

Abstract
The social and medical costs of the biological aging process are high and will rise rapidly in coming decades, creating an enormous challenge to societies worldwide. In recent decades, researchers have expanded their understanding of the underlying deleterious structural and physiological changes (aging damage) that underlie the progressive functional impairments, declining health, and rising mortality of aging humans and other organisms and have been able to intervene in the process in model organisms, even late in life. To preempt a global aging crisis, we advocate an ambitious global initiative to translate these findings into interventions for aging humans, using three complementary approaches to retard, arrest, and even reverse aging damage, extending and even restoring the period of youthful health and functionality of older people.
INTRODUCTION
Age is the greatest risk factor for most major chronic diseases in the industrialized world and to an increasing degree in the developing world. After adolescent development, functionality declines progressively with age (1), and mortality rates increase exponentially, doubling roughly every 7 to 8 years after puberty. This exponentiality manifests as a progressive, roughly synchronous rise in the incidence of disease, disability, and death from chronic diseases beginning after midlife (examples in Fig. 1) and suggests a causal—rather than a casual—relationship.
Fig. 1. Chronic diseases and aging.

The incidence of major chronic diseases rises exponentially with age, as shown: cardiovascular disease (blue squares) [data from (32)], cancer (red diamonds) [data from (32)], AD (gray squares) [data from (33)], and influenza-associated hospitalization (green triangles) [data from (34)]. Incidence rates are normalized to the first data point.
The physiological basis of these phenomena lies in the progressive lifelong accumulation of deleterious changes in the structure of the body at the molecular, cellular, and tissue levels. These changes (aging damage) arise primarily as damaging side effects of normal metabolism, aggravated by environmental toxins and unhealthy lifestyle. Aging damage contributes to pathology either directly (by impairing the function of specific biomolecules) or indirectly [by eliciting cellular or systemic responses that generally serve near-term protective functions but ultimately are deleterious (2, 3)]. As damage accumulates, organisms suffer progressively diminished functionality, homeostasis, and plasticity, reducing the capacity to survive and recover from environmental challenge. These changes both contribute etiopathologically to specific age-related diseases and increase the organism’s vulnerability to other insults that contribute to them, leading to increasing morbidity and mortality.
The surprising conclusion from the past two decades of research on biological aging is that aging is plastic: Within a species, maximum life span is not fixed but can be increased by dietary manipulation [particularly calorie restriction (CR) (4)] or genetic manipulation [particularly dampened insulin/insulin-like growth factor–1 signaling (IIS) (5)]. These interventions generally reduce the generation, enhance the repair, and/or increase the tolerance of the molecular and cellular damage of aging. Although our ability to assess “health span” in model organisms remains incomplete (6), these interventions generally preserve “youthful” functionality in regard to tested parameters and reduce the incidence of age-related disease.
There have long been calls (7, 8) for greater efforts to translate this research into clinical interventions to expand the healthy, productive period of human life. By targeting the aging damage that is responsible for the age-related rise in disease vulnerability, such interventions would reduce the incidence of most, if not all, age-related diseases in unison, by modulating the underlying biology that drives them all, rather than treating each in isolation, as in conventional medicine. To date, however, investments in such research by the National Institutes of Health (NIH) and its international equivalents have been disproportionately low relative to their potential return; for example, the NIH $28 billion budget allocates <0.1% (7)—perhaps as little as $10 million—to research on biological aging. Contrast this allocation with the costs of medical care for today’s aged, such as the current Medicare budget of $430 billion, and with projected outlays many times that number to treat future increases in the diseases of aging.
Calls for an intensive agenda of research on the biology of aging have particular salience today because of two converging trends: one demographic and one scientific. Demographically, we are entering a period of unprecedented global aging, as the ratio of retired elderly to younger workers increases dramatically within the next decades in both developing and industrialized nations (9). Age-related disease and disability greatly increase medical costs, even when adjusted for survivorship, and are major determinants of the decline in productivity and labor force participation after midlife. Thus, the results of biological aging are both a rise in social costs and a decrease in a national workforce’s ability to produce the goods and services necessary to meet those costs. The costs of global aging to individuals and societies are therefore high and are projected to inflate into an unprecedented economic and social challenge in coming decades.
