

Highlights
-
•
We show stress induced acceleration of platelets shape changes.
-
•
Time-lapse AFM imaging visualizes two phases of filopodia extrusion and contraction.
-
•
SMFS reveals a close to 100 percent activity of the integrin αIIbβ3.
Keywords: Platelets, Single molecule force spectroscopy, Atomic force microscopy, Integrin αIIbβ3
Abstract
Platelets are essential in hemostasis. Upon activation they undergo a shape-change accompanied with receptor presentation. Atomic force microscopy (AFM) imaging and single molecule force spectroscopy (SMFS) were used as powerful tools for exploring morphological changes as well as receptor activities of platelets. Imaging time series was accomplished with and without fixation steps at the single platelet level. Hereby the response of mechanical stimulation of the platelet by the AFM cantilever tip was directly observed. We demonstrate that living and fixed platelets develop filopodia after a short activation time followed by their disappearance including cellular bleb formation. Thereafter a second filopodia formation (filopodia extrusion) was observed; those filopodia subsequently disappeared again, and finally platelets detached from the support due to cell death. We determined the influence of mechanical stress on the chronology of morphological changes of platelets and demonstrated shear force induced filopodia formation. Through recordings over several hours, topographical AFM images over the full platelet lifetime – from early activation up to apoptosis – are presented. SMFS measurements on living platelets allowed determining the activation state of the most prominent membrane receptor integrin αIIbβ3 at all different phases of activation. αIIbβ3 was fully activated, independent of the morphological state.
1. Introduction
Platelets, small anucleate blood particles with an approximate size of 3.6 × 0.7 μm, are of key importance for hemostasis and thrombosis, innate immune defense, and wound healing, and are involved in metastasis [1]. They are generated in the bone marrow and are degraded in the reticuloendothelial system after about 9–12 days [2]. Platelets remain in an inactive state when circulating in the blood, whereas when activated they form a hemostatic plug in areas of endothelial damage in order to close the vessel wall [3].
Extracellular matrix proteins exposed in such areas interact with glycoproteins of the platelet membrane (adhesion). Binding of biochemical agonists to their receptors on the platelet surface induces a signaling cascade (secretion) resulting in platelet activation, which again is strongly related to the activation of the most numerous platelet integrin αIIbβ3. Platelet-platelet-interaction (aggregation) and platelet interaction with plasma coagulation factors is facilitated through αIIbβ3. Thereby, a dramatic shape change is required to expose the relevant receptor [1]. Discoid and non-adherent platelets develop spike-like filopodia and then spread to sticky discs [4–8]. This process of adhesion and aggregation is of main importance for preventing or healing lethal diseases. Exploring detailed topographical changes as well as following the receptor activity in living platelets contributes to a more detailed understanding of platelet physiology.
Due to its nanometer resolution, AFM is a versatile tool to investigate shape changes of activated platelets. Non-activated platelets are difficult to visualize with the AFM [6,7] since they either do not adhere to glass slides or are activated immediately after attachment as a result of this contact. Nonetheless, several pioneering studies using AFM imaging and force probe techniques have been conducted over the last years: Morphological changes of activated platelets under near physiological conditions were successfully investigated over a time period of 30 min [6,7]. AFM studies investigating biophysical properties of platelets revealed elastic moduli between 1–50 kP [9–11]. Force mapping of specific ligand-receptor binding across platelets was used to probe the binding behavior of isolated platelet receptors [12]. Molecular interaction forces of the most abundant integrin αIIbβ3 directly revealed its affinity to fibrinogen as well as to RGD (Arg-Gly-Asp peptide sequence) using micromanipulation, optical tweezers, and the force volume mode [13–22]. Moreover, the interaction forces between an RGD modified AFM tip and the GPIIb/IIIa receptor (integrin αIIbβ3) on human platelets [23] and between the GPIIb/IIIa receptor reconstituted in a supported lipid bilayer and fibrinogen using AFM tapping mode [25] were explored. Finally, changes of the binding affinity during interactions between the disintegrin rhodostomin and αIIbβ3 in living cells were monitored using optical tweezers [26].
In this study we developed an optimized platelet immobilization assay and used a gentle imaging technique avoiding artefacts. Early stages of activation and their related shape changes over a time period of several hours were investigated following the whole spectrum of shape changes from activation over blebbing, and filopodia extrusion up to apoptosis related cell shrinkage.
In addition, we performed single molecule force spectroscopy to gain insights into functional properties of platelets. Measurements on living platelets allowed exploring activation states of the platelet membrane receptor GPIIb/IIIa.
2. Experimental design
2.1. Materials
2.1.1. Chemicals
All chemicals were used in the highest available quality. 3-Aminopropyl-triethoxy silane (APTES; SigmaAldrich, Vienna, Austria) was distilled at low pressure and stored under argon in sealed crimp vials over silica gel (to avoid polymerisation) at −20 °C. MilliQ (Millipore, Massachusetts, USA). Purified water was used for all aqueous solutions. Triethylamine (TEA, SigmaAldrich, Vienna, Austria) was stored under argon in the dark to avoid amine oxidation. The heterobifunctional crosslinker Acetal-PEG-NHS (α-(2-[4-(Dimethoxymethyl)-benzoyl]-aminopropyl)-ω-(2-[4-(N-succinimidyl-oxycarbonyl)-butanoylamido]-propyl)-poly(oxyethylene)-800) was used as described in Wildling et al. [27]. Chloroform was purchased from J.T. Baker (Griesheim, Germany), and argon and N2 from Linde Gas GmbH (Stadl-Paura, Austria). Glass slides were obtained from VWR International (Vienna, Austria). NaCNBH3 and ethanolamine.HCl was purchased from SigmaAldrich, (Vienna, Austria).
