombetastatin A4 | 30 ± 1 | 69 ± 2 | 74 ± 2 |
| 8 | 12 ± 1 | 47 ± 1 | 51 ± 1 |
| 5 | 0 ± 1 | 0 ± 2 | 0 ± 2 |
Although the number of antileishmanial hits identified from this screen was rather disappointing and the level of activity observed from the majority of these hits was also less than desired, the screen successfully identified the interesting antimicrotubule compounds 5, 8, 23 and 25, the identity and purity of which were confirmed by LCMS. In addition, the structural similarity of some of the hits may indicate new classes of molecules which have the potential to be optimized to produce improved lead compounds.
Conclusion
Our screening efforts have identified one compound with good activity against Leishmania tubulin assembly, 5. Compound 5 shows threefold selectivity for Leishmania tubulin over mammalian tubulin. This selectivity could improve through the systematic synthesis of analogues. While this compound also showed activity against L. donovani axenic amastigotes, cell cycle analysis revealed that inhibition of tubulin may not be the primary cause of activity. Despite the questions over the mechanism of action of compound 5 against amastigotes, this is still a promising compound with potential as an antileishmanial lead.
Compound 8 was identified as an inhibitor of mammalian tubulin assembly. Further investigations revealed its promising activity against a number of cancer cell lines. Two analogues of compounds 8, 23 and 25 were later identified from the library as compounds with increased potency against mammalian tubulin, and compound 23 also showed excellent activity against several cancer cell lines. These compounds are structurally similar to E7010 (28) and T-138067 (29), two compounds currently in clinical trials. Compounds 28 and 29 bind at the colchicine binding site, suggesting the same is true of compound 8 and its analogues. This is supported by the observed inhibition of MTPT binding by compound 8. The SAR information surrounding compound 8 and the identification of the more potent analogues provides a good framework around which to design SAR studies. There is still some potential for improvement in the tolerance to cycles of treatment for ABT-751 (39) and clinical effectiveness for T-138067 (35,38). Despite the structural similarities between ABT-751 and T-138067, the former compound appears to be more effective in cancer trials. Compounds 8, 23 and 25 may thus provide structural clues for the discovery of tubulin-targeted anticancer agents that might possess subtle differences in pharmacokinetic properties, tumor cell potency and host cell toxicity compared with ABT-751 and T-138067. Such differences could translate into improved efficacy against some forms of cancer.
Acknowledgments
This work was funded by NIH Grant R01 AI 061021 (to K.A.W.). The contribution of D.L.S. was supported by the intramural research program of the National Institute of Child Health and Human Development, NIH. We also thank Dr Chenglong Li for helpful discussions.
Footnotes
References
- 1.Pellegrini F, Budman DR. Review: tubulin function, action of antitubulin drugs, and new drug development. Cancer Invest. 2005;23:264–273. doi: 10.1081/cnv-200055970. [DOI] [PubMed] [Google Scholar]
- 2.Zhou J, Giannakakou P. Targeting microtubules for cancer chemotherapy. Curr Med Chem Anticancer Agents. 2005;5:65–71. doi: 10.2174/1568011053352569. [DOI] [PubMed] [Google Scholar]
- 3.Martin RJ, Robertson AP, Bjorn H. Target sites of anthelmintics. Parasitology. 1997;114:S111–S124. [PubMed] [Google Scholar]
- 4.Downing KH, Nogales E. Tubulin structure: insights into microtubule properties and functions. Curr Opin Struct Biol. 1998;8:785–791. doi: 10.1016/s0959-440x(98)80099-7. [DOI] [PubMed] [Google Scholar]