Scientifically, this phenomenon coincides with the first robust reports of effective interventions into the biological aging of mammals that are already in late middle age when treatment begins. In 2004, CR was first shown to extend life span in mice as old as 19 months (10), which is broadly equivalent to the current average age of postwar “baby boomers.” And 2009 saw the first demonstration of pharmacological intervention into the biological aging of similaraged mice, with preliminary evidence of delays in cancer incidence and other changes in gross pathology (11).
Intervention in the degenerative aging process need only lead to a simple delay in the appearance of age-related disability and rising medical costs in order to alleviate the projected social costs and challenge of global demographic aging. This alone would increase the ratio between productive workers of all ages and the dependent frail elderly, simultaneously expanding the resources available to bear the costs of supporting a subpopulation of frail elderly and reducing the relative size of that subpopulation during the critical period of demographic transition. The benefit to be gained from intervention in biological aging would be even greater, however, if it were able to not only delay the onset but reduce the absolute ultimate burden of age-related disease. Preliminary evidence from animal models of retarded age-related degeneration [for example, (12, 13)] and the identification of human subpopulations characterized by extreme survivorship with surprisingly little morbidity (14) (possibly indicative of a phenotype of slow biological aging) suggest that such intervention might have this even more beneficial effect. Whether it would actually do so, however, is uncertain.
Preliminary glimpses of the benefit to be anticipated from therapeutics targeting the underlying degenerative aging process can be gleaned from two studies performed a quarter-century apart (15, 16). Recently, Manton et al. (15) demonstrated that, by improving the health of older adults, investment in conventional medical technology in the late 20th century buffered projected declines in labor force productivity and thereby contributed significantly to economic growth. Such investment thereby constrained the growth of health care costs as a share of gross domestic product, effectively paying for itself; the authors provide analysis to suggest that ongoing investments can be projected to continue to do so. Economic modeling performed independently in the 1980s (16) indicated that even greater economic benefits can be expected from interventions that successfully slow the rate of biological aging. But this analysis is probably an underestimate, because it preceded and does not factor in the rapid rise in dependency ratios that lies ahead today, the alleviation of which represents a significant part of the benefits now projected to be realized by expanding investment in even conventional medical technology (15). Incorporating this new demographic challenge into the analysis of the economic impact of interventions targeting the underlying degenerative aging process would clearly substantially amplify the benefits to be expected.
In light of these convergent scientific and demographic phenomena, we advocate an intensive, dedicated, and focused R&D agenda by developed and rapidly developing nations globally, to devise interventions to restore and maintain the health and functionality of humans in late middle age and older.
RESEARCH ROADMAP
Our consensus is that a realistic path toward this goal exists, by targeting age-associated changes that, based on existing research, are known or thought to be important primary components of human age-related degeneration and thus drivers of vulnerability to age-related disease. Here we outline such an agenda, focusing on targets that are likely to be biomedically tractable, even later in life, and would make efficient use of intellectual, capital, and temporal resources.
We propose a global biological aging research agenda focused on the detailed understanding of the following overlapping core age changes and developing therapies for decelerating, arresting, and reversing them: (i) the loss of proliferative homeostasis, (ii) neurodegeneration, (iii) somatic mutations in both nuclear and mitochondrial DNA, (iv) nonadaptive alterations in gene expression, (v) immunosenescence, (vi) nonadaptive inflammation, and (vii) alterations of the extracellular milieu. See the supporting online material (SOM) for brief elucidation.
To ameliorate age-related changes, we identify three broad modes of intervention that should be exploited in addition to ongoing conventional, disease-centered medical innovation: (i) reduction in exposure to environmental toxins and amelioration of other risk factors through improved public health; (ii) modulation of metabolic pathways contributing to age-related changes; and (iii) a more broadly conceived regenerative medicine, to embrace the repair, removal, or replacement of existing aging damage or its decoupling from its pathological sequelae.
The relative potential of these interventions and their combination is portrayed in Fig. 2, presented in terms of their ability to deliver the 7-year postponement of the onset of age-related degeneration identified recently by four prominent gerontologists (including one of us, R.N.B.) as a realistic medium-term goal of biomedical gerontology (7).
Fig. 2. Postponing degeneration.