2.1.2. AFM cantilever
For Mac-Mode imaging magnetically coated cantilevers (MAC levers, Type VI, Agilent, Chandler, AZ, USA) with 0.292 N m−1 nominal spring constant and 43 kHz resonant frequency), and for MRFS experiments non-conductive Silicon Nitride MSCT tips (Brucker Corporation, MA, USA, C-cantilever) with 0.010 N m−1 nominal spring constant were used. The actual spring constant was determined according to Hutter et al. [28] using the thermal noise method.
2.1.3. Buffers
Citric acid solution: 1% (w/v) citric acid was dissolved in ultrapure water
Thyrode-buffer: 3 mM Hepes, 4 mM NaH2PO4, 137 mM NaCl, 2.6 mM KCl, 1 mM MgCl2, (pH 7.3 adjusted with NaOH)
PBS: 150 mM NaCl and 5 mM NaH2PO4 (pH 7.4 adjusted with NaOH)
2.2. Tip chemistry
The tip functionalization protocol was optimized regarding reproducibility and stability. The protocol consists of three main steps and is described in detail in the following sections. As a first step the silicon nitride tip material has to be converted into a chemically addressable surface (2.2.1.), followed by the binding of a distensible heterobifunctional crosslinker (2.2.2.), and, finally, by the coupling of the ligand protein to the outer linker end (2.2.3.).
2.2.1. Aminofunctionalization
Commercial silicon–nitride cantilever (Section 2.1.2.) were washed with chloroform (3*5 min incubation) and dried in a gentle nitrogen gas stream immediately before further treatment. The APTES functionalization was performed as described previously [24]: A desiccator (5 L) was flooded with argon gas to remove air and moisture. Then two small plastic trays (e.g. the lids of Eppendorf reaction vials) were placed inside the desiccator. 30 μL of APTES and 10 μL of triethylamine were separately pipetted into two trays and the AFM tips were placed close to the trays on a clean, inert surface (Teflon). The desiccator was closed and flooded with argon for one minute. After 120 min of incubation APTES and triethylamine was removed and the desiccator was again flooded with argon for 5 min. The tips were left inside for 2 days in order to cure the APTES coating [24,29].
2.2.2. Coupling of Acetal-PEG-NHS
The linker coupling was performed as described in Wildling et al. [27]: In brief, APTES functionalized AFM tips were incubated in 0.5 mL of a 1 mg/mL solution of Acetal-PEG-NHS (see Section 2.1.1.) in chloroform containing 0.5% (v/v) of TEA as catalyst. Subsequently, the tips were rinsed in chloroform (3×) und dried in a gently stream of nitrogen. Immediately before ligand coupling, the acetal group was deprotected by incubation of the Acetal-PEG-NHS functionalized tips in citric acid solution (Section 2.1.3) for 20 min, followed by washing in water (3 × 5 min) and drying as described above.
2.2.3. Coupling of the monoclonal antibodies against CD41 (integrin αIIb) and activated αIIbβ3
Functionalized cantilever were mounted astrally on a piece of parafilm in a petri dish, and a drop of 100 μl of 0.15 mg/ml protein solution – either anti mouse CD41 (clone MWReg30, BD Biosciences, NJ, USA) or PAC-1 (clone PAC-1, BD Biosciences, NJ, USA) antibody – was placed on the tips. This allowed the monoclonal antibodies (mAbs) to covalently bind to the aldehyde functions via their lysine residues. Finally, 2 μl 1 M NaCNBH3 were added to the drop and allowed to react for two hours. Subsequently, 5 μl 1 M ethanolamine (in MilliQ water) were added in order to passivate unreacted aldehyde groups. Finally, the tips were washed 3 times in PBS and stored at 4 °C in PBS until further use.
2.3. Platelet immobilization
Glass slides were cleaned with ethanol and chloroform in an ultrasonic bath (1. 90% ETOH:10% CHCl3, 2. 50% ETOH:50% CHCl3, 3. 10% CHCl3:90% ETOH) for five minutes each followed by drying in a nitrogen gas stream. Fresh human blood was taken from a healthy adult volunteer and directly applied on a cleaned glass slice in a liquid fluid cell. All platelets used in this study were taken from the same person.
2.3.1. Unfixed platelets
After incubating a blood droplet for 45–60 s the sample was rinsed with thyrode buffer, until erythrocytes were completely washed away. The washing procedure was kept as fast as possible and imaging was started immediately thereafter to follow early activation steps.
