

Abstract
The central processes of primary nociceptors form synaptic connections with the second-order nociceptive neurons located in the dorsal horn of the spinal cord. These synapses gate the flow of nociceptive information from the periphery to the CNS, and plasticity at these synapses contributes to centrally mediated hyperalgesia and allodynia. Although exocytosis and synaptic plasticity are controlled by Ca2+ at the release sites, the mechanisms underlying presynaptic Ca2+ signalling at the nociceptive synapses are not well characterized. We examined the presynaptic mechanisms regulating Ca2+ clearance following electrical stimulation in capsaicin-sensitive nociceptors using a dorsal root ganglion (DRG)/spinal cord neuron co-culture system. Cytosolic Ca2+ concentration ([Ca2+]i) recovery following electrical stimulation was well approximated by a monoexponential function with a τ∼2 s. Inhibition of sarco-endoplasmic reticulum Ca2+-ATPase did not affect presynaptic [Ca2+]i recovery, and blocking plasmalemmal Na+/Ca2+ exchange produced only a small reduction in the rate of [Ca2+]i recovery (∼12%) that was independent of intracellular K+. However, [Ca2+]i recovery in presynaptic boutons strongly depended on the plasma membrane Ca2+-ATPase (PMCA) and mitochondria that accounted for ∼47 and 40%, respectively, of presynaptic Ca2+ clearance. Measurements using a mitochondria-targeted Ca2+ indicator, mtPericam, demonstrated that presynaptic mitochondria accumulated Ca2+ in response to electrical stimulation. Quantitative analysis revealed that the mitochondrial Ca2+ uptake is highly sensitive to presynaptic [Ca2+]i elevations, and occurs at [Ca2+]i levels as low as ∼200–300 nm. Using RT-PCR, we detected expression of several putative mitochondrial Ca2+ transporters in DRG, such as MCU, Letm1 and NCLX. Collectively, this work identifies PMCA and mitochondria as the major regulators of presynaptic Ca2+ signalling at the first sensory synapse, and underlines the high sensitivity of the mitochondrial Ca2+ uniporter in neurons to cytosolic Ca2+.
Key points
The first sensory synapse formed between the central processes of primary afferents and dorsal horn neurons plays an important role in controlling the flow of nociceptive information from the periphery to the CNS, and plasticity at this synapse contributes to centrally mediated pain hypersensitivity.
Although exocytosis and synaptic plasticity are regulated by presynaptic Ca2+, the mechanisms underlying presynaptic Ca2+ signalling at the first sensory synapse are not well understood.
In this study we show that the plasma membrane Ca2+-ATPase and mitochondria are the major regulators of presynaptic Ca2+ signalling in capsaicin-sensitive dorsal root ganglion neurons accounting for ∼47 and ∼40% of presynaptic Ca2+ clearance, respectively.
Quantitative analysis of changes in cytosolic and mitochondrial Ca2+ concentrations demonstrates that the mitochondrial Ca2+ uniporter is highly sensitive to cytosolic Ca2+ at this synapse.
These results help us understand presynaptic mechanisms at the first sensory synapse.
Introduction
Primary nociceptive neurons send their central processes to the dorsal horn of the spinal cord where they form glutamatergic synaptic connections with the second-order nociceptive neurons (Woolf & Salter, 2006; Kuner, 2010). This synaptic connection, often referred to as the first sensory synapse, plays a critical role in controlling the flow of nociceptive information from the periphery into the CNS. Inhibiting transmission at this synapse with blockers of voltage-gated Ca2+ channels or opioids produces analgesic effects (Cao, 2006; McGivern, 2006; Heinke et al. 2011), whereas facilitation of sensory transmission through various forms of synaptic plasticity (including wind-up, central sensitization and long-term potentiation) contributes to the pain hypersensitivity associated with inflammation or nerve injury (Ji et al. 2003; Woolf & Salter, 2006; Kuner, 2010; Ruscheweyh et al. 2011). The mechanisms responsible for various forms of synaptic plasticity at the first sensory synapse are complex and not fully understood.
Presynaptic Ca2+ is the principal regulator of neurotransmitter release and synaptic plasticity that acts via multiple Ca2+-sensing proteins to control almost every aspect of the synaptic vesicle life cycle. For example, Ca2+ triggers synchronous transmitter release via the low-affinity Ca2+ sensors synaptotagmins 1, 2 and 9 (Sugita et al. 2002; Xu et al. 2007), whereas asynchronous and spontaneous transmitter release are mediated by the high-affinity Ca2+ sensor Doc2 (Groffen et al. 2010; Yao et al. 2011). Endocytotic retrieval of synaptic vesicles is stimulated by Ca2+/calcineurin-dependent dephosphorylation of dynamin-1 and other endocytotic proteins (Cousin & Robinson, 2001; Clayton & Cousin, 2009). Residual presynaptic [Ca2+]i accumulated during repeated electrical stimulation contributes to short-term synaptic plasticity by controlling the size of the release-ready pool of synaptic vesicles (Zucker & Regehr, 2002; Neher & Sakaba, 2008) through Ca2+-dependent activation of Munc13 and protein kinases A and C (PKA and PKC; Turner et al. 1999; Junge et al. 2004; Sørensen, 2004). To carry out these versatile synaptic functions, presynaptic Ca2+ signals must be precisely organized in time and space, which is achieved through the coordinated actions of Ca2+ channels, buffers and transporters. Therefore, identifying the specific components of presynaptic Ca2+ machinery is essential for understanding mechanisms of synaptic transmission and plasticity at any given synapse.
In spite of the key role of the first sensory synapse in transmitting sensory information and processing pain, the mechanisms that control presynaptic cytosolic Ca2+ concentration ([Ca2+]i) at this synapse are not known. Here we used cytosolic and mitochondrial Ca2+ imaging to examine mechanisms of presynaptic [Ca2+]i clearance at the synapses formed between dorsal root ganglion (DRG) and spinal cord (SC) neurons in culture. We focused our analysis on a subset of nociceptive DRG neurons that express TRPV1 (transient receptor potential vanilloid 1) receptors because of their well-established role in acute and chronic pain (Caterina & Julius, 2001; Szallasi et al. 2007). Our data demonstrate that plasma membrane Ca2+-ATPase (PMCA) and mitochondria are the major regulators of presynaptic [Ca2+]i at this synapse.
Methods
Cell culture
DRG/SC co-cultures were prepared as previously described (Medvedeva et al. 2008, 2009). In brief, newborn (postnatal days 0–2) Sprague–Dawley rats were killed by decapitation with sharp scissors, according to a protocol approved by the University of Iowa Institutional Animal Care and Use Committee and in accordance with the guidelines of the National Institutes of Health. Every effort was made to minimize the number of animals used. DRG were dissected from the thoracic and lumbar segments and incubated in pronase E dissolved in DMEM (1 mg ml−1) containing 20 mm Hepes (pH 7.4) and penicillin–streptomycin (100 U ml−1 and 100 μg ml−1, respectively) for 7 min at 37°C in a 10% CO2 incubator. Ganglia were washed twice in cold DMEM containing Hepes (20 mm; pH 7.4) and then mechanically dissociated by trituration with flame-constricted Pasteur pipettes of decreasing diameter. The spinal cord was dissected into small segments (∼0.5 mm) and then digested in DMEM containing 0.025% trypsin, Hepes (20 mm, pH 7.4) and penicillin–streptomycin (100 U ml−1 and 100 μg ml−1, respectively) for 8 min at 37°C in a 10% CO2 incubator. SC segments were then washed with cold complete DMEM medium supplemented with 5% heat-inactivated horse serum, 5% fetal bovine serum and penicillin–streptomycin (100 U ml−1 and 100 μg ml−1, respectively) and dissociated using the procedure described above for DRG dissociation. Suspensions of SC and DRG cells were plated onto 25 mm glass coverslips coated with poly-l-ornithine and laminin. Cells were grown in DMEM supplemented with 5% heat-inactivated horse serum, 5% fetal bovine serum, insulin (6 μg ml−1) and penicillin–streptomycin (100 U ml−1 and 100 μg ml−1, respectively) in a 10% CO2 incubator at 37°C. After 48 h, 5 μm cytosine β-d-arabinofuranoside (AraC) was added to cultures for 24 h. Two days later, cells were again treated with 5 μm AraC for 24 h. Cultures were grown for 10–16 days before use; 50% of the culture medium was replaced every 5–6 days.