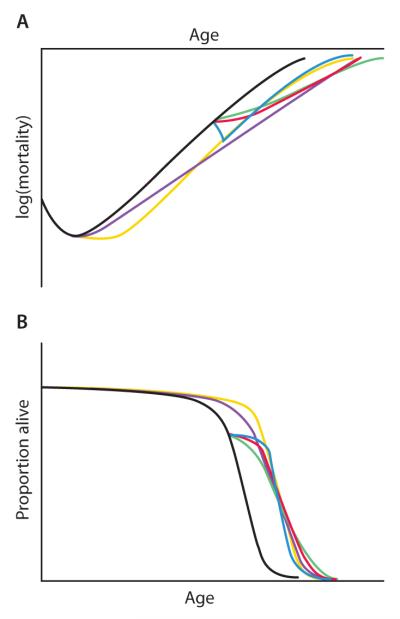
This illustration compares the trajectories of the proposed modalities of intervention necessary to achieve the target (7) of a 7-year average postponement of the onset of age-related degeneration, depicted in terms of (A) an exponential rise in mortality rates and (B) survival. The black trajectories indicate scenarios if no interventions are applied. Nutritional and other public health interventions would need to be applied aggressively and from an early age (ideally prenatally) (yellow trajectories). Metabolic interventions applied from an early age would suffice even if they only mildly slowed the rate of accumulation of aging damage (purple). Metabolic interventions applied only from middle age would need to at least halve this rate—a daunting challenge (green). Late-onset regenerative therapies would postpone biological aging by substantially reversing the initial level of aging damage and then allowing it to continue as normal, but they would also be challenging to implement comprehensively enough (blue). A combination of more modest implementations of the late-onset metabolic and regenerative approaches seems most tractable and could lead to an equal or greater extension of healthy productive life (red).
Public health and medical advancements
There remains substantial room to improve healthy life expectancy through improvements in public health and lifestyles (17), medical control of disease risk factors, and traditional disease-oriented medicine. However, we note their limitations in the late-middle-aged cohorts in whom intervention is most urgent. These improvements are most effective when applied relatively early in life, especially during development (18); in later life, the effect of environmental influences declines (19). In fact, age-related changes lead to paradoxical relationships between disease risk factors and outcomes in the elderly: The relationship between well-established risk factors—such as overweight, hypertension, and hyperinsulinemia—and adverse outcomes often declines in magnitude or even reverses relative to their relationship in younger people (20). The causes and implications of these changes are often unclear. Some may be the result of “reverse causation,” in which the causal relationship between two closely associated phenomena is mistakenly taken to be the reverse of what it actually is; for example, mild overweight in older adults is associated with longer life expectancy, which may not indicate a protective effect of excess weight but rather that thinness in older adults is often the result of medical conditions that themselves cause weight loss (such as cancer, chronic obstructive pulmonary disease, or depression) or of the cachexia (wasting syndrome) and sarcopenia (the loss of muscle mass, strength, and function) of aging (21). But others may represent genuine age-related changes in the causal relationship between a risk factor, its underlying metabolic basis, and clinical disease. This uncertainty creates potential for unintentional worsening of patient health through mismanagement of the risk factor. Improvements in public health and conventional medicine will therefore contribute primarily to the future health of currently young people rather than people already in late middle age and beyond.
Modulation of the metabolic determinants of aging damage
Interventions that mimic the modulation of metabolic pathways influencing the rate at which aging damage accumulates in model organisms—such as pharmacological mimetics of CR and down-regulation of IIS—have thus far received more attention than alternative routes to postponing human age-related degeneration (5). This avenue is undoubtedly promising, but we note possible limitations. Many of these promising interventions have been demonstrated in model organisms with simpler signaling systems than those of humans; the inbred laboratory strains of model species that have dominated research to date may create experimental artifacts; and whereas life-span extension is readily quantitated, effects on age-related functional decline (reduced health span) are difficult to assess and characterization is limited (6). Accordingly, the benefits of even faithfully translated interventions in the health and functionality of aging humans remain uncertain.