2.3.2. Fixed platelets
Eight samples were prepared in parallel with the same drop of blood. In detail, immobilized platelets were fixed with 4% paraformaldehyde diluted with thyrode buffer for 15 min after various times of incubation in thyrode buffer (15 min, 30 min, 1 h 30 min, 1 h 45 min, 2 h, 2 h 15 min, 2 h 30 min, 3 h). Finally, the samples were again rinsed with thyrode buffer 10–20 times to remove paraformaldehyde, and finally stored at 4 °C until further use.
2.4. AFM imaging
Magnetic AC (MAC) mode topography imaging was used for monitoring platelet adherence to glass. The resonance frequency was determined and the actuation frequency was set 0.5 kHz below this value. The scanning frequency was set to 1 Hz. Each sample was imaged on at least five different areas. So as to accurately determine filopodia areas the image size was reduced and the platelets were imaged in successive parts. All AFM measurements were performed in thyrode buffer on a Pico SPM II 5500 (Agilent Chandler, AZ, USA). Typically, a large area scanner (max. scan size ∼100 × 100 μm) was used.
2.5. Force spectroscopy
For SMFS measurements tips functionalized with CD41 or PAC-1 mAb as described in Section 2.2.3. were used. Since the activation cascade and the related shape change of the blood platelets is immediately initiated after taking the blood sample from the test person, extreme care was taken to start the measurements as fast as possible (within a few minutes). Therefore all measuring settings were adjusted before sample preparation. The functionalized cantilever tip was positioned directly over a platelet by using a CCD-camera coupled to the AFM. SMFS measurements were performed for more than 5 h. About 1300 curves were recorded at the same pulling velocity (800 nm/s) within one measurement session (∼20 min). The position of the tip relative to the investigated platelet was changed every 200 FDCs to statistically avoid position dependent artefacts. The measurements were performed on both, on the central body as well as on the outer regions. No experiments were performed on the very outer filopodia regions. A 20 min break between sessions allowed to minimize the external force on the platelets, thereby lowering shear force induced activation and apoptosis behavior of the spreading platelets.
So as to proof the specificity of the molecular interaction, block experiments were performed. For this purpose purified αIIbβ3 receptors (0.1 mg/ml) were suspended in buffer and the mAb functionalized tips were incubated for 2–3 h within this solution.
2.6. Data evaluation
2.6.1. Platelet shape changes
All recorded images were flattened with Gwyddion in order to correct for sample tilts and scanner-nonlinearities. So as to quantify shape changes of platelets during activation, the images were analyzed using ImageJ [30]. A total platelet area (contact region of the platelet with the glass support) and the filopodia area (contact region of the filopodia with the glass support) were determined. The height of the filopodia (lower than 200 nm) and the shape of the spike like extrusions were used to distinguish between the platelet base body area and the filopodia area. For improved visibility of the appearing filopodia, the background was subtracted (area with structures lower than 4 nm) and dyed in green.
For unfixed platelets, at least three different platelets from three different blood drops were imaged over time. From these, an arithmetical mean and standard deviation of the percentage of filopodia area relative to the total platelet area was calculated at each point in time. In the case of fixed platelets, each sample (according to Section 2.3.2.) was imaged at different positions to yield images of at least ten different platelets for each point in time. Three such time series were taken to calculate mean and standard deviation of the maxima for filopodia extrusion and blebbing. The maximum of the first and second filopodia extrusion period was determined by the appearance of the highest relative filopodia area and the maximum of blebbing by the highest number of blebs in all time series.
2.6.2. SMFS data
The probability of complex formation and the most probable unbinding force was investigated recording force-distance-cycles (FDCs) as detailed in the results section. Such FDCs allow following single molecule antibody/receptor unbinding events. The ratio between FDCs showing an unbinding event relative to the total number of FDCs reflects the binding probability. In order to determine the most likely unbinding force, unbinding events were evaluated and unbinding forces and their standard deviation were used to construct Gaussians of unitary area. The sum of these calculations yielded the probability density function of unbinding forces, from which the maximum reflects the most probable unbinding force.
3. Results and discussion
In contrast to non activated platelets, which are round shaped and non adhesive, activated platelets undergo a dramatic change in the cytoskeleton and release supporting substances for blood clotting [2,5]. Platelets are also known to contain a large number of membrane receptors embedded in the cellular membrane that play a major role in the platelet activation process. They are responsible for adhesion and for promoting signalling cascades, resulting in morphological changes of the platelet. Optimizing the methodology by using a gentle AFM imaging technique, we were able to visualize the chronological shape changes of activated platelets. Due to the lack of specific binding partners (e.g. von Willebrand-factor, collagen, fibrinogen) for the physiological activation cascade, changes are slowed down. This allowed us following membrane receptor properties as a function of platelet morphology. At different activation states we investigated the grade of activation of integrin αIIbβ3 by performing force spectroscopy experiments using PAC-1 or CD41 antibodies. CD41 mAbs bind to the active as well as to the inactive conformation of the integrin receptor αIIbβ3, while the PAC-1 mAbs only binds to activated integrin αIIbβ3 [19].
3.1. Morphological changes of blood platelets
Platelets in the blood circulation of healthy persons are commonly not activated, however, blood drawing may already cause shear stress, resulting in activation of platelets. Pricking the finger initiates platelet activation and the time period between blood drawing and imaging contributes to the total time of activation. In earlier studies [6,7] resuspended platelet pellets from fresh human platelet concentrates were used. Here we followed the activation cascade starting at the earliest possible time. Hence, blood samples from volunteers prepared directly before measuring were used. In addition, by applying gently MAC-Mode imaging, artifacts due to gentle indentation forces were avoided. We followed the stages of activation and observed related shape changes in time series with living and fixed blood platelets.