Electrophysiological recordings
The methods for monitoring whole-cell Ca2+ currents and excitatory postsynaptic currents (EPSCs) were similar to those previously described by our group (Medvedeva et al. 2008, 2009). Whole-cell patch-clamp recordings were obtained using a Axopatch 200B patch-clamp amplifier and a Digidata 1322A analog-to-digital converter (Molecular Devices, Union City, CA, USA). Data were collected (filtered at 2 kHz and sampled at 5 kHz) and analysed using the pClamp 9 software (Molecular Devices). Patch pipettes were pulled from borosilicate glass (Narishige, New York, USA; 3–5 mΩ) on a Sutter Instruments (Novato, CA, USA) P-87 micropipette puller. For EPSC recordings, spinal cord neurons were voltage-clamped at –60 mV using patch pipettes that were filled with the following solution (mm): 125 potassium gluconate, 10 KCl, 3 Mg-ATP, 1 MgCl2, 5 EGTA and 10 Hepes (pH 7.25 adjusted with KOH; 290 mosmol kg−1 with sucrose). The standard extracellular recording solution contained (mm): 140 NaCl, 5 KCl, 1.3 CaCl2, 0.4 MgSO4, 0.5 MgCl2, 0.4 KH2PO4, 0.6 Na2HPO4, 3 NaHCO3, 10 glucose and 10 Hepes (pH 7.4 with NaOH; 310 mosmol kg−1 with sucrose). The extracellular solution also contained 10 μm bicuculline and 2 μm strychnine to block inhibitory postsynaptic currents mediated by GABAA and glycine receptors, respectively. Evoked EPSCs in SC neurons were elicited every 4 s using a glass extracellular stimulating electrode (0.2–0.4 ms pulse) positioned near the cell body of a nearby DRG neuron. For Ca2+ current recordings, extracellular solution contained (mm): 115 choline chloride, 30 TEACl, 1.3 CaCl2, 1 MgCl2, 10 glucose, 10 Hepes and 1 μm tetrodotoxin (pH 7.4 with TEAOH; 310 mosmol kg−1 with sucrose); and the patch-pipette solution had the following composition (mm): 135 caesium gluconate, 3 Mg-ATP, 1 MgCl2, 10 EGTA and 10 Hepes (pH 7.25 adjusted with CsOH; 290 mosmol kg−1 with sucrose). Voltage-gated Ca2+ currents were evoked by step depolarizations from –60 to +10 mV (100 ms, every 20 s).
Measurements of [Ca2+]i in axonal boutons
The protocols for monitoring presynaptic [Ca2+]i were similar to those we have previously described (Medvedeva et al. 2008, 2009). Cells were placed in a flow-through chamber mounted on the stage of an inverted IX-71 microscope (Olympus, Tokyo, Japan). For [Ca2+]i measurements, DRG neurons were loaded with the low affinity (Kd∼ 5.5 μm) Ca2+ indicator Fura-FF (200 μm) or, in some experiments involving small-amplitude [Ca2+]i measurements (see Figs 4D, E and 8C, D) the high-affinity (Kd = 225 nm) Ca2+ indicator Fura-2 (200 μm), via patch pipettes by holding neurons in the whole-cell configuration for 3–5 min; recordings began within 15–20 min of the pipette withdrawing. Only neurons that had membrane potential more negative than –50 mV immediately after patch membrane rupture were used for further experimentation. Fluorescence of Fura-FF or Fura-2 was alternately excited at 340 nm (12 nm bandpass) and 380 nm (12 nm) using a Polychrome IV monochromator (TILL Photonics, Gräfelfing, Germany) via a 40× oil-immersion objective (NA = 1.35, Olympus). Emitted fluorescence was collected at 510 (80) nm using an IMAGO CCD camera (TILL Photonics). Pairs of 340/380 nm images were sampled at 10 Hz. The fluorescence ratio (R = F340/F380) was converted to [Ca2+]i according to the formula: [Ca2+]i = Kdβ(R–Rmin)/(Rmax–R) (Grynkiewicz et al. 1985). The dissociation constants (Kd) used for Fura-2 and Fura-FF were 225 and 5500 nm, respectively (Molecular Probes Handbook). Rmin, Rmax and β were determined by applying 10 μm ionomycin in Ca2+-free buffer (1 mm EGTA) and saturating Ca2+ (1.3 mm Ca2+). For Fura-2, calibration constants were: Rmin = 0.23, Rmax = 2.70 and β = 4.75. For Fura-FF, calibration constants were: Rmin = 0.21, Rmax = 2.25 and β = 5.30. Fluorescence was corrected for background, as determined prior to loading of cells with the indicators.
Figure 4. The rate of presynaptic Ca2+ clearance is not affected by inhibition of SERCA.
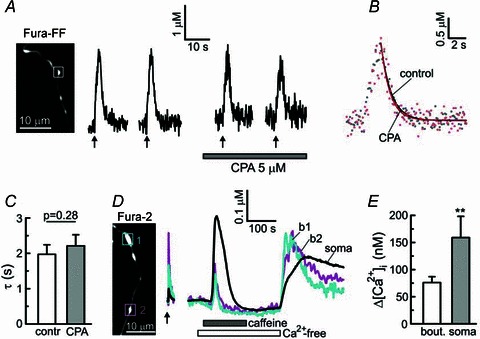
A, [Ca2+]i transients in axonal bouton (white box) were evoked by trains of action potentials (10 Hz for 2 s) in the absence or presence (5 μm) of the SERCA inhibitor cyclopiazonic acid (CPA). The CPA treatment started 3 min prior to the third stimulation. B, [Ca2+]i transients generated in the absence (grey dots) or presence of 5 μm CPA (red dots) are superimposed. The [Ca2+]i traces were offset along the y-axis to facilitate comparison. Traces obtained by monoexponential fitting of the corresponding [Ca2+]i data points are shown by continuous lines. C, plot summarizing time constants for [Ca2+]i recovery in controls (contr) and in the presence of 5 μm CPA (paired Student's t test, n = 18 boutons/4 cells). D, [Ca2+]i changes recorded from two axonal boutons (b1 and b2, squares) and the cell body of a Fura-2-loaded DRG neuron in response to electrical stimulation (6 Hz for 5 s; arrow) or the ryanodine receptor activator caffeine (10 mm). E, comparison of the amplitudes of [Ca2+]i responses to 10 mm caffeine in axonal boutons (n = 27 boutons/5 cells) and somata (n = 5) of DRG neurons. **P < 0.01, unpaired Student's t test.
Figure 8. Quantification of mitochondrial Ca2+ uptake as a function of [Ca2+]i elevation.
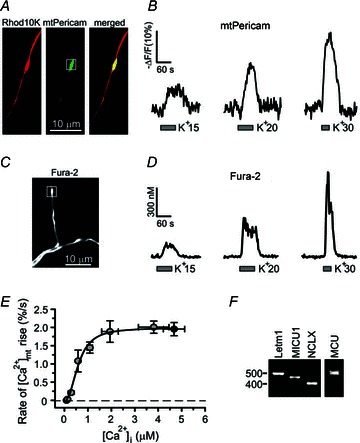
A, DRG neurons were transfected with mtPericam (green) and subsequently loaded with Rhod10K to identify presynaptic boutons. B, [Ca2+]mt changes in axonal boutons (white box) were evoked by depolarization of incremental magnitude using solutions containing 15 mm KCl, 20 mm KCl or 30 mm KCl. C, axonal distribution of Fura-2 in a resting DRG neuron. D, [Ca2+]i changes in axonal boutons (white box) recorded in Fura-2-loaded DRG neurons using the same stimulation protocol as in B. E, the initial rate of the [Ca2+]mt increase is plotted as a function of peak [Ca2+]i elevation in response to K+-induced depolarization or TRPV1 stimulation. [Ca2+]i and [Ca2+]mt changes were evoked by applying 10, 15, 20, 30, 50 or 90 mm KCl (30 s, grey; n = 8–24 boutons from 4–8 cells) or 1 μm capsaicin (30 s; black; n = 16 boutons/5 cells for [Ca2+]i and n = 15 boutons/4 cells for [Ca2+]mt), as described in B and D. [Ca2+]i was measured using Fura-2 (10, 15, 20, 30 and 50 mm KCl) or Fura-FF (90 mm KCl and 1 μm capsaicin); [Ca2+]mt was measured using mtPericam. The initial rate of rise in [Ca2+]mt was calculated by dividing half of the [Ca2+]mt amplitude by the time required to reach half of the [Ca2+]mt peak. The data were fitted using the Hill equation (Origin 7 software), which produced a K0.5 = 606 nm and a Hill coefficient = 2.3. F, RT-PCR analysis of neuron-enriched DRG culture shows that several putative molecular components of mitochondrial Ca2+ transport, Letm1, MCU, MICU1 and NCLX, are expressed in DRG neurons.
After dye loading and patch pipette withdrawal, intact DRG neurons were stimulated with trains of action potentials using extracellular field stimulation as previously described (Usachev & Thayer, 1999). In brief, field potentials were generated by passing current between two platinum electrodes via a Grass SS stimulator and a stimulus isolation unit (Quincy, MA, USA). Trains of 1 ms pulses were delivered at 10 Hz. The stimulus voltage threshold sufficient to elicit a detectable increase in [Ca2+]i from neuronal processes and axonal boutons was determined before beginning an experiment, and the stimulus voltage for further experimentation was set at 20 V higher than the threshold voltage. An increase of the stimulus voltage above the threshold did not lead to a change in the amplitude of the resulting [Ca2+]i transients. For the experiments involving substitution of intracellular K+ with Cs+ to examine the role of K+-dependent Na+/Ca2+ exchange (see Fig. 2E), DRG neurons were stimulated under the whole-cell voltage-clamp mode by applying 20 5-ms voltage pulses from a holding potential of –60 mV to +10 mV at a rate of 10 Hz.