- 5.Kline-Smith SL, Walczak CE. Mitotic spindle assembly and chromosome segregation: refocusing on microtubule dynamics. Mol Cell. 2004;15:317–327. doi: 10.1016/j.molcel.2004.07.012. [DOI] [PubMed] [Google Scholar]
- 6.Amos LA, Schlieper D. Microtubules and maps. Adv Protein Chem. 2005;71:257–298. doi: 10.1016/S0065-3233(04)71007-4. [DOI] [PubMed] [Google Scholar]
- 7.Wilson L, Miller HP, Farrell KW, Snyder KB, Thompson WC, Purich DL. Taxol stabilization of microtubules in vitro: dynamics of tubulin addition and loss at opposite microtubule ends. Biochemistry. 1985;24:5254–5262. doi: 10.1021/bi00340a045. [DOI] [PubMed] [Google Scholar]
- 8.Skoufias DA, Wilson L. Mechanism of inhibition of microtubule polymerization by colchicine: inhibitory potencies of unliganded colchicine and tubulin-colchicine complexes. Biochemistry. 1992;31:738–746. doi: 10.1021/bi00118a015. [DOI] [PubMed] [Google Scholar]
- 9.Himes RH. Interactions of the catharanthus (Vinca) alkaloids with tubulin and microtubules. Pharmacol Ther. 1991;51:257–267. doi: 10.1016/0163-7258(91)90081-v. [DOI] [PubMed] [Google Scholar]
- 10.David-Pfeuty T, Simon C, Pantaloni D. Effect of antimitotic drugs on tubulin GTPase activity and self-assembly. J Biol Chem. 1979;254:11696–11702. [PubMed] [Google Scholar]
- 11.Lubega GW, Ochola OK, Prichard RK. Trypanosoma brucei: anti-tubulin antibodies specifically inhibit trypanosome growth in culture. Exp Parasitol. 2002;102:134–142. doi: 10.1016/s0014-4894(03)00035-3. [DOI] [PubMed] [Google Scholar]
- 12.Ngô H, Tschudi C, Gull K, Ullu E. Double-stranded RNA induces mRNA degradation in Trypanosoma brucei. Proc Natl Acad Sci USA. 1998;95:14687–14692. doi: 10.1073/pnas.95.25.14687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chan MM, Triemer RE, Fong D. Effect of the anti-microtubule drug oryzalin on growth and differentiation of the parasitic protozoan Leishmania mexicana. Differentiation. 1991;46:15–21. doi: 10.1111/j.1432-0436.1991.tb00861.x. [DOI] [PubMed] [Google Scholar]
- 14.Werbovetz KA, Sackett DL, Delf n D., Bhattacharya G, Salem M, Obrzut T, Rattendi D, Bacchi C. Selective anti-microtubule activity of N1-phenyl-3,5-dinitro-N4,N4-di-n-propylsulfanilamide (GB-II-5) against kinetoplastid parasites. Mol Pharmacol. 2003;64:1325–1333. doi: 10.1124/mol.64.6.1325. [DOI] [PubMed] [Google Scholar]
- 15.Bhattacharya G, Herman J, Delf n D., Salem M, Barszcz T, Mollet M, Riccio G, Brun R, Werbovetz KA. Synthesis and Antitubulin Activity of N1- and N4-Substituted 3,5-dinitro sulfanilamides against African trypanosomes and Leishmania. J Med Chem. 2004;47:1823–1832. doi: 10.1021/jm0304461. [DOI] [PubMed] [Google Scholar]
- 16.George TG, Johnsamuel J, Delfn DA, Yakovich A, Mukherjee M, Phelps MA, Dalton JT, Sackett DL, Kaiser M, Brun R, Werbovetz KA. Antikinetoplastid antimitotic activity and metabolic stability of dinitroaniline sulfonamides and benzamides. Bioorg Med Chem. 2006;14:5699–5710. doi: 10.1016/j.bmc.2006.04.017. [DOI] [PubMed] [Google Scholar]
- 17.George TG, Endeshaw MM, Morgan RE, Mahasenan KV, Delfn DA, Mukherjee MS, Yakovich AJ, Fotie J, Li C, Werbovetz KA. Synthesis, biological evaluation, and molecular modeling of 3,5-substituted-N1-phenyl-N4,N4-di-n-butylsulfanilamides as antikinetoplastid antimicrotubule agents. Bioorg Med Chem. 2007;15:6071–6079. doi: 10.1016/j.bmc.2007.06.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu D, George TG, Hurh E, Werbovetz KA, Dalton JT. Pre-systemic metabolism prevents in vivo antikinetoplastid activity of N1,N4-substituted 3,5-dinitro sulfanilamide, GB-II-150. Life Sci. 2006;79:1081–1093. doi: 10.1016/j.lfs.2006.03.028. [DOI] [PubMed] [Google Scholar]
- 19.Dawson P, Gutteridge W, Gull KA. Comparison of the interaction of anhelmintic benzimidazoles with tubulin isolated from mammalian tissue and the parasitic nematode Ascaridia galli. Biochem Pharmacol. 1984;33:1069–1074. doi: 10.1016/0006-2952(84)90515-x. [DOI] [PubMed] [Google Scholar]