In order to quantify morphological changes, platelet regions were divided into a cellular base body and filopodia. In Fig. 1 (left part) the cellular shape of the same live platelet is shown before (Fig. 1A) and at the maximum (D) of filopodia formation. An increase in filopodia formation is clearly visible from Fig. 1A–D. The cross sections of the platelet at early state (Fig. 1B) and at the maximum of filopodia formation (E) visualize the height profile of the cellular body (blue line) and of the filopodia (red line), respectively. The height of the filopodia was significant lower, typically in the range of 100 nm. Discriminating the cellular base body (Fig. 1C + F, blue) from filopodia (Fig. 1C + F), red) allowed quantifying changes in filopodia formation. In the following sections morphological changes of platelets over the whole life time and the influence of shear force are shown. Thereby, equally prepared live platelet samples were either imaged over a long time period (Section 3.1.1.) or platelet samples were fixed after different time points (Section 3.1.2.).
Fig. 1.

(A) and (D). AFM images showing the formation of pseudopodia of adhered, unfixed platelets. Scan range: 10 μm. The background area characterized by structures lower than 4 nm was removed and colored in green to make the development of the filopodia more visible. (B) and (E). Cross section of the platelets as indicated in (A) and (D). (C) and (F). Scheme of platelet analysis showing the base body in blue and the filopodia in red. The percentage of filopodia area to the total platelet area at earlier state (A–C) was 8% and increased to 24% at completed filopodia formation (D–F).
3.1.1. Adhering platelets (without fixation)
The observed shape change during activation of adhering platelets is depicted in Fig. 2. Two different typical time series are shown (upper blue and lower green frame). MAC mode AFM images visualized the formation (Fig. 2A) of filopodia, visible as small branches protruding from the cellular body (highlighted with arrows in the figure). This represents the first activation period of the platelet. The maximum of the first filopodia extrusion was reached 25 ± 8 min after incubation (Fig. 2, right plot, a). At this time the ratio between filopodia and cell body area was 28%, representing the highest value within the whole activation process. Subsequently, the filopodia started to disappear (arrows from Fig. 2A and B). These morphological changes were accompanied by the appearance of round shaped structures protruding out of the cellular membrane, also termed as “blebbing” of cells (highlighted in Fig. 2B with squares), which is typical for activated platelets [31]. This blebbing reached a maximum at 75 ± 10 min after platelet activation (Fig. 2B) and is expected to be the basis for the formation of platelet-derived microparticles (PMP, for a review see [32]). PMPs are a heterogeneous population of vesicles, typically smaller than 1 μm and known to be derived from stimulated cells (e.g. shear force activated platelets).
Fig. 2.
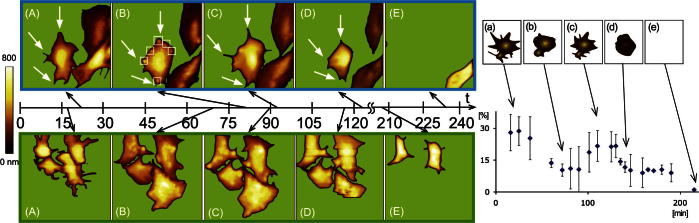
Left: Visualization of shape changes of unfixed platelets during activation imaged using AFM (one with blue and one with green frame). Distinctive states were picked out and shown as topography images: (A) activation and 1st filopodia extrusion, (B) blebbing, (C) 2nd filopodia extrusion, (D) cell shrinkage, (E) detachment. The white arrows in the time series with the blue frame indicate the development and disappearance of the filopodia over time. The white squares in (B) point out the state of “blebbing”. Scan range: 15 μm. Right: Corresponding diagram of the percentage of pseudopodia area to total platelet area (y-axis) plotted against time (x-axis). The maximum of the first filopodia extrusion was reached 25 ± 8 min after incubation. The second filopodia extrusion state reached maximum after about 1 h 53 ± 12 min after activation. The illustrations on top of the diagram accentuate the distinctive states of shape change again: (a) activation and 1st filopodia extrusion (b) blebbing, (c) 2nd filopodia extrusion, (d) cell shrinkage and (e) detachment.
Subsequently, a second filopodia extrusion period appeared typically 1 h 53 ± 12 min after activation. Depicted by the arrows in Fig. 2C, filopodia extruded on the same positions as in the first period. In contrast to the previous activation, blebbing was abundant and the amount of filopodia formed was somewhat less pronounced (22% relative to the cellular body) (Fig. 2C). This second filopodia extrusion period is most probably related to apoptosis. Despite apoptosis of platelets [33] was reported to occur separate from activation [34,35], our study indicates that activation and apoptosis may be strongly related and activation can directly merge into platelet apoptosis, which is in good agreement with work published earlier [31,36]. The next morphological shape change was the contraction of the platelets accompanied by disappearance of the filopodia (Fig. 2D). Finally the cell detached from the support (Fig. 2E).