Figure 2. The rate of presynaptic [Ca2+]i recovery is slightly reduced by inhibition of the plasma membrane Na+/Ca2+ exchanger.
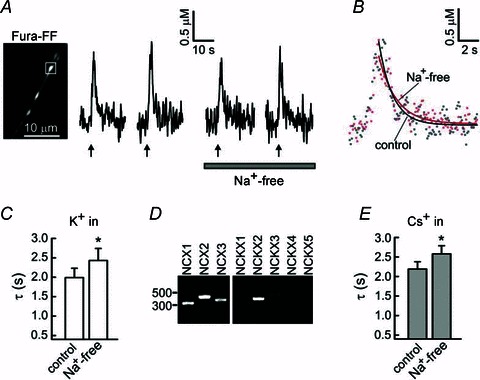
A, [Ca2+]i transients in an axonal bouton (white box) were evoked by trains of action potentials (20 action potentials at 10 Hz for each train; arrows) using extracellular field stimulation under control conditions and after equimolar substitution of extracellular Na+ with Li+ (Na+-free). B, superimposition of [Ca2+]i transients obtained under control (grey dots) and Na+-free (red dots) conditions, for the axonal bouton in A. Continuous lines are the result of the monoexponential fitting procedure. C, summary of time constants (τ) describing presynaptic [Ca2+]i recovery in control conditions and after substitution of extracellular Na+ with Li+ (Na+-free). Experiments were performed as in A. The value of τ was calculated for each [Ca2+]i transient. Time constants for the control condition were obtained by averaging τ for the first and second [Ca2+]i transients, and those for the Na+-free condition were obtained by averaging τ for the third and fourth [Ca2+]i transients. *P < 0.05; paired Student's t test; n = 15 boutons/5 cells D, RT-PCR analysis of NCX1–3 and NCKX1–5 expression in neuron-enriched rat DRG culture. E, summary of time constants describing presynaptic [Ca2+]i recovery obtained under control and Na+-free (equimolar substitution with Li+) conditions in the absence of intracellular K+ (equimolar substitution with Cs+ in the patch pipette). The experimental protocol was similar to that described in A, except that the cells were stimulated under the whole-cell mode of patch-clamp (voltage-clamp), by 20 voltage steps from –60 to +10 mV, for 5 ms each and applied at 10 Hz. The control time constants were obtained by averaging τ for the first and second [Ca2+]i transients, and the Na+-free time constants were obtained by averaging τ for the third and fourth [Ca2+]i transients. *P < 0.05, paired Student's t test.
Analysis of the rate of [Ca2+]i clearance
The recovery phase of each [Ca2+]i transient was fitted with a monoexponential function defined by the formula [Ca2+]i = [Ca2+]i0+Aexp(–(t–t0)/τ) using a non-linear, least-squares curve fitting algorithm (Origin 7 software), where [Ca2+]i0 is the basal [Ca2+]i level, A is the amplitude of the [Ca2+]i response, t0 is the time at the peak [Ca2+]i and τ is the time constant describing [Ca2+]i recovery. For a monoexponential [Ca2+]i recovery process the rate of [Ca2+]i clearance (–d[Ca2+]i/dt) is a linear function of [Ca2+]i that can be determined by the formula: –d[Ca2+]i/dt = k[Ca2+]i, where the rate constant k = 1/τ. To calculate the contribution of each specific Ca2+ transporting system, τ and k were calculated for each presynaptic [Ca2+]i transient under control conditions (τcontrol, kcontrol) and after inhibition of the Ca2+ transporter (τinhibit, kinhibit) of interest. The rate constants were averaged, and the partial contribution of a specific Ca2+ transport was calculated as:
All the data are presented as mean ± SEM.
Measurements of mitochondrial Ca2+ concentrations ([Ca2+]mt) in axonal boutons
[Ca2+]mt was measured using the mitochondria-targeted Ca2+ indicator mtPericam (Nagai et al. 2001) transferred to DRG neurons using lentivirus as previously described (Medvedeva et al. 2008). To visualize axonal boutons, DRG neurons overexpressing mtPericam were loaded with tetramethylrhodamine 10K dextran (Rhod10K; Invitrogen, Carlsbad, CA, USA) via patch pipettes (200 μm). mtPericam fluorescence was excited at 410 (12) nm via a 40× oil-immersion objective (NA = 1.35, Olympus) and measured at 530 (44) nm (sampling rate = 5 Hz). At 410 nm, a decrease in mtPericam fluorescence corresponds to the [Ca2+]mt increase. [Ca2+]mt changes were presented as
where F is the current fluorescence intensity and F0 is the fluorescence intensity in the resting cell. The initial rate of [Ca2+]mt increase was determined by dividing half of the [Ca2+]mt amplitude by the time that was required to reach one half of [Ca2+]mt peak.
Reagents
Fura-2, Fura-FF and Rhod10K were obtained from Invitrogen; capsaicin was from Tocris (Ellisville, MO, USA); ionomycin was from Calbiochem (San Diego, CA, USA); agatoxin IVA was from Bachem (Torrance, CA, USA); and ω-conotoxin GVIA was from Alomone Labs (Jerusalem, Israel). All other reagents were purchased from Sigma (St Louis, MO, USA).
RT-PCR analysis
Enriched neuronal DRG cultures were prepared as previously described (Usachev et al. 2002). In brief, DRG cultures were grown in the presence of AraC (5 μm) for 5–6 days to eliminate non-neuronal cells. To provide trophic support for this glial cell-deficient culture, fresh culture medium was mixed with an equal volume of medium conditioned by DRG cultures grown in the absence of AraC for 4–6 days. These growth conditions yield cultures containing ∼75% of neurons and do not alter [Ca2+]i homeostasis in DRG neurons (Usachev et al. 2002). Total RNA was isolated using the RNase easy mini kit (Qiagen, Valencia, CA, USA) and reverse transcribed and amplified using the one-step RT-PCR kit (Qiagen) according to the manufacturer's protocols. RT-PCR reactions were performed (Hybaid PCR Sprint Thermal Cycler, MA, USA) under the following conditions: 50°C for 30 min for reverse transcription, followed by 95°C for 15 min for initial PCR activation, then 30 cycles of 95°C for 1 min, 55°C for 1 min and 72°C for 1 min, and 72°C for 10 min. Amplified transcripts were separated by electrophoresis on 1% agarose gels and detected using ethidium bromide. The sets of PCR primers are listed in Table 1.
Table 1.
Primer sequences
| Target gene (accession no.) | Primer sequence |
|---|---|
| NCX1 | F: 5′-CTTCGTCCCACCTACAGAAT-3′ |
| (NM_019268.2) | R: 5′-TGGTAGATGGCAGCAATGGA-3′ |
| NCX2 | F: 5′-TGCCATCCTGCTTTGACTAC-3′ |
| (NM_078619.1) | R: 5′-GTCAACAGTGTGACCGAGAA-3′ |
| NCX3 | F: 5′-GAAACATGCAGCAGAGCAAG-3′ |
| (NM_078620.1) | R: 5′-GACATTGCTCAGTCTCACGA-3′ |
| NCKX1 | F: 5′-CACCTTCCTGGGATCCATCATC-3′ |
| (NM_020090.1) | R: 5′-CGATCTTCTAACATCACACTGATC-3′ |
| NCKX2 | F: 5′-TTATCATGTGGTGGGAAAGC-3′ |
| (NM_031743.2) | R: 5′-GCTTTTTCTCTGAACCTCCC-3′ |
| NCKX3 | F: 5′-GACGTTTGCTTCCTCTACACTGT-3′ |
| (NM_053505.1) | R: 5′-AACTCCGTCATGATGGAGAAA-3′ |
| NCKX4 | F: 5′-GGTGTGGCTGGTGACTATTATTG-3′ |
| (NM_001108051.1) | R: 5′-CGTTGCTTCCGATGGTGTTAGAG-3′ |
| NCKX5 | F: 5′-CTTTCTCTAAGTTTACATGGTCTTAGT-C-3′ |
| (NM_001107769.1) | R: 5′-CTTTCGGTCTAGTTTCCAGCC-G-3′ |
| Letm1 | F: 5′-AATAAGCTGCCCTCCACATTTG-3′ |
| (NM_001005884.1) | R: 5′-TGTTTCAGCTGGCCCTTCAG-3′ |
| NCLX | F: 5′-AGTCACCGCAGCCAAGTTCTT-C-3′ |
| (NM_001017488.1) | R: 5′-AGTGCAGATGATGACCGTGACC-3′ |
| MICU1 | F: 5′-ACCCAATGAGAAGCAGCCAG-3′ |
| (NM_199412.1) | R: 5′-ACGTCATGCTGCAGTTTACGC-3′ |
| MCU | F: 5′-AGTACGGTTGTGCCCTCTGATG-3′ |
| (NM_001106398.1) | R: 5′-AGTGGTCCTCTTCTCCGCTTTC-3′ |
Results
DRG neurons were co-cultured with spinal cord neurons for 10–16 days as previously described, to enable formation of sensory synapses in vitro (Medvedeva et al. 2008, 2009). Synapses formed by DRG and SC neurons grown in co-culture possess many important characteristics of the first sensory synapse, such as: (1) EPSCs are inhibited by the antagonists of 2-amino-3-(3-hydroxy-5-methyl-isoxazol-4-yl)propanoic acid (AMPA)-type glutamate receptors (Gruner & Silva, 1994; Gu & Macdermott, 1997; Bao et al. 1998; Medvedeva et al. 2008; Heinke et al. 2011); (2) N-type voltage-gated Ca2+ channels (CaV2.2) play the predominant role in controlling evoked glutamate release (Fig. 1A and B; Gruner & Silva, 1994; Bao et al. 1998; Heinke et al. 2004; Cao, 2006); (3) a large proportion of these synapses are sensitive to TRPV1 agonists such as capsaicin and NADA that produce dramatic increases in the frequency of spontaneous EPSCs (Fig. 1C–E; Yang et al. 1998; Huang et al. 2002; Sikand & Premkumar, 2007; Medvedeva et al. 2008); and (4) these synapses demonstrate wind-up and long-term potentiation (LTP)-like forms of synaptic plasticity (Vikman et al. 2001; Ji et al. 2003; Woolf & Salter, 2006). Consequently, the DRG/SC co-culture system has been widely used as a reliable tool for studying the first sensory synapse under highly defined recording conditions (Gu & Macdermott, 1997; Vikman et al. 2001; Tsuzuki et al. 2004; Sikand & Premkumar, 2007; Medvedeva et al. 2008, 2009).