- 20.Lacey E. Mode of action of benzimidazoles. Parasitol Today. 1990;6:112–115. doi: 10.1016/0169-4758(90)90227-u. [DOI] [PubMed] [Google Scholar]
- 21.Robinson MW, McFerran N, Trudgett A, Hoey L, Fairweather I. A possible model of benzimidazole binding to ****betatubulin disclosed by invoking an inter-domain movement. J Mol Graph Model. 2004;23:275–284. doi: 10.1016/j.jmgm.2004.08.001. [DOI] [PubMed] [Google Scholar]
- 22.Yakovich AJ, Ragone FL, Alfonzo JD, Sackett DL, Werbovetz KA. Leishmania tarentolae: purification and characterization of tubulin and its suitability for antileishmanial drug screening. Exp Parasitol. 2006;114:289–296. doi: 10.1016/j.exppara.2006.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baldwin J, Michnoff CH, Malmquist NA, White J, Roth MG, Rathod PK, Phillips MA. High-throughput screening for potent and selective inhibitors of Plasmodium falciparum dihydroorotate dehydrogenase. J Biol Chem. 2005;280:21847–21853. doi: 10.1074/jbc.M501100200. [DOI] [PubMed] [Google Scholar]
- 24.St. George S, Bishop JV, Titus RG, Selitrennikoff CP. Novel compounds active against Leishmania major. Antimicrob Agents Chemother. 2006;50:474–479. doi: 10.1128/AAC.50.2.474-479.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Mackey ZB, Baca AM, Mallari JP, Apsel B, Shelat A, Hansell EJ, Chiang PK, Wolff B, Guy KR, Williams J, McKerrow JH. Discovery of trypanocidal compounds by whole cell HTS of Trypanosoma brucei. Chem Biol Drug Des. 2006;67:355–363. doi: 10.1111/j.1747-0285.2006.00389.x. [DOI] [PubMed] [Google Scholar]
- 26.Weisman JL, Liou AP, Shelat AA, Cohen FE, Guy RK, DeRisi JL. Searching for new antimalarial therapeutics amongst known drugs. Chem Biol Drug Des. 2006;67:409–416. doi: 10.1111/j.1747-0285.2006.00391.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Baniecki ML, Wirth DF, Clardy J. High-throughput Plasmodium falciparum growth assay for malaria drug discovery. Antimicrob Agents Chemother. 2007;51:716–723. doi: 10.1128/AAC.01144-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Martyn DC, Jones DC, Fairlamb AH, Clardy J. High-throughput screening affords novel and selective trypanothione reductase inhibitors with anti-trypanosomal activity. Bioorg Med Chem Lett. 2007;17:1280–1283. doi: 10.1016/j.bmcl.2006.12.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Werbovetz KA, Brendle JJ, Sackett DL. Purification, characterization and drug susceptibility of tubulin from Leishmania. Mol Biochem Parasitol. 1999;98:53–65. doi: 10.1016/s0166-6851(98)00146-7. [DOI] [PubMed] [Google Scholar]
- 30.Salem MM, Werbovetz KA. Antiprotozoal compounds from Psorothamnus polydenius. J Nat Prod. 2005;68:108–111. doi: 10.1021/np049682k. [DOI] [PubMed] [Google Scholar]
- 31.Hastie SB. Interactions of colchicine with tubulin. Pharmacol Ther. 1991;51:377–401. doi: 10.1016/0163-7258(91)90067-v. [DOI] [PubMed] [Google Scholar]
- 32.Hu L, Li Z, Li Y, Qu J, Ling Y-H, Jiang J, Boykin DW. Synthesis and structure-activity relationships of carbazole sulf-onamides as a novel class of antimitotic agents against solid tumors. J Med Chem. 2006;49:6273–6282. doi: 10.1021/jm060546h. [DOI] [PubMed] [Google Scholar]
- 33.Yoshino HB, Ueda N, Niijima J, Sugumi H, Kotake Y, Koyanagi N, Yoshimatsu K, Asada M, Watanabe T, Nagaau T, Tsukahara K, Iijima A, Kitoh K. Novel sulfonamides as potential, systemically active antitumor agents. J Med Chem. 1992;35:2496–2497. doi: 10.1021/jm00091a018. [DOI] [PubMed] [Google Scholar]
- 34.Fox E, Maris JM, Widemann BC, Meek K, Goodwin A, Goodspeed W, Kromplewski M, Fouts ME, Medina D, Cho SY, Cohn SL, Krivoshik A, Hagey AE, Adamson PC, Balis FM. A phase 1 study of ABT-751, an orally bioavailable tubulin inhibitor, administered daily for 7 days every 21 days in pediatric patients with solid tumors. Clin Cancer Res. 2006;12:4882–4887. doi: 10.1158/1078-0432.CCR-06-0534. [DOI] [PubMed] [Google Scholar]