3.1.2. Fixed platelets
Experiments with fixed platelets were performed to evaluate the influence of permanent scanning forces to unfixed, adhering and living platelets. Similar to the time series of unfixed platelets, distinctive states of activation, blebbing and filopodia extrusion were observed. The different states developed somewhat delayed and the second formation of filopodia (filopodia extrusion) appeared to be weaker (Fig. 3). Percentages of pseudopodia area to total platelet area are depicted in Fig. 3 (right). Platelets fixed 15 min after immobilization showed formation of filopodia (Fig. 3A). Whereas the lower left platelet in this image (white arrow) clearly developed filopodia, the cell directly above (gray arrow) did not form significant filopodia. Cells fixed about 25 ± 5 min after incubation showed a clear maximum (∼25%) of filopodia formation (Fig. 3B). The state of blebbing (Fig. 3C) was observed after 1 h 10 ± 28 min, although the visibility of the formed “bubbles” was weaker compared to unfixed platelets at the same stage. Subsequently filopodia were extruded again (Fig. 3F, G and c) after 2 h 15 ± 21 min. The second filopodia formation period showed only one-third of filopodia when compared to the first period. It has to be noted that the absence of further stimulants, which are known to promote the activation cascade (e.g. vWF or collagen) significantly slowed down the morphological changes compared to platelet shape changes using vWF bound platelets investigated previously by Kuwahara under high shear flow [8].
Fig. 3.
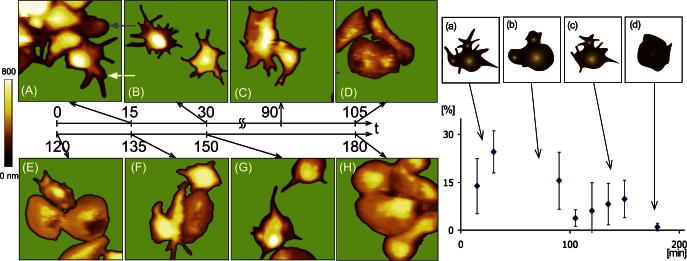
Left: Platelet images of the fixed samples at eight different times. (B) State of activation and 1st filopodia extrusion, (C) state of blebbing, (F and G) filopodia extrusion. Scan range: 15 μm. Right: Corresponding diagram of the percentage of pseudopodia area to total platelet area (y-axis) plotted against time (x-axis). The maximum of the first filopodia formation was visible after about 25 ± 5 min after incubation and the maximum of the second filopod extrusion was observed after 2 h 15 ± 21 min. The scheme on top of the diagram accentuates the distinctive states of shape change.
3.1.3. Comparison
We observed two phases of filopodia formation in the lifetime of an activated platelet. The first filopodia extrusion period is caused by platelet activation, whereas the second one is most likely related to the beginning of apoptosis. The phase of blebbing, which can be seen as the preliminary state of PNP release, mainly occurred after the first activation and no blebs on the cellular membrane were observed after the second filopodia extrusion period.
Indicative from the plots (right side) in Figs. 2 and 3, the phases of morphological changes appeared to occur retarded on fixed platelets when compared to the live samples. Whereas the first period in filopodia formation appeared at the same time (∼25 min), respectively, all other phases of the fixed cells were delayed by a factor of 1.3–1.4. This most likely reveals the influence of mechanical shear stress due to the imaging tip. The difference in the time sequence of morphological changes between fixed and unfixed samples in platelet-activation was also reported earlier [7]. Moreover, fixed platelet samples also showed a lower grade of filopodia extrusions. This was mainly evident in the second phase; fixed platelets showed 10 ± 5% relative filopodia area (Fig. 3c) and unfixed platelets up to 21 ± 5% filopodia area (Fig. 2c). Apparently, permanent scanning forces on the cells partially enhanced filopodia extrusion. The possibility that fixed cells protrude filopodia into all dimensions, whereas scanned live cells are forced to protrude them only into two dimensions can be excluded since no enhanced filopodia formation in the third dimension on fixed cells was observed. A further difference was found in the overall lifetime of platelets. Living platelets did not detach significantly from the support when kept untouched, but were removed after about three hours of permanent imaging. The latter finding is most likely a result from cantilever-tip accelerated apoptosis.
3.2. SMFS on αIIbβ3 integrin receptors on live platelets
In addition to morphological studies, bio-functional properties of receptors were investigated using a force-probe mode of AFM. Thereby, single molecule force spectroscopy experiments were performed to explore the functional binding states of the platelet membrane receptors. The goal of this study was to determine the activation state of the most prominent platelet integrin αIIbβ3. Time dependent force spectroscopy measurements with the antibodies CD41 or PAC-1 tethered to the AFM tip by a long polyethylene glycol chain (Acetal-PEG-NHS) were performed on adhered live blood platelets during the various activation periods as schematically shown in Fig. 4A. A well established functionalization protocol was used to ensure that statistically only one single antibody is covalently bound to the outer tip apex (which is the contact region with the cell). The tip was amino-functionalized with APTES [24,29] followed by a two step coupling of CD41 or PAC-1 mAb [27] as described in Section 2.2.
Fig. 4.