Additionally, the modulation of metabolic pathways typically imposes substantial side effects in model organisms, such as impaired immunity, low bone mass, vulnerability to cold, and lower fertility. Rapamycin, a likely CR mimetic because its inhibitory effects on a nutrient-sensing pathway parallel those of CR and several longevity mutations, was recently shown (11) to extend life span in mice when first administered late in life. This drug is an immunosuppressant, induces hyperlipidemia in humans (which would only modestly affect mouse life span, because wild-type mice are not susceptible to atherosclerosis), and might interfere with normal brain function—none of which were assessed in the recent report.
Finally, even if interventions that favorably modulate the metabolic origins of aging damage can be fully translated to humans and any deleterious side effects mitigated, there remains the progressively reduced efficacy of such interventions the later in life they are initiated. These interventions decelerate age-related decline but cannot arrest or reverse its course (22). Thus, even assuming full human translatability, a rough extrapolation from results to date (22) suggests that a CR mimetic might extend human life expectancy by 25 years beyond the 85-year life expectancy that would otherwise result from “aging as usual” if begun at weaning but only 9.3 years if begun at age 54.
Regenerative therapies
A third mode of intervention in the degenerative aging process is to directly target age-related changes themselves, rather than their environmental and metabolic determinants. This is the goal of regenerative medicine, a term often limited to cell therapy and tissue engineering: replacing lost cells and tissues with versions that are new and structurally youthful to restore function. We propose to broaden its scope to include conceptually similar interventions targeting other age-related changes.
Where they are possible, regenerative therapies would have the advantage of being effective even after youthful functionality has been lost. This feature also implies simpler and more rapid clinical testing, because any effects will necessarily be more immediate and direct (23). Regenerative therapies are thus especially attractive because they have effects even when initiated late in life, when the body has already accumulated extensive age-related changes (23, 24).
Regenerative therapies, too, would have limitations. Their effects would necessarily be segmental, specifically affecting changes linked to the particular damage that a given therapy repairs. Further, it is unclear whether such therapies could be developed to address all age-related changes, although proofs of concept exist and other potential interventions can be foreseen from existing developments (25, 26).
It is possible that therapies of different types might be used complementarily. Whereas regenerative therapies are segmental, metabolic interventions (especially CR) are highly pleiotropic, decelerating many, if not all, degenerative aging processes. The two approaches could thus be synergistic, with metabolic interventions decelerating age-related degeneration systemically and regenerative therapies used to restore functionality in particular tissues more fully. If regenerative therapies strengthen the weakest links in the chain of age-related changes decelerated by metabolic modulation, a disproportionate increase in healthy life span might result (Fig. 2).
POLICY PRIORITIES
Funding
Recognizing the potential of this research agenda to avert enormous economic, social, and human costs, we advocate that substantial new investments be made by governments, while engaging and facilitating the participation of the biomedical industry. A previous proposal that included one of us (R.N.B.) as an author suggested that the United States invest $3 billion annually (<1% of the current Medicare budget) in a broadly similar agenda (7); we suggest that this funding level is inadequate to deliver interventions in time to avert demographic crisis. We therefore urge a larger investment, targeted specifically to late-life interventions, matched by other developed and developing nations in proportion to the means and demographic urgency of each.
Regulatory changes
Because they would reverse existing age-related changes, the effects of regenerative therapies may be so rapid as to be amenable to direct testing for their effects on specific diseases in time frames similar to those of conventional medicines (23), allowing their evaluation in clinical trials within existing regulatory frameworks. However, new regulatory structures will also need to be developed for the unique features of this class of medicines, especially for interventions targeting modulation of the metabolic determinants of the rate of accumulation of aging damage, whose effects will be more global and will emerge more gradually.
Regulatory agencies such as the U.S. Food and Drug Administration (FDA) should be charged with developing new guidelines for testing interventions that do not necessarily target a single specific disease but that retard, arrest, or reverse the structural degeneration and loss of functionality associated with aging. Preliminary meetings exploring a subset of such issues have occurred between geriatricians and FDA officials (27); they will need to be expanded into interdisciplinary working bodies drawing in experts in the basic biology of aging (particularly experimentalists with extensive experience in lifelong interventional studies in mammals) and translational medicine. The ability of an agent to extend life span and health span in mammalian models, based on evidence of a broad spectrum of health effects in rodent models with robust historical controls, should be evaluated as sufficient preclinical evidence of efficacy for clinical trials.