Figure 1. Characterization of synaptic transmission and action potential-evoked presynaptic [Ca2+]i transients in DRG/SC co-culture.
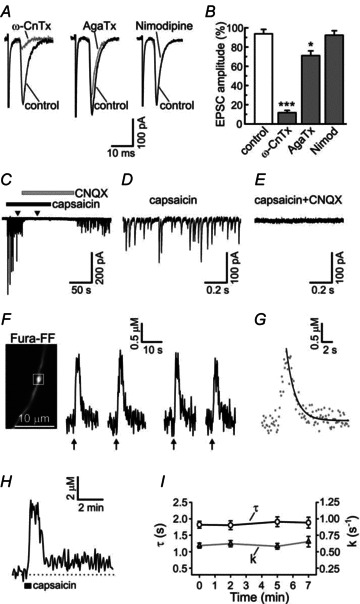
A, evoked EPSCs were recorded in DRG/SC co-culture as previously described (Medvedeva et al. 2008, 2009). The N-type voltage-gated Ca2+ channel (VGCC) inhibitor ω-conotoxin GVIA (1 μm; ω-CnTx) nearly completely abolished the EPSCs (n = 7), whereas the P/Q-type VGCC inhibitor agatoxin IVA (200 nm; AgaTx) produced a much smaller effect (n = 5). The EPSCs were not affected by the L-type VGCC inhibitor nimodipine (5 μm; n = 7). The superimposed EPSC traces represent recordings obtained before (control; black) and during the application of VGCC inhibitors (grey) for individual synaptic pairs. B, plot summarizing the effects of VGCC inhibitors (grey bars) or vehicle control (white bar) on EPSC amplitudes. To quantify the effects of VGCC inhibitors, 10 EPSC traces obtained in the presence of a given VGCC inhibitor (3 min of treatment) were averaged and normalized to the mean amplitude of the first 10 EPSC for each synaptic pair. *P < 0.05, ***P < 0.001, one-way ANOVA with Dunnett's post test (relative to control). C, capsaicin (1 μm) strongly increases the frequency of spontaneous EPSCs. This effect is reversibly inhibited by the AMPA receptor antagonist CNQX (10 μm). Enhanced glutamate release continues long after the capsaicin washout as a result of prolonged mitochondria-dependent presynaptic [Ca2+]i plateau (Medvedeva et al. 2008). D and E, two short segments of EPSC recordings from the experiment described in are shown using an expanded time scale during the treatments with capsaicin alone (D) or capsaicin combined with CNQX (E). The times corresponding to these segments are shown by arrowheads in C. F, presynaptic [Ca2+]i transients evoked by repeated trains of action potentials in capsaicin-sensitive DRG neurons. Presynaptic [Ca2+]i transients were evoked by trains of action potentials (2 s at 10 Hz for each train; arrows) applied at the time points t = 0, 2, 5 and 7 min. Image on the left shows the distribution of Fura-FF fluorescence (λex = 380 nm) in an unstimulated cell. [Ca2+]i recording was made from the axonal bouton indicated by the white box. G, the second [Ca2+]i transient from the experiment in F is shown on an expanded time scale (grey dots), and the monoexponential function obtained by fitting the recovery phase of this [Ca2+]i transient is shown by a continuous line. H, [Ca2+]i response to the TRPV1 agonist capsaicin (1 μm, 30 s), recorded from the axonal bouton in F. [Ca2+]i rapidly recovered to a new steady-state level ([Ca2+]i plateau), and remained elevated for an additional 5–15 min, as previously described (Medvedeva et al. 2008). I, summary of time constants (τ; open circles) and the rate constants (k; grey triangles) calculated for each presynaptic [Ca2+]i transient, using the same experimental protocol as in F. Data are for 27 presynaptic boutons/6 cells. Time and rate constants did not change significantly during the time course of the experiments; repeated one-way ANOVA with Bonferroni post test.
[Ca2+]i transients in presynaptic boutons of DRG neurons were measured using the low-affinity fluorescent Ca2+ indicator Fura-FF (Kd∼ 5.5 μm). This dye is ideally suited for monitoring [Ca2+]i changes predicted to be in the micromolar range, while the low Ca2+ affinity of the indicator minimizes its influence on Ca2+ buffering processes (Neher & Augustine, 1992). Fura-FF was loaded into small to medium-sized DRG neurons (18–32 μm) via a patch pipette, which enabled visualization of multiple axonal boutons along the processes (Figs 1–6). We have previously shown that these structures are associated with the presynaptic proteins synaptophysin and bassoon as well as the postsynaptic marker PSD95 (Medvedeva et al. 2008, 2009). [Ca2+]i transients were evoked by a train of 20 action potentials applied at 10 Hz, consistent with firing patterns of primary sensory neurons in vivo (Meyer & Campbell, 1981; Slugg et al. 2000). Such stimulation produced [Ca2+]i elevation from the basal level of 0.12 ± 0.05 μm to a peak of 2.14 ± 0.12 μm (n = 79 boutons/24 cells; Fig. 1F). After reaching its maximum, [Ca2+]i rapidly recovered to the prestimulus level. For the majority of axonal boutons (79 of 84 tested), [Ca2+]i recovery was well approximated using a monoexponential function with a time constant τ = 2.02 ± 0.11 s (n = 79 boutons/24 cells; Fig. 1G). Each cell was stimulated four times as described in Fig. 1F, and τ and the rate constant (k = 1/τ) for each [Ca2+]i transient were calculated. The first two stimuli were used as controls, and treatments directed at inhibiting various Ca2+ transporters were applied during the third and fourth stimuli. The contribution of a specific Ca2+ clearance mechanism was calculated as (kcontrol–kinhibit)/kcontrol100%, where kcontrol is the average rate constant in the control, and kinhibit is the average rate constant under conditions when a specific Ca2+ transporter is inhibited. In the absence of treatment, τ and k did not change significantly throughout the course of the experiment (Fig. 1I). Functional expression of TRPV1 was determined at the end of each experiment by applying 1 μm capsaicin (30 s; Fig. 1H), and only capsaicin-responding cells (mean presynaptic [Ca2+]i elevation ≥ 100% above baseline) were used for further analysis. We used the described approach to determine the roles of plasma membrane Na+/Ca2+ exchangers, PMCA, sarco-endoplasmic reticulum Ca2+-ATPase (SERCA) and mitochondria in clearing Ca2+ from the presynaptic boutons of DRG neurons.
Figure 6. The rate of presynaptic [Ca2+]i recovery is significantly reduced by inhibition of the mitochondrial uniporter.