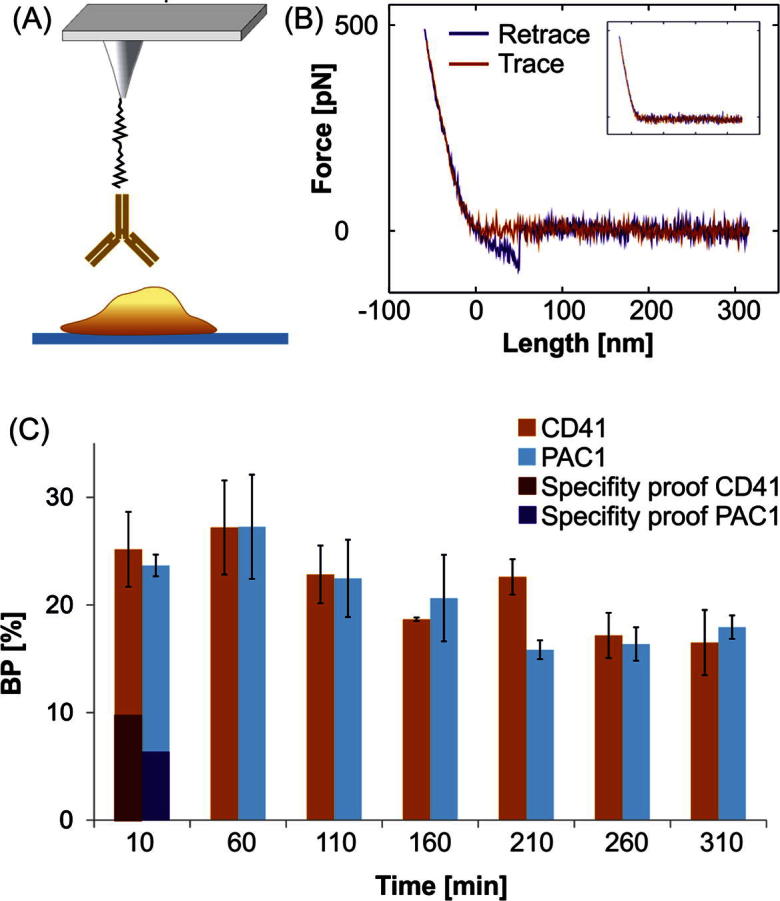
(A) Configuration for SMFS measurements. A mAb (CD41 or PAC-1) modified AFM tip is positioned above a live platelet (containing membrane embedded αIIbβ3 receptors). (B) Typical force distance cycle for MRFS experiments. An unbinding event with an unbinding length of ∼50 nm and a rupture force of ∼60 pN is shown. In the blocking experiment (inset) no specific unbinding event occurred. (C) Binding probability of CD41 and the PAC1 mAb to αIIbβ3 receptors in the membrane of live platelets on glass plotted against time.
The probability of complex formation and the rupture force was investigated recording force-distance-cycles (FDCs). Such FDCs allow following single molecule recognition events as shown in Fig. 4B: In the beginning of a measurement cycle the tip is far away from the cell surface (Fig. 4B red curve, right side). A cantilever comes closer and reaches the point of contact and starts to bend until a previously set force limit (typically 300–600 pN) is reached. During the whole contact time the tip tethered (CD41 or PAC-1) mAb can form a complex with its cognate receptor. Subsequently the retraction period (Fig. 4B), blue curve) is started. When the tip detaches from the cell surface, the cantilever reaches its resting position and the deflection goes back to zero. If a ligand-receptor complex is formed, further withdrawing of the tip from the cell surface causes a cantilever bending towards the surface accompanied by a stretching of the cellular membrane and the linker, until the antibody on the tip dissociates from the receptor in the membrane. This can be seen as an unbinding event (Fig. 4B), blue curve) in the FDC. In contrast, by avoiding complex formation (e.g. by blocking the receptor binding sites) the unbinding events disappear as depicted in the inset of Fig. 4B.
Our results show minor changes in the binding activity and no significant change of the interaction strength of both antibodies for integrin αIIbβ3 over time. The mean interaction forces of CD41 and PAC-1 were 60 to 80 pN and 45–65 pN, respectively, irrespective of the activation period of the platelets. In addition, the unbinding length (distance from point of contact to unbinding, c.f. Fig. 4B, blue curve) remained almost the same (40–50 nm), indicating that the platelet elasticity was independent of its activation state.
From temporal changes of the binding probability (BP) (cf. 1.6.2.) between mAbs and the platelet, predictions for the number densities and/or binding epitope accessibilities of αIIbβ3 integrins in the platelet membrane can be made. As shown in figure Fig. 4C only minor changes were observed for both antibodies, CD41 (light red bars) and PAC-1 (light blue bars). The maximum binding probability of 26% was reached after one hour, which correlates with the blebbing period. Subsequently the BP decreases to values of about 20% or lower. These variations are most probably caused by the morphological changes of the platelets over time that might influence the accessibility of the integrin binding epitope due to local variations in the membrane topology. Remarkably, PAC-1 only senses the active form of the integrin αIIbβ3 whereas CD41 complexes with αIIbβ3 independent on its activation state. Fig. 4C clearly shows that at all investigated times (i.e. in all phases of platelet activation) the BP of PAC-1 (light blue bar) and CD41 (light red bar) are almost equal. This indicates that at all investigated time points all αIIbβ3 integrins are in their activated form. Thus, once activated, the αIIbβ3 integrin remains in its active conformation during the whole activation process until apoptosis. In addition, we could not see significant differences depending on the investigated position on the platelet (central part or filopodia region).