For human testing, new surrogate outcomes will need to be designed that would offer evidence for parallel effects without necessitating a measurement of life span, such as the panels of nonspecific deficits used in cohort frailty studies (1), reducing the acceleration of total mortality rate over the course of 8 years (7), and the cautious use of metabolic changes observed in animal models that are thought to be mechanistically important to the observed deceleration of the rate of biological aging.
We also advocate that regulatory agencies charge interdisciplinary panels with identifying age-related dysfunctions that are sufficiently well characterized to merit consideration as new licensable therapeutic indications (that is, medical conditions for which regulatory bodies will approve effective therapies for marketing). A pressing example is sarcopenia, which occurs even in master athletes and in which loss of mass is only one relatively reversible element. Sarcopenia is a major contributor to age-related frailty and adverse outcomes, ranging from loss of activities of daily living to institutionalization, fracture risk, and increased mortality. It is estimated to cost the United States $18.5 billion ($11.8 billion to $26.2 billion) per year (~$1.5% of total health care expenditures) in direct medical costs alone (28). Exercise and supplemental energy and protein consumption can increase muscle mass to a limited extent but do not address the degradation of myocyte and neuromuscular unit structure. Beyond this, clinicians can at best resort to non–evidence-based off-label use of medications, risky and minimally effective hormone therapies, or unregulated, putatively ergogenic dietary supplements. Yet because sarcopenia is not a licensable indication, no incentive exists to develop therapies specifically targeting it. New treatments targeting determinants of sarcopenia other than loss of muscle mass could greatly benefit the health and functionality of older adults, and expert panels should explore this and other causes of age-related disability as possible new licensable indications.
We also advocate efforts to include more people over the age of 65 in clinical trials. Older adults are the largest consumers of prescription medications and have the highest prevalence of the diseases for which many drugs are indicated. Yet they are sorely underrepresented in clinical trials and are often perversely excluded from trials precisely because of their burden of other age-related disease. For example, an analysis of 3470 community-living older adults with possible or probable Alzheimer’s disease (AD) found that >90% would be precluded from participation in either of two trials for cholinesterase inhibitors, the main drug class approved to treat AD symptoms (29). Extrapolation of the results of trials performed in younger adults into older patients is fraught with potential artifacts because there are substantial differences in drug pharmacokinetics and in the range and severity of adverse reactions, because of primary and secondary age-related changes.
This exclusion of older people is a major problem in conventional medicine testing and will almost preclude the testing of agents whose purpose is to retard, delay, or reverse age-related changes in late life. In addition to implementing comprehensive reforms to address weaknesses in the existing system (proposals from the American Geriatrics Society and the American Association for Geriatric Psychiatry merit consideration), we advocate that trials of therapeutics specifically targeting biological aging or new indications for age-related diseases should be required to undergo testing in persons 50 years old and above, with significant representation of people over 65, beginning no later than in phase II trials.
CONCLUSIONS AND BEGINNINGS
We therefore advocate the development and implementation of all three forms of intervention in age-related degeneration discussed above, but with emphasis on metabolic and regenerative interventions, and on the most aggressive schedule possible, bearing in mind the urgency of the demographic challenge before us. We cannot be certain of success. Nor can the full range of social impacts, positive and negative, of a dramatic increase in healthy human life span be known with certainty in advance.
One obvious and quantifiable challenge that would result from a rapid decline in late-life mortality would be upward pressure on global population. Contrary to what is widely assumed, however, the net effect should be relatively minor. Because the effect on global population of adding each additional entire human life span (and one future parent) to the world is greater than the effect of adding some fraction of a life span onto each extant life, the effect of birth rates on population growth is much greater than the effect of late-life death rates. Without intervention in biological aging, the emerging global shift into sub-replacement fertility is likely to lead to the stabilization and later ongoing shrinkage of world population at ~9 billion in the 2050–2070 range (9). Demographic modeling in the contemporary Swedish population of the effect of a reduction in the rate of acceleration of mortality of the same magnitude (50%) as is required to achieve our proposal finds that population would continue to decline over the next century. In fact, even a much more radical intervention into age-related mortality than we envision, in which the rate of age-related mortality is arrested at the equivalent level of today’s 50-year-olds, would result in a surprisingly low increase of 35% in global population over the critical 50-year period of concern for global demographic aging (30). And of course, fertility rates themselves can be the subject of policy decisions, both directly and indirectly.