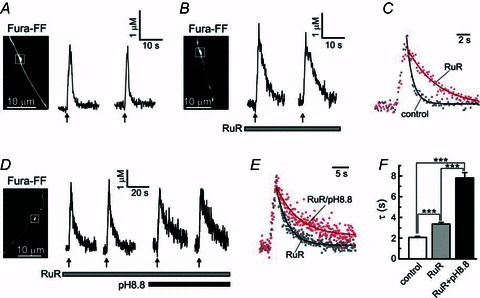
A and B, action potential-evoked (10 Hz for 2 s; arrows) [Ca2+]i transients were recorded in control cells (A) and cells loaded with the MCU inhibitor ruthenium red (RuR; B). In the latter case, 10 μm RuR was included in the patch pipette solution, along with 200 μm Fura-FF. White boxes indicate axonal boutons from which recordings were made. C, control [Ca2+]i trace (grey dots) is superimposed with a [Ca2+]i trace recorded in the presence of 10 μm RuR (red dots). [Ca2+]i transients were normalized to facilitate comparison of the recovery kinetics. Continuous lines indicate the plots obtained by monoexponential fitting of the corresponding [Ca2+]i traces. D, [Ca2+]i transients were studied in cells containing 10 μm RuR under normal (pH = 7.4) or basic (pH = 8.8) extracellular pH. [Ca2+]i responses were induced by trains of action potentials (10 Hz for 2 s, arrows) generated by extracellular field stimulation. E, superimposition of [Ca2+]i transients obtained under normal (pH = 7.4; grey dots) or elevated (pH = 8.8) extracellular pH (red dots), for the same cell as shown in D. Continuous lines represent mononexponential fitting of the corresponding [Ca2+]i data points. F, summary of the effects of RuR and high pH on the time constant of [Ca2+]i recovery. The data were obtained from experiments such as those shown in A, B and D. ***P < 0.001, one-way ANOVA with Bonferroni post test; n = 79 boutons/24 cells for control; n = 33 boutons/7 cells for RuR only; n = 16 boutons/5 cells for RuR + pH 8.8.
Presynaptic Ca2+ clearance in DRG neurons depends to a small extent on plasma membrane Na+/Ca2+ exchange
Plasma membrane Na+/Ca2+ exchangers are low-affinity Ca2+ transporters that are expressed in excitable and non-excitable cells (Blaustein & Lederer, 1999; Lytton, 2007). Two large families of the exchangers have been identified: the K+-independent Na+/Ca2+ exchangers (NCX proteins), and the K+ -dependent Na+/Ca2+ exchangers (NCKX proteins). The NCX transporters utilize the transmembrane Na+ gradient to extrude 1 Ca2+ ion from the cell in exchange for 3 or 4 Na+ ions moved into the cell, whereas the NCKX transporters extrude 1 Ca2+ and 1 K+ ion in exchange for 4 Na+ ions (Blaustein & Lederer, 1999; Lytton, 2007). We blocked plasma membrane Na+/Ca2+ exchange by replacing extracellular Na+ with Li+. This treatment produced a small but significant slowing of presynaptic [Ca2+]i recovery (Fig. 2). In these experiments, the time constant of [Ca2+]i recovery increased from 1.99 ± 0.24 s in control to 2.43 ± 0.31 s in Na+-free external solution (n = 15 boutons/5 cells; P < 0.05, paired Student's t test). The corresponding rate constants were 0.59 ± 0.06 and 0.52 ± 0.07 s−1, respectively (n = 15 boutons/5 cells; P < 0.05, paired Student's t test). The overall contribution of plasma membrane Na+/Ca2+ exchange to presynaptic Ca2+ clearance in DRG neurons was ∼12%. Using the high-affinity Ca2+ indicator Fura-2 we found that the Na+/Ca2+ exchanger also controls presynaptic [Ca2+]i at rest, as replacement of extracellular Na+ with Li+ resulted in a small but significant [Ca2+]i elevation from 107 ± 4 to 134 ± 8 nm (n = 20 boutons/6 cells; P < 0.001, paired Student's t test; data not shown).
The NCX protein family comprises three isoforms (NCX1–3) and the NCKX family five (NCKX1–5). With the exception of NCKX1 (rod-specific isoform), all the NCX and NCKX isoforms are found in the brain (Annunziato et al. 2004; Lytton, 2007). DRG neurons express NCX2 and NCX3, whereas NCX1 is found primarily in satellite cells in the DRG (Persson et al. 2010). Which NCKX isoforms are expressed in DRG neurons is unknown. Using RT-PCR, we detected transcripts of NCX1, NCX2, NCX3 and NCKX2 in neuron-enriched DRG cultures (Fig. 2D). A very weak band corresponding to NCKX3 was also detected. Given the presence of some NCKX isoforms in DRG neurons, we examined whether presynaptic Ca2+ extrusion mediated by Na+/Ca2+ exchange depends on K+. Substitution of intracellular K+ with Cs+ inhibits NCKX2 and NCKX3 by 80 and 95%, respectively (Lee et al. 2007b). Therefore, we replaced K+ with an equimolar amount of Cs+ in the patch pipette solution (Fig. 2E). Since action potentials could not be evoked under these conditions, we stimulated the neurons by applying 20 short (5 ms) depolarization pulses (from –60 to +10 mV) at 10 Hz using the patch clamp technique. In the absence of intracellular K+, the slowing of [Ca2+]i recovery induced by Na+ removal was similar to that observed in the presence of intracellular K+ (compare graphs in Fig. 2C and E), suggesting that NCKX exchangers do not significantly contribute to Ca2+ clearance.
Presynaptic Ca2+ clearance in DRG neurons is highly dependent on PMCA
PMCAs are key components of Ca2+ extrusion in central and peripheral neurons (Benham et al. 1992; Usachev et al. 2002; Wanaverbecq et al. 2003; Empson et al. 2007; Gover et al. 2007b). Four major PMCA isoforms (1–4) have been identified (Carafoli, 1991; Strehler & Zacharias, 2001), and we previously showed that PMCA2 and PMCA4 are the major isoforms expressed in DRG neurons (Usachev et al. 2002). In addition, strong PMCA4 immunoreactivity was detected in the superficial layers of the dorsal horn (Tachibana et al. 2004), the area where the majority of TRPV1-positive central processes terminate (Guo et al. 1999; Cavanaugh et al. 2011). Here we examined the functional significance of PMCAs in presynaptic boutons of DRG neurons. Ca2+ extrusion by PMCA requires the countertransport of protons, and thus can be blocked by raising extracellular pH (Carafoli, 1991; Schwiening et al. 1993; Usachev et al. 2002; Wanaverbecq et al. 2003). Using Fura-2, we found that blocking PMCA by raising extracellular pH to 8.8 led to baseline [Ca2+]i elevation from 98 ± 5 to 178 ± 12 nm (n = 29 boutons/6 cells; P < 0.001, Student's t test; data not shown). Fura-FF-based measurements determined that PMCA inhibition markedly slowed [Ca2+]i recovery in the axonal boutons of DRG neurons (Fig. 3). The time constant increased from 2.01 ± 0.19 s in control to 4.29 ± 0.53 s at pH 8.8 (n = 27 boutons/7 cells; P < 0.001, paired Student's t test). The corresponding rate constants were 0.60 ± 0.06 and 0.32 ± 0.03 s−1, respectively (n = 27 boutons/7 cells; P < 0.001, paired Student's t test). The contribution of PMCA to presynaptic [Ca2+]i clearance was ∼47%.
Figure 3. The rate of presynaptic [Ca2+]i recovery is markedly slowed by inhibition of PMCA.
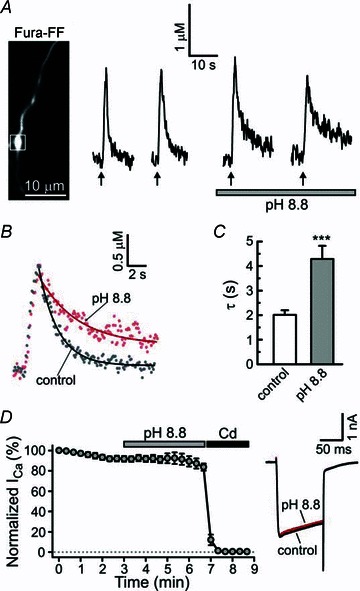
A, [Ca2+]i transients were evoked by trains of 20 action potentials at 10 Hz (arrows) using extracellular field stimulation under control conditions and after raising extracellular pH to 8.8 to block PMCA. The white box indicates an axonal bouton from which [Ca2+]i traces were recorded. B, superimposition of control [Ca2+]i (grey dots) with that obtained under conditions of elevated extracellular pH (red dots). Continuous lines show the results of monoexponential fitting procedure for the corresponding traces. C, plot summarizing τ obtained from seven independent experiments similar to that shown in A. Control time constants were obtained by averaging τ for the first and second [Ca2+]i responses, and the time constants for pH = 8.8 were obtained by averaging τ for the third and fourth [Ca2+]i responses. ***P < 0.001, paired Student's t test, n = 27 boutons/7 cells. D, raising extracellular pH to 8.8 does not affect the amplitude of voltage-activated Ca2+ currents (ICa). Plot (left) summarizing ICa recordings (100 ms depolarizations from –60 to +10 mV) in DRG neurons (n = 6) in control and following treatments with either pH 8.8 or a blocker of voltage-gated Ca2+ channels, Cd2+ (500 μm). The ICa amplitudes were normalized to the amplitude of the first ICa response recorded for each cell. The right panel shows a superimposition of representative ICa traces in control conditions (black) and following extracellular pH increase to 8.8 (red).