- 35.Berlin JD, Venook A, Bergsland E, Rothenberg M, Lockhart AC, Rosen L. Phase II trial of T138067, a novel microtubule inhibitor, in patients with metastatic, refractory colorectal carcinoma. Clin Colorectal Cancer. 2008;7:44–47. doi: 10.3816/CCC.2008.n.006. [DOI] [PubMed] [Google Scholar]
- 36.Gelmon KA, Belanger K, Soulieres D, Britten C, Chia S, Charpentier D, Chi K, Powers J, Walsh W, Seymour L. A phase I study of T900607 given once every 3 weeks in patients with advanced refractory cancers; National Cancer Institute of Canada Clinical Trials Group (NCIC-CTG) IND 130. Invest New Drugs. 2005;23:445–453. doi: 10.1007/s10637-005-2904-2. [DOI] [PubMed] [Google Scholar]
- 37.Fox E, Maris JM, Widemann BC, Goodspeed W, Goodwin A, Kromplewski M, Fouts ME, Medina D, Cohn SL, Krivoshik A, Hagey AE, Adamson PC, Balis FM. A phase I study of ABT-751, an orally bioavailable tubulin inhibitor, administered daily for 21 days every 28 days in pediatric patients with solid tumors. Clin Cancer Res. 2008;14:1111–1115. doi: 10.1158/1078-0432.CCR-07-4097. [DOI] [PubMed] [Google Scholar]
- 38.Kirby S, Gertler SZ, Mason W, Watling C, Forsyth P, Aniagolu J, Stagg R, Wright M, Powers J, Eisenhauer EA. Phase 2 study of T138067-sodium in patients with malignant glioma: trial of the National Cancer Institute of Canada Clinical Trials Group. Neuro Oncol. 2005;7:183–188. doi: 10.1215/S1152851704000602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mauer AM, Cohen EE, Ma PC, Kozloff MF, Schwartzberg L, Coates AI, Qian J, Hagey AE, Gordon GB. A phase II study of ABT-751 in patients with advanced non-small cell lung cancer. J Thorac Oncol. 2008;3:631–636. doi: 10.1097/JTO.0b013e318174e01f. [DOI] [PubMed] [Google Scholar]
- 40.Shan B, Medina JC, Santha E, Frankmoelle WP, Chou T-C, Learned RM, Narbut MR, Stott D, Wu P, Jaen JC, Rosen T, Timmermans PB, Beckmann H. Selective, covalent modification of β-tubulin residue Cys-239 by T138067, an antitumor agent with in vivo efficacy against multidrug-resistant tumors. Proc Natl Acad Sci USA. 1999;96:5686–5691. doi: 10.1073/pnas.96.10.5686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yoshimatsu K, Yamaguchi A, Yoshino H, Koyanagi N, Kitoh K. Mechanism of action of E7010, an orally active sulfonamide antitumor agent: inhibition of mitosis by binding to the colchicine site of tubulin. Cancer Res. 1997;57:3208–3213. [PubMed] [Google Scholar]
- 42.Owa T, Yokoi A, Yamazaki K, Yoshimatsu K, Yamori T, Nagasu T. Array-based structure and gene expression relationship study of antitumor sulfonamides including N-[2-[(4-hydroxyphenyl)amino]-3-pyridinyl]-4-methoxybenzenesulfonamide and N-(3-chloro-7-indolyl)-1,4-benzenedisulfonamide. J Med Chem. 2002;45:4913–4922. doi: 10.1021/jm0201060. [DOI] [PubMed] [Google Scholar]
- 43.Owa T, Okauchi T, Yoshimatsu K, Sugi NH, Ozawa Y, Nagasu T, Koyanagi N, Okabe T, Kitoh T, Yoshino H. A focused compound library of novel N-(7-indolyl)benzenesulfonamides for the discovery of potent cell cycle inhibitors. Bioorg Med Chem Lett. 2000;10:1223–1226. doi: 10.1016/s0960-894x(00)00219-5. [DOI] [PubMed] [Google Scholar]
- 44.Banerjee M, Poddar A, Mitra G, Surolia A, Owa T, Bhattacharyya B. Sulfonamide drugs binding to the colchicine site of tubulin: thermodynamic analysis of the drug–tubulin interactions by isothermal titration calorimetry. J Med Chem. 2005;48:547–555. doi: 10.1021/jm0494974. [DOI] [PubMed] [Google Scholar]
- 45.Liou JP, Hsu KS, Kuo CC, Chang CY, Chang JY. A novel oral indoline-sulfonamide agent, N-[1-(4-methoxybenzenesulfonyl)-2,3-dihydro-1H-indol-7-yl]-isonicotinamide (J30), exhibits potent activity against human cancer cells in vitro and in vivo through the disruption of microtubule. J Pharmacol Exp Ther. 2007;323:398–405. doi: 10.1124/jpet.107.126680. [DOI] [PubMed] [Google Scholar]