But it should also be emphasized that the social challenge posed by overpopulation is not determined by sheer numbers but by a variety of factors within the sphere of public policy, such as the efficiency of resource use and the equity of resource distribution, as well as the rate of economic growth. Moreover, the predictable early expansion of productive capacity resulting from intervention in biological aging will increase the resource pool available to meet the population and other challenges to which such intervention may contribute and with which it will interact.
This and other potential impacts of intervention in the degenerative aging process must be the subject of open, early, and serious public dialogue; in our view, such challenges should be met under the broad approach called the “vigilance principle” (31): that action should be taken for the greater social good based on current knowledge, acknowledging uncertainty surrounding its possible future ramifications (positive and negative) and monitoring such consequences actively. The resilience and adaptability exhibited by human cultures throughout history should be recognized and engaged, with more specific policy-based remedies applied judiciously in cases in which organic social response proves insufficient to mitigate specific deleterious effects that actually (rather than hypothetically) emerge.
In the case of late-life intervention in human age-related degeneration, what we can be certain of today is that a policy of aging as usual will lead to enormous humanitarian, social, and financial costs. Efforts to avert that scenario are unequivocally merited, even if those efforts are costly and their success and full consequences uncertain. To realize any chance of success, the drive to tackle biological aging head-on must begin now.
Supplementary Material
Acknowledgments
The authors thank A. Foster for yeoman’s work in preparing and repeatedly revising the figures for this paper.
Funding: C.E.F. is funded by the Ellison Medical Foundation and the National Institute on Aging. G.M.M. is funded by NIH grant R24CA078088.
Dedicated to the memory of our coauthor and friend, Robert N. Butler, a giant in his lifelong contributions who epitomized the value of healthy human longevity through tireless advocacy for aging research to the very end. Among his many accomplishments, Dr. Butler was the first director of the National Institute on Aging. We are privileged to have shared the creation of this publication with him and take comfort that it represents so much of what he worked for. He will be sorely missed.
Footnotes
Origin of paper: This meeting report is the result of a workshop held on 7 and 8 August 2009, sponsored and organized by the Life-Star Institute, a 501(c)3 nonprofit organization whose World Health Initiative is dedicated to mitigating the enormous economic, social, and human costs of global aging by ensuring that therapies are developed and made available globally which restore physiological (including cognitive) function in already-mature people, on the unprecedented schedule and scale dictated by the imminent surge in the world’s elder demographic.
Competing interests: M.J.R. is a research assistant to A.D.N.J. de G., an employee of SENS Foundation, and a member of the Calorie Restriction Society. R.N.B. has a financial interest in BioTime, Inc. A.D.N.J. de G. is the chief science officer for SENS Foundation, a charity dedicated to furthering the development of regenerative medicine solutions to the problems of age-related ill health. C.E.F. has a financial relationship with Acumen Pharmaceuticals and a paid consulting relationship with the Ellison Medical Foundation and the CureAlzheimer Foundation. G.M.M. is the scientific director of the American Federation for Aging Research, the chair of the Scientific Advisory Board of and a paid consultant for the Ellison Medical Foundation, and a consultant for Icogenix Corporation and LifeSpan BioSciences. K.M.P. is a member of the board of directors of SENS Foundation. B.J.L. is the acting chair of SENS Foundation Board and chief executive officer of the LifeStar Institute.