We also found that PMCA blockade resulted in a mild but significant increase of the amplitude of electrically evoked presynaptic [Ca2+]i responses from 1.83 ± 0.14 μm in control to 2.25 ± 0.19 μm at pH 8.8 (n = 27 boutons/7 cells; P < 0.01, paired Student's t test). Raising extracellular pH to 8.8 did not have any significant effect on voltage-gated Ca2+ currents in DRG neurons, as determined by whole-cell patch-clamp recordings (Fig. 3D). Thus, PMCA strongly contributes to multiple aspects of presynaptic [Ca2+]i signalling in DRG neurons, including maintenance of low [Ca2+]i at rest, regulation of the [Ca2+]i clearance rate and setting the amplitude of [Ca2+]i elevation in response to action potentials.
Presynaptic Ca2+ clearance does not depend on SERCA
SERCA is a high-affinity Ca2+ pump that serves to lower cytosolic Ca2+ and replenish intracellular Ca2+ stores for subsequent Ca2+ release via the ryanodine or inositol 1,4,5-trisphosphate (IP3) receptors (Henzi & MacDermott, 1992; Pozzan et al. 1994; Thomas & Hanley, 1994). Three SERCA isoforms (1–3) have been identified, two of which (SERCA2 and SERCA3) are present in the brain (Mata & Sepulveda, 2005; Vangheluwe et al. 2009). SERCA plays an important role in regulating [Ca2+]i in the somata of DRG and trigeminal neurons, and its contribution to [Ca2+]i clearance can be 50% and higher, depending on the stimulation protocol and sensory neuron population (Shmigol et al. 1994; Usachev & Thayer, 1999; Lu et al. 2006; Gover et al. 2007a). We tested the effects of a selective SERCA inhibitor, cyclopiazonic acid (CPA, 5 μm), on [Ca2+]i in the presynaptic boutons of DRG neurons. Application of 5 μm CPA did not affect the recovery kinetics of presynaptic [Ca2+]i transients (Fig. 4A–C), and the time constants were 1.97 ± 0.27 and 2.21 ± 0.31 s in control and in the presence of CPA, respectively (n = 18 boutons/5 cells; P = 0.28, paired Student's t test). The corresponding rate constants were 0.66 ± 0.08 and 0.62 ± 0.09 s−1, respectively (P = 0.39, paired Student's t test). Similar results were obtained using another SERCA inhibitor, thapsigargin (Tg) (Thomas & Hanley, 1994; Thayer et al. 2002). Treatment with 1 μm Tg did not significantly change the time constant, which was 2.18 ± 0.17 s in the absence and 2.13 ± 0.19 s in the presence of 1 μm Tg (n = 16 boutons/4 cells; P = 0.72, paired Student's t test; data not shown). Thus, Ca2+ clearance in the presynaptic boutons of DRG neurons is independent of SERCA.
To test whether functional endoplasmic reticulum (ER) Ca2+ stores are present in the axonal boutons of DRG neurons, we applied an activator of Ca2+ release from ryanodine-sensitive stores, caffeine, in a Ca2+-free buffer. To improve the detection of potentially small [Ca2+]i changes in response to caffeine, we used a high-affinity Ca2+ indicator, Fura-2 (Kd = 225 nM), in these experiments. Application of 10 mm caffeine produced a small but detectable Ca2+ release in presynaptic boutons with an amplitude of 76 ± 11 nm (n = 27 boutons/5 cells; Fig. 4D and E). This value was significantly smaller than that for the caffeine-induced [Ca2+]i responses in the somata of DRG neurons (159 ± 39 nm; n = 5; P < 0.01, non-paired Student's t test). In contrast, [Ca2+]i transients induced by electrical stimulation were larger in the axonal boutons than those in the somata (Fig. 4D; arrow). Notably, after caffeine-induced depletion of the ER Ca2+ stores, switching from a Ca2+-free to normal solution containing 1.3 mm Ca2+ led to a pronounced [Ca2+]i overshoot in both presynaptic boutons and somata. This transient [Ca2+]i elevation is probably the result of activation of store-operated Ca2+ channels, as previously described for DRG neurons (Usachev & Thayer, 1999; Lu et al. 2006; Gemes et al. 2011). Moreover, depleting Ca2+ stores with 5 μm CPA or 1 μm Tg induced small but significant presynaptic [Ca2+]i elevations from 106 ± 3 to 152 ± 5 nm for CPA (n = 38 boutons/7 cells; P < 0.001 paired Student's t test) and from 116 ± 8 to 134 ± 7 nm for Tg (n = 14 boutons/4 cells; P < 0.001 paired Student's t test), consistent with the functional presence of store-operated Ca2+ channels in presynaptic boutons of DRG neurons.
Mitochondria regulate the amplitude and duration of presynaptic [Ca2+]i transients in DRG neurons
Presynaptic terminals and boutons are densely packed with mitochondria that support high energy demand at the synapses (Hollenbeck, 2005; Ly & Verstreken, 2006). Mitochondria also contribute to presynaptic Ca2+ signalling and synaptic plasticity at several central synapses, as well as at the neuromuscular junction (Billups & Forsythe, 2002; David & Barrett, 2003; Jonas, 2004; Garcia-Chacon et al. 2006; Lee et al. 2007a). We recently reported that mitochondria control TRPV1-mediated prolonged elevations of presynaptic [Ca2+]i and glutamate release at the first sensory synapse (Medvedeva et al. 2008). Here we examined whether mitochondria are also involved in shaping presynaptic [Ca2+]i transients in response to brief trains of action potentials. Ca2+ uptake by mitochondria is mediated by the mitochondrial Ca2+ uniporter (MCU), and the mitochondrial membrane potential ΔΨmt (∼–150 mV) provides the driving force for the Ca2+ uptake (Bernardi, 1999; Nicholls & Budd, 2000; Thayer et al. 2002). Mitochondrial Ca2+ uptake can be blocked by depolarizing mitochondria with a protonophore, carbonyl cyanide p-trifluoromethoxyphenylhydrazone (FCCP). We examined the effects of 1 μm FCCP on presynaptic [Ca2+]i transients induced by electrical stimulation (10 Hz for 2 s). FCCP treatment was combined with application of the F1,F0 ATP-synthase inhibitor oligomycin (2 μm) to prevent ATP depletion via the reverse mode of the synthase (Nicholls & Ward, 2000; Toescu & Verkhratsky, 2003). Using Fura-2, we found that FCCP+oligomycin increased baseline presynaptic [Ca2+]i from 126 ± 6 to 163 ± 7 nm (n = 17 boutons/4 cells; P < 0.001 Student's t test). Fura-FF-based measurements revealed that FCCP produced a marked slowing of [Ca2+]i recovery in the axonal boutons of DRG neurons (Fig. 5). The time constants were 2.11 ± 0.21 s in control and 4.60 ± 0.39 s after the FCCP treatment (n = 17 boutons/5 cells; P < 0.001, paired Student's t test). The corresponding rate constants were 0.54 ± 0.05 and 0.25 ± 0.02 s−1, respectively (n = 17 boutons/5 cells; P < 0.001, paired Student's t test). We also found that the amplitudes of the [Ca2+]i responses increased from 2.17 ± 0.24 μm in control to 3.45 ± 0.42 μm in the presence of FCCP (n = 17 boutons/5 cells; P < 0.01, paired Student's t test). Whole-cell patch-clamp recordings showed that FCCP+oligomycin treatment reduced voltage-gated Ca2+ currents by ∼30% in DRG neurons (Fig. 5D), probably as a result of reduced Ca2+ buffering and, consequently, increased Ca2+-dependent inhibition of voltage-gated Ca2+ channels (Hernandez-Guijo et al. 2001). Overall, our data suggest that mitochondria play an important role in buffering presynaptic Ca2+ influx and clearing [Ca2+]i elevations after periods of neuronal activity.
Figure 5. Inhibition of mitochondrial uptake by FCCP strongly reduces the rate of presynaptic Ca2+ clearance.
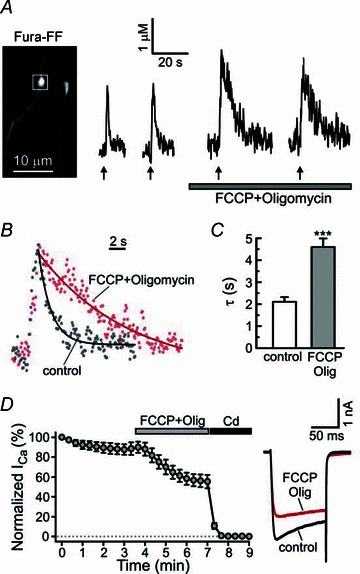
A, electrically evoked [Ca2+]i transients (10 Hz, 2 s; arrows) were recorded from axonal boutons of capsaicin-sensitive DRG neurons (image on the left) in control and during treatment with the mitochondrial protonophore FCCP (1 μm) and F1,Fo ATP-synthase inhibitor oligomycin B (2 μm). B, superimposition of [Ca2+]i traces in control (grey dots; second trace in A) conditions and in the presence of 1 μm FCCP + 2 μm oligomycin (red dots; third trace in A). Plots obtained by monoexponential fitting of [Ca2+]i recovery are shown by continuous lines. C, plot summarizing the time constant data for all the experiments as those shown in A. ***P < 0.001, paired Student's t test. D, treatment with 1 μm FCCP + 2 μm oligomycin decreased the amplitude of voltage-activated Ca2+ currents (ICa) in DRG neurons. Amplitudes of ICa (depolarization from –60 to +10 mV for 100 ms) were normalized to the amplitude of the first evoked ICa for each cell (n = 5). The effect of FCCP + oligomycin was significant starting from t = 5 min (P < 0.001; repeated one-way ANOVA with Dunnett's post test relative to control ICa taken at t = 3 min). ICa was completely blocked by 500 μm Cd2+. Plot on the right shows superimposition of representative ICa traces in control conditions (black) and after 2 min of treatment using FCCP + oligomycin (red).