The specifity of the antibody-integrin interaction was verified by blocking experiments. In order to inhibit complex formations of the tip tethered mAbs with integrins embedded in the platelet membrane, the functionalized tips were incubated in a buffer containing αIIbβ3 receptors as described in article 2.5. Live platelet samples as well as fixed platelets showed a significant lower binding probability by using these previously blocked tips. For live platelets the BP decreased from 28–9% for CD41 (Fig. 4C, dark red bar) and from 22–7% for PAC-1 (dark blue bar). These numbers are typical for SMFS measurement on cellular surfaces and verify the specificity of the investigated interactions. The remaining non-specific binding can be explained by unspecific tip-surface adhesion.
4. Conclusion
Unfixed, adhering and living platelets were probed with the AFM under near physiological conditions with high resolution. During activation unfixed platelets went through a sequence of morphological changes, i.e. activation with subsequent disappearance of filopodia followed by a 2nd phase with filopodia formation finally ending in apoptosis. These events of shape changes were more accelerated even by gentle AFM imaging compared to cells fixed after different activation times. Compared to cells fixed after different activation times, those shape changes were accelerated even by gentle AFM imaging. In addition to the observation of structural changes, single molecule force spectroscopy was utilized to probe membrane receptors at the single molecule level. By investigating the integrin αIIbβ3 using two different specific antibodies we showed that αIIbβ3 remains in an activated state during the entire activation process, independent of morphological shape changes. Thus SMFS has the potential to investigate membrane receptors in various activation states by using its cognetive binding molecule attached to the AFM tip. Potentially, by varying the force loading rates, kinetic (e.g. koff) and energetic parameters (e.g. xβ) of the complex can be determined thus making AFM to a powerful tool in platelet research.
Acknowledgments
This work was supported by the MNT-era.net project IntelliTip (FFG 823980) and the FWF (Project I 767-B11). We thank Christian Rankl for his support in data evaluation and Hermann Gruber for providing heterobifunctional crosslinker, and Beatrix Riedl and Elisabeth Köberl for fruitful discussion.
Footnotes
This is an open-access article distributed under the terms of the Creative Commons Attribution-NonCommercial-No Derivative Works License, which permits non-commercial use, distribution, and reproduction in any medium, provided the original author and source are credited.
References
- 1.Jurk K., Kehrel B.E. Seminars in Thrombosis and Hemostasis. 2005;31:381–392. doi: 10.1055/s-2005-916671. [DOI] [PubMed] [Google Scholar]
- 2.Michelson A.D. Academic Press, Elsevier Inc.; London, Waltham, San Diego: 2013. Platelets. [Google Scholar]
- 3.Quinn M. Humana Press; Totowa, New Yersey: 2005. Platelet Physiology. [Google Scholar]
- 4.Kickler T.S. Transfusion Alternatives in Transfusion Medicine. 2005:79–85. [Google Scholar]
- 5.Bearer E.L. Humana Press; Totowa, New Yersey: 2005. Structure-Function of the Platelet Cytoskeleton. [Google Scholar]
- 6.Fritz M., Radmacher M., Gaub H.E. Biophysical Journal. 1994;66:1328–1334. doi: 10.1016/S0006-3495(94)80963-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fritz M., Radmacher M., Gaub H.E. Experimental Cell Research. 1993;205:187–190. doi: 10.1006/excr.1993.1074. [DOI] [PubMed] [Google Scholar]
- 8.Kuwahara M., Sugimoto M., Tsuji S.F., Matsui H., Mizuno T., Miyata S., Yoshioka A. Arteriosclerosis, Thrombosis, and Vascular Biology. 2002:329–334. doi: 10.1161/hq0202.104122. [DOI] [PubMed] [Google Scholar]
- 9.Radmacher M., Fritz M., Kacher C.M., Cleveland J.P., Hansma P.K. Biophysical Journal. 1996;70:556–567. doi: 10.1016/S0006-3495(96)79602-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee I., Marchant R.E. Colloids and Surfaces B: Biointerfaces. 2000;19:357–365. doi: 10.1016/s0927-7765(00)00144-2. [DOI] [PubMed] [Google Scholar]
- 11.Walch M., Ziegler U., Groscurth P. Ultramicroscopy. 2000;82:259–267. doi: 10.1016/s0304-3991(99)00135-7. [DOI] [PubMed] [Google Scholar]