SUPPORTING ONLINE MATERIAL www.sciencetranslationalmedicine.org/cgi/content/full/2/40/40cm21/DC1 SOM Text References
REFERENCES AND NOTES
- 1.Searle SD, Mitnitski A, Gahbauer EA, Gill TM, Rockwood K. A standard procedure for creating a frailty index. BMC Geriatr. 2008;8:24. doi: 10.1186/1471-2318-8-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Campisi J, Vijg J. Does damage to DNA and other macromolecules play a role in aging? If so, how? J. Gerontol. A Biol. Sci. Med. Sci. 2009;64A:175–178. doi: 10.1093/gerona/gln065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Finch CE. Inflammation, Nutrition, and Aging in the Evolution of Lifespans. Academic Press; Burlington, MA: 2007. The Biology of Human Longevity. [Google Scholar]
- 4.Weindruch R, Walford RL. The Retardation of Aging and Disease by Dietary Restriction. Charles C. Thomas; Springfield, IL: 1988. [Google Scholar]
- 5.Kenyon C. The plasticity of aging: Insights from long-lived mutants. Cell. 2005;120:449–460. doi: 10.1016/j.cell.2005.02.002. [DOI] [PubMed] [Google Scholar]
- 6.Kirkland JL, Peterson C. Healthspan, translation, and new outcomes for animal studies of aging. J. Gerontol. A Biol. Sci. Med. Sci. 2009;64A:209–212. doi: 10.1093/gerona/gln063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Olshansky SJ, Perry D, Miller RA, Butler RN. In pursuit of the longevity dividend. Scientist. 2006;20:28–36. [Google Scholar]
- 8.Lockett BA. Setting the federal agenda for health research: The case of the National Institute on Aging. J. Health Polit. Policy Law. 1984;9:63–80. doi: 10.1215/03616878-9-1-63. [DOI] [PubMed] [Google Scholar]
- 9.Lutz W, Sanderson W, Scherbov S. [accessed 12 August 2009];IIASA’s 2007 Probabilistic World Population Projections. IIASA World Population Program Online Data Base of Results. 2008 Available online at http://www.iiasa.ac.at/Research/POP/proj07/index.html?sb=5.
- 10.Dhahbi JM, Kim HJ, Mote PL, Beaver RJ, Spindler SR. Temporal linkage between the phenotypic and genomic responses to caloric restriction. Proc. Natl. Acad. Sci. U.S.A. 2004;101:5524–5529. doi: 10.1073/pnas.0305300101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter CS, Pahor M, Javors MA, Fernandez E, Miller RA. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature. 2009;460:392–395. doi: 10.1038/nature08221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vergara M, Smith-Wheelock M, Harper JM, Sigler R, Miller RA. Hormone-treated snell dwarf mice regain fertility but remain long lived and disease resistant. J. Gerontol. A Biol. Sci. Med. Sci. 2004;59:1244–1250. doi: 10.1093/gerona/59.12.1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shimokawa I, Higami Y, Hubbard GB, McMahan CA, Masoro EJ, Yu BP. Diet and the suitability of the male Fischer 344 rat as a model for aging research. J. Gerontol. 1993;48:B27–B32. doi: 10.1093/geronj/48.1.b27. [DOI] [PubMed] [Google Scholar]
- 14.Willcox BJ, Willcox DC, Ferrucci L. Secrets of healthy aging and longevity from exceptional survivors around the globe: Lessons from octogenarians to supercentenarians. J. Gerontol. A Biol. Sci. Med. Sci. 2008;63:1181–1185. doi: 10.1093/gerona/63.11.1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Manton KG, Gu XL, Ullian A, Tolley HD, Headen AE, Jr., Lowrimore G. Long-term economic growth stimulus of human capital preservation in the elderly. Proc. Natl. Acad. Sci. U.S.A. 2009;106:21080–21085. doi: 10.1073/pnas.0911626106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kotlikoff LJ. In: Aging: Biology and Behavior. March J, McGaugh JL, Kiesler SB, editors. Academic Press; New York: 1982. pp. 97–114. [Google Scholar]
- 17.Mokdad AH, Marks JS, Stroup DF, Gerberding JL. Actual causes of death in the United States, 2000. JAMA. 2004;291:1238–1245. doi: 10.1001/jama.291.10.1238. [DOI] [PubMed] [Google Scholar]