MCU-mediated Ca2+ transport can be blocked directly by ruthenium red (RuR; Bernardi, 1999; Thayer et al. 2002). An important advantage of this approach is that it does not disrupt either the mitochondrial electrochemical gradient or the ΔΨmt. RuR is membrane impermeable and thus was delivered to the cell through a patch pipette (10 μm) along with Fura-FF (see Methods). The amplitudes of action potential-evoked [Ca2+]i transients were significantly larger in RuR-loaded boutons (4.63 ± 0.45 μm; 33 boutons/7 cells) than in control cells (2.02 ± 0.12 μm; 79 boutons/24 cells; P < 0.001, unpaired Student's t test). As shown in Fig. 6, treatment with RuR also led to a marked slowing of presynaptic [Ca2+]i recovery. The time constants were 2.06 ± 0.10 s in control cells (n = 79 boutons/24 cells) and 3.35 ± 0.16 s in RuR-treated cells (n = 33 boutons/7 cells; P < 0.001, unpaired Student's t test). The corresponding rate constants were 0.58 ± 0.03 s−1 in control cells (n = 79 boutons/24 cells) and 0.35 ± 0.02 s−1 in RuR-loaded cells (n = 33 boutons/7 cells; P < 0.001, unpaired Student's t test). Thus, mitochondria blunt presynaptic [Ca2+]i elevations and play important role in presynaptic Ca2+ clearance in DRG neurons. The mitochondrial contribution to [Ca2+]i recovery process ranges between 40% (based on RuR experiments) and 54% (based on FCCP experiments).
It has been proposed that the Ca2+ accumulated in mitochondria can stimulate several Ca2+-dependent dehydrogenases and thereby enhance the production of ATP (Hajnoczky et al. 1995; Denton, 2009). Thus, the inhibition of mitochondrial Ca2+ uptake could potentially slow ATP-dependent transport, such as PMCA-mediated Ca2+ efflux. We therefore tested the role of PMCA in RuR-treated neurons. Inhibiting PMCA by raising extracellular pH to 8.8 produced a more than two-fold increase in the time constant, from 3.44 ± 0.20 s in RuR only-treated cells to 7.80 ± 0.53 s after a combined RuR + pH 8.8 treatment (n = 16 boutons/5 cells; P < 0.001, paired Student's t test; Fig. 6D-F). The magnitude of the pH 8.8 effect in RuR-treated cells was similar to the pH 8.8 effect in control cells (Fig. 3), indicating that PMCA-mediated Ca2+ transport was independent of the RuR-sensitive mitochondrial Ca2+ uptake under the described experimental conditions. We were unable to perform similar experiments in FCCP-treated cells because the combined application of FCCP and pH 8.8 destabilized presynaptic [Ca2+]i in DRG neurons (n = 14 boutons/3 cells; data not shown).
Properties of Ca2+ uptake by presynaptic mitochondria in DRG neurons
Given that mitochondria significantly contribute to the amplitude and time course of changes in presynaptic [Ca2+]i, we next examined the properties of Ca2+ transport by presynaptic mitochondria in capsaicin-sensitive DRG neurons. In these experiments, we directly monitored [Ca2+]mt using a mitochondria-targeted Ca2+ indicator, mtPericam (Nagai et al. 2001), as previously described (Medvedeva et al. 2008). DRG neurons transfected with mtPericam were subsequently loaded with Rhod10K through a patch pipette to enable visualization of axonal varicosities (Fig. 7A). Stimulating DRG neurons with trains of action potentials produced a rapid [Ca2+]mt elevation. Only 3–5 action potentials were required to produce detectable Ca2+ uptake by mitochondria (Fig. 7B). Comparison of the time courses of [Ca2+]i and [Ca2+]mt showed that mitochondrial Ca2+ uptake developed with a 1–2 s delay relative to the presynaptic [Ca2+]i rise. For example, in response to a stimulus with 20 action potentials (10 Hz, 2 s), [Ca2+]i and [Ca2+]mt reached their peak values within 1.9 ± 0.1 s (79 boutons/24 cells) and 3.5 ± 0.2 s (n = 22 boutons/5 cells; P < 0.001, unpaired Student's t test compared to the [Ca2+]i rising time) after beginning of the stimulation, respectively. In addition, Ca2+ removal from mitochondria occurred at a markedly slower rate (half recovery time = 9.2 ± 0.8 s; n = 22 boutons/5 cells) than Ca2+ clearance from the presynaptic cytosol (Fig. 7D). The fact that [Ca2+]mt remained elevated long after [Ca2+]i has fully recovered suggests that mitochondria serve as an integrator of consecutive presynaptic [Ca2+]i spikes produced by repeated neuronal stimulation.
Figure 7. Ca2+ changes in presynaptic mitochondria ([Ca2+]mt) monitored using mitochondria-targeted Ca2+ indicator mtPericam.
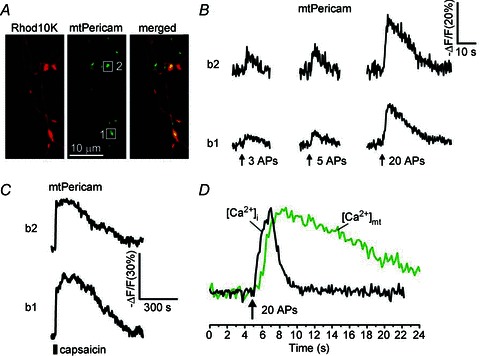
A, DRG neurons were transfected with mtPericam (green) and subsequently loaded with rhodamine 10K dextran (Rhod10K; 200 μm; red) via a patch pipette to facilitate the visualization of axonal boutons. B, mitochondrial Ca2+ changes were evoked by 3, 5 or 20 action potentials (10 Hz) and recorded from two axonal boutons (b1 and b2) indicated by white boxes in A. C, [Ca2+]mt responses to 1 μm capsaicin (30 s) for the same axonal boutons as in B. D, comparison of typical [Ca2+]i (black) and [Ca2+]mt (green) changes induced by 20 action potentials (10 Hz for 2 s). The [Ca2+]mt trace was obtained by averaging traces from five axonal boutons of the cell depicted in A. The [Ca2+]i trace was generated by averaging traces from four axonal boutons of a different cell loaded with Fura-FF. The traces were normalized along the vertical axis.
Next we addressed the important and widely debated question of how mitochondrial Ca2+ uptake depends on [Ca2+]i. The reported values for the Ca2+ affinity of MCU cover a wide range (0.5–20 μm), depending on the preparation, cell type and method of monitoring mitochondrial Ca2+ uptake (Gunter & Pfeiffer, 1990; Bernardi, 1999; Spat et al. 2008). In neuronal preparations, high Ca2+ sensitivity of MCU (K0.5∼0.4–0.5 μm) was reported for presynaptic mitochondria at nerve–muscular junctions of the fruit-fly and lizard (David, 1999; Chouhan et al. 2010). In contrast, mitochondrial Ca2+ uptake in the cell body of bullfrog sympathetic neurons was estimated to have a much lower Ca2+ affinity (K0.5∼ 10 μm; Colegrove et al. 2000). Comparable quantifications in mammalian neurons are lacking. To determine the dependence of mitochondrial Ca2+ uptake on [Ca2+]i in axonal boutons of DRG neurons, we compared [Ca2+]i and [Ca2+]mt elevations induced by depolarizing pulses of incremental amplitude (extracellular buffers containing 10, 15, 20, 30, 50 or 90 mm KCl; Fig. 8), as well as by treating cells with 1 μm capsaicin, which is known to produce large presynaptic [Ca2+]i and [Ca2+]mt elevations in DRG neurons (Medvedeva et al. 2008). To facilitate detection of small presynaptic [Ca2+]i changes in response to mild to moderate depolarization (10–50 mm KCl), we used the high-affinity Ca2+ indicator Fura-2, while the low-affinity Ca2+ indicator Fura-FF was employed for monitoring [Ca2+]i produced by the stronger stimuli such as 90 mm KCl and 1 μm capsaicin. Since the excitation spectra of Fura-2 (Fura-FF) and mtPericam overlap significantly, the measurements of [Ca2+]i and [Ca2+]mt were carried out in parallel experiments. Figure 8 shows representative presynaptic [Ca2+]i and [Ca2+]mt traces in response to incremental depolarization. [Ca2+]i changes in axonal boutons were detectable in response to 10 mm KCl, whereas triggering [Ca2+]mt response required 15 mm KCl to produce a stronger depolarization. Analysis of the initial rate of the [Ca2+]mt rise as a function of presynaptic [Ca2+]i elevations showed that Ca2+ uptake by mitochondria was induced by a [Ca2+]i elevation as little as 200–300 nm (K0.5∼ 600 nm); the rate of [Ca2+]mt rise (VMCU) progressively increased with the [Ca2+]i increase, until VMCU reached its maximum once [Ca2+]i began to exceed ∼2 μm (Fig. 8E). Thus, the mitochondrial Ca2+ uptake mechanism in axonal boutons of DRG neurons is highly sensitive to cytosolic Ca2+. Moreover, the Hill coefficient was 2.3, suggesting that MCU activation by Ca2+ is cooperative (Fig. 8E).