- 12.Holland N.B., Siedlecki C.A., Marchant R.E. Journal of Biomedical Materials Research. 1999;45:167–174. doi: 10.1002/(sici)1097-4636(19990605)45:3<167::aid-jbm2>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 13.Sung K., Frojmovic M., O’Toole T., Zhu C., Ginsberg M., Chien S. Blood. 1993;81:419–423. [PubMed] [Google Scholar]
- 14.Goldsmith H.L., McIntosh F.A., Shahin J., Frojmovic M.M. Biophysical Journal. 2000;78:1195–1206. doi: 10.1016/S0006-3495(00)76677-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Litvinov R.I., Shuman H., Bennett J.S., Weisel J.W. Proceedings of the National Academy of Sciences. 2002;99:7426–7431. doi: 10.1073/pnas.112194999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Litvinov R.I., Bennett J.S., Weisel J.W., Shuman H. Biophysical Journal. 2005;89:2824–2834. doi: 10.1529/biophysj.105.061887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Agnihotri A., Soman P., Siedlecki C.A. Colloids and Surfaces B. 2009;71:138–147. doi: 10.1016/j.colsurfb.2009.01.019. [DOI] [PubMed] [Google Scholar]
- 18.Weisel J.W., Shuman H., Litvinov R.I. Current Opinion in Structural Biology. 2003;13:227–235. doi: 10.1016/s0959-440x(03)00039-3. [DOI] [PubMed] [Google Scholar]
- 19.Arya M., López J.A., Romo G.M., Cruz M.A., Kasirer-Friede A., Shattil S.J., Anvari B. Journal of Thrombosis and Haemostasis. 2003;1:1150–1157. doi: 10.1046/j.1538-7836.2003.00295.x. [DOI] [PubMed] [Google Scholar]
- 20.Litvinov R.I., Barsegov V., Schissler A.J., Fisher A.R., Bennett J.S., Weisel J.W., Shuman H. Biophysical Journal. 2011;100:165–173. doi: 10.1016/j.bpj.2010.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Litvinov R.I., Nagaswami C., Vilaire G., Shuman H., Bennett J.S., Weisel J.W. Blood. 2004;104:3979–3985. doi: 10.1182/blood-2004-04-1411. [DOI] [PubMed] [Google Scholar]
- 22.Litvinov R.I., Vilaire G., Li W., DeGrado W.F., Weisel J.W., Bennett J.S. Biochemistry. 2006;45:4957–4964. doi: 10.1021/bi0526581. [DOI] [PubMed] [Google Scholar]
- 23.Lee I.M. Surface Science. 2001;491:433–443. [Google Scholar]
- 24.Ebner A., Hinterdorfer P., Gruber H.J. Ultramicroscopy. 2007;107:922–927. doi: 10.1016/j.ultramic.2007.02.035. [DOI] [PubMed] [Google Scholar]
- 25.Hussain A.A., Siedlecki C.A. Langmuir. 2005;21:6979–6986. doi: 10.1021/la046943h. [DOI] [PubMed] [Google Scholar]
- 26.Hsieh C.F., Chang Bo-Jui, Pai Chyi-Huey, Chen Hsuan-Yi, Chi Sien, Hsu Long, Tsai Jin-Wu, Lin Chi-Hung, Chang Bo-Jui, Pai Chyi-Huey, Chen Hsuan-Yi, Long Sien Chi, Tsai Jin-Wu, Lin Chi-Hung. Optical Trapping and Optical Micromanipulation. 2004;5514:12–25. [Google Scholar]
- 27.Wildling L., Unterauer B., Zhu R., Rupprecht A., Haselgrùbler T., Rankl C., Ebner A., Vater D., Pollheimer P., Pohl E.E., Hinterdorfer P., Gruber H.J. Bioconjugate Chemistry. 2011;22:1239–1248. doi: 10.1021/bc200099t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hutter J.L., Bechhoefer J. Review of Scientific Instruments. 1993;64:1868–1873. [Google Scholar]
- 29.Lyubchenko Y., Shlyakhtenko L., Harrington R., Oden P., Lindsay S. Proceedings of the National Academy of Sciences. 1993;90:2137–2140. doi: 10.1073/pnas.90.6.2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.<http://rsbweb.nih.gov/ij/index.html>.
- 31.Leytin V., Allen D.J., Mykhaylov S., Mis L., Lyubimov E.V., Garvey B., Freedman J. Biochemical and Biophysical Research Communications. 2004;320:303–310. doi: 10.1016/j.bbrc.2004.05.166. [DOI] [PubMed] [Google Scholar]
- 32.Siljander P.R.M. Thrombosis Research. 2011;127(Suppl. 2):S30–S33. doi: 10.1016/S0049-3848(10)70152-3. [DOI] [PubMed] [Google Scholar]
- 33.Vanags D.M., Orrenius S., Aguilar-Santelises M. British Journal of Haematology. 1997;99:824–831. doi: 10.1046/j.1365-2141.1997.4813284.x. [DOI] [PubMed] [Google Scholar]
- 34.Wolf B.B., Goldstein J.C., Stennicke H.R., Beere H., Amarante-Mendes G.P., Salvesen G.S., Green D.R. Blood. 1999;94:1683–1692. [PubMed] [Google Scholar]
- 35.Leytin V., Allen D.J., Mutlu A., Mykhaylov S., Lyubimov E., Freedman J. British Journal of Haematology. 2008;142:494–497. doi: 10.1111/j.1365-2141.2008.07209.x. [DOI] [PubMed] [Google Scholar]
- 36.Özgen Ü., Özerol Elif, Aminci Mehmet. Turkish Journal of Hematology. 2007;24:171–176. [PubMed] [Google Scholar]