- 18.Barker DJ. The developmental origins of well-being. Philos. Trans. R. Soc. London Ser. B. 2004;359:1359–1366. doi: 10.1098/rstb.2004.1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hjelmborg J. v. B., Iachine I, Skytthe A, Vaupel JW, McGue M, Koskenvuo M, Kaprio J, Pedersen NL, Christensen K. Genetic influence on human lifespan and longevity. Hum. Genet. 2006;119:312–321. doi: 10.1007/s00439-006-0144-y. [DOI] [PubMed] [Google Scholar]
- 20.Abbott RD, Curb JD, Rodriguez BL, Masaki KH, Yano K, Schatz IJ, Ross GW, Petrovitch H. Age-related changes in risk factor effects on the incidence of coronary heart disease. Ann. Epidemiol. 2002;12:173–181. doi: 10.1016/s1047-2797(01)00309-x. [DOI] [PubMed] [Google Scholar]
- 21.Hu FB. In: Obesity Epidemiology. Hu FB, editor. Oxford Univ. Press; New York: 2008. pp. 38–52. [Google Scholar]
- 22.Rae M. It’s never too late: Calorie restriction is effective in older mammals. Rejuvenation Res. 2004;7:3–8. doi: 10.1089/154916804323105026. [DOI] [PubMed] [Google Scholar]
- 23.Hadley EC, Lakatta EG, Morrison-Bogorad M, Warner HR, Hodes RJ. The future of aging therapies. Cell. 2005;120:557–567. doi: 10.1016/j.cell.2005.01.030. [DOI] [PubMed] [Google Scholar]
- 24.Phoenix CR, de Grey ADNJ. A model of aging as accumulated damage matches observed mortality patterns and predicts the life-extending effects of prospective interventions. Age (Omaha) 2007;29:133–189. doi: 10.1007/s11357-007-9038-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.de Grey AD, Ames BN, Andersen JK, Bartke A, Campisi J, Heward CB, McCarter RJ, Stock G. Time to talk SENS: Critiquing the immutability of human aging. Ann. N. Y. Acad. Sci. 2002;959:452–462. doi: 10.1111/j.1749-6632.2002.tb02115.x. discussion 463–465. [DOI] [PubMed] [Google Scholar]
- 26.Rae MJ. In: The Future of Aging: Pathways to Human Life Extension. Fahy GM, West M, Coles LS, Harris SB, editors. Springer Verlag; New York: 2010. pp. 806–826. [Google Scholar]
- 27.Bhasin S, Espeland MA, Evans WJ, Ferrucci L, Fried LP, Gill TM, Pahor M, Studenski S, Guralnik J, Nayfield S, Romashkin S, Perlstein R, Burke L, Parks M. Working Group on Functional Outcome Measures for Clinical Trials, Indications, labeling, and outcomes assessment for drugs aimed at improving functional status in older persons: A conversation between aging researchers and FDA regulators. J. Gerontol. A Biol. Sci. Med. Sci. 2009;64:487–491. doi: 10.1093/gerona/gln042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Janssen I, Shepard DS, Katzmarzyk PT, Roubenoff R. The healthcare costs of sarcopenia in the United States. J. Am. Geriatr. Soc. 2004;52:80–85. doi: 10.1111/j.1532-5415.2004.52014.x. [DOI] [PubMed] [Google Scholar]
- 29.Schneider LS, Olin JT, Lyness SA, Chui HC. Eligibility of Alzheimer’s disease clinic patients for clinical trials. J. Am. Geriatr. Soc. 1997;45:923–928. doi: 10.1111/j.1532-5415.1997.tb02960.x. [DOI] [PubMed] [Google Scholar]
- 30.Gavrilov LA, Gavrilova NS. Demographic consequences of defeating aging. Rejuvenation Res. 2010;13:329–334. doi: 10.1089/rej.2009.0977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brand S. Whole Earth Discipline: An Ecopragmatist Manifesto. Viking Penguin; New York: 2009. [Google Scholar]
- 32.Driver JA, Djoussé L, Logroscino G, Gaziano JM, Kurth T. Incidence of cardiovascular disease and cancer in advanced age: Prospective cohort study. BMJ. 2008;337:a2467. doi: 10.1136/bmj.a2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Evans DA, Bennett DA, Wilson RS, Bienias JL, Morris MC, Scherr PA, Hebert LE, Aggarwal N, Beckett LA, Joglekar R, Berry-Kravis E, Schneider J. Incidence of Alzheimer disease in a biracial urban community: Relation to apolipoprotein E allele status. Arch. Neurol. 2003;60:185–189. doi: 10.1001/archneur.60.2.185. [DOI] [PubMed] [Google Scholar]
- 34.Thompson WW, Weintraub E, Shay DK, Brammer L, Cox N, Fukuda K. Age-specific estimates of US influenza-associated deaths and hospitalisations. Proc. Int. Conf. Options Control Influenza. 2004;V:316–320. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.