Finally, we examined the expression of several recently identified components of mitochondrial Ca2+ transport. MCU has been proposed to mediate mitochondrial Ca2+ uptake (Baughman et al. 2011; De Stefani et al. 2011), whereas the Na+/Ca2+/Li+ exchanger (NCLX) is probably responsible for mitochondrial Na+/Ca2+ exchange (Palty et al. 2010). Leucine zipper EF hand-containing transmembrane protein (Letm1) was identified as a mitochondrial Ca2+/H+ antiporter that can contribute to both mitochondrial Ca2+ uptake and release, depending on the mitochondrial Ca2+ concentration (Jiang et al. 2009). In addition, the mitochondrial calcium uptake 1 (MICU1) has been proposed to function as an essential regulator of mitochondrial Ca2+ uptake (Perocchi et al. 2010; Mallilankaraman et al. 2012). RT-PCR analysis of RNA extracted from neuron-enriched DRG cultures demonstrated the presence of all four transcripts (Fig. 8F).
Discussion
The present study identifies mechanisms of presynaptic [Ca2+]i regulation in an in vitro model of the first sensory synapse. This synapse controls nociceptive transmission from the periphery to the CNS, and plasticity at this synapse contributes to bidirectional modulation of pain processing (Ji et al. 2003; Woolf & Salter, 2006; Kuner, 2010; Ruscheweyh et al. 2011). We found that presynaptic [Ca2+]i in DRG is at ∼100 nm at rest, and that this low baseline [Ca2+]i is maintained by Ca2+ extrusion via PMCA and NCX, as well by Ca2+ buffering into intracellular Ca2+ organelles. Presynaptic [Ca2+]i transients induced by brief electrical stimulation rapidly recovered to the baseline with a time constant of ∼2 s. The ER Ca2+ pumps did not significantly contribute to presynaptic [Ca2+]i clearance at this synapse under the described stimulation conditions. On the other hand, PMCA-mediated Ca2+ extrusion and Ca2+ uptake by presynaptic mitochondria had major impacts on the rate of [Ca2+]i clearance, accounting for ∼47 and 40%, respectively, of the [Ca2+]i recovery. Ca2+ imaging in presynaptic mitochondria revealed a high sensitivity of MCU to [Ca2+]i elevations, with 3–5 action potentials being sufficient to produce detectable Ca2+ uptake by presynaptic mitochondria. Quantification of the rate of mitochondrial Ca2+ uptake as a function of [Ca2+]i yielded a K0.5 of ∼600 nm, and a Hill coefficient of 2.3. These values are consistent with the high Ca2+ affinity of presynaptic MCU (K0.5∼ 0.4–0.5 μm) reported for non-mammalian synapses (David, 1999; Chouhan et al. 2010). In addition, the Hill coefficient of 2.3 is similar to the previously reported value of ∼2 for isolated mitochondria and intact cells (Gunter & Pfeiffer, 1990; Colegrove et al. 2000), and suggests that Ca2+ cooperatively binds to and activates MCU in the presynaptic mitochondria of DRG neurons.
Our finding that presynaptic [Ca2+]i recovers in DRG neurons with τ∼ 2 s is in good agreement with the [Ca2+]i recovery rates reported for presynaptic boutons in the hippocampus (1–3 s) (Scotti et al. 1999; Scott & Rusakov, 2006) and neurohypophysial axonal terminals (1–2 s) (Lee et al. 2002), although faster [Ca2+]i clearance kinetics have been observed in presynaptic terminals of Purkinje and cerebellar granule cells (τ∼ 200–300 ms) (Mintz et al. 1995; Regehr & Atluri, 1995). Our data are also in good agreement with the observations in human nociceptive C-fibres, where electrically evoked [Ca2+]i transients recovered to baseline with a half-recovery time of ∼2–3 s (Mayer et al. 1999). However, a 5–10-fold slower rate of [Ca2+]i clearance was reported for sensory terminals of the rat cornea (Gover et al. 2007b), suggesting that Ca2+ clearance mechanisms at these sensory terminals originating from the trigeminal ganglia appear to be quite different from those in the peripheral and central processes of DRG neurons. This may not be surprising given that mitochondria do not participate in [Ca2+]i clearance from corneal terminals (Gover et al. 2007b), but significantly impact presynaptic [Ca2+]i recovery in DRG neurons (Figs 5 and 6). It is also possible that use of the high-affinity Ca2+ indicator Oregon Green BAPTA-1 dextran (Kd∼170 nm) to measure [Ca2+]i in the corneal terminals (Gover et al. 2007b) might have also contributed to a slowed rate of [Ca2+]i recovery in that work (Neher & Augustine, 1992; Kirischuk et al. 1999).
Our finding that SERCA does not significantly contribute to presynaptic Ca2+ clearance in DRG neurons (Fig. 4) is somewhat surprising given that SERCA plays major roles in setting the duration and amplitude of [Ca2+]i responses in the somata of DRG and trigeminal sensory neurons (Usachev & Thayer, 1999; Lu et al. 2006; Gover et al. 2007a; Rigaud et al. 2009). Just the same, our data are consistent with a negligible role of SERCA in peripheral sensory terminals of the cornea (Gover et al. 2007b), suggesting that the impact of SERCA on [Ca2+]i regulation differs substantially between the cell body and axons in sensory neurons. However, we cannot rule out the possibility that SERCA blockade led to enhanced function of PMCA or other Ca2+ transporters, thereby masking the effects of SERCA inhibitors. It is likely that the presynaptic boutons of primary sensory neurons contain functional intracellular Ca2+ stores associated with the ER. First, the SERCA inhibitor Tg was shown to modulate glutamate release at the first sensory synapse (Tsuzuki et al. 2004). Second, we found that an activator of ryanodine receptors, caffeine, induced Ca2+ release from the intracellular stores in presynaptic boutons of DRG neurons (Fig. 4D). Although SERCA pumps do not contribute significantly to the rate of presynaptic [Ca2+]i recovery, Ca2+ stores are involved in the maintenance of low [Ca2+]i at rest, as the SERCA inhibitors CPA and Tg produced 20–50 nm elevation of the [Ca2+]i baseline. The latter could be explained either by activation of Ca2+ influx through presynaptic store-operated Ca2+ channels (Usachev & Thayer, 1999; Gemes et al. 2011) or the role of SERCA pumps in controlling presynaptic [Ca2+]i at rest.
Na+/Ca2+ exchange of the plasma membrane is another mechanism that can contribute to Ca2+ clearance. Based on our RT-PCR data (Fig. 2D), DRG neurons express three Na+/Ca2+ exchanger isoforms that are K+-independent (NCX1, NCX2 and NCX3) and one that is K+-dependent (NCKX2). The functional analysis showed that plasma membrane Na+/Ca2+ exchange regulated the resting [Ca2+]i and contributed approximately 12% to presynaptic Ca2+ clearance in DRG neurons (Fig. 2), and that this transport was probably mediated by the NCX, but not NCKX, isoforms (Fig. 2E). Only limited information is available about the specific roles of NCX and NCKX at various synapses. NCKX was shown to be the major mechanism mediating Ca2+ extrusion from axonal terminals of the rat neurohypophysis (Lee et al. 2002), whereas both NCX and NCKX contribute to presynaptic [Ca2+]i clearance at the calyx of Held synapse (Kim et al. 2005). Notably, deletion of NCX2 results in increased paired-pulsed facilitation of synaptic transmission and enhanced LTP in the CA1 region of the hippocampus (Jeon et al. 2003), whereas deletion of NCKX2 leads to a profound loss of the hippocampal LTP (Li et al. 2006). Thus, it is likely that the NCX and NCKX isoforms play distinct roles at various synapses.
Identification of PMCA as a key regulator of presynaptic [Ca2+]i at the sensory synapses is in agreement with the major role of PMCA in controlling [Ca2+]i recovery in the somata of DRG and trigeminal sensory neurons (Benham et al. 1992; Usachev et al. 2002; Gover et al. 2007a), as well as in peripheral sensory terminals innervating the cornea (Gover et al. 2007b). PMCA is a high-affinity (0.1–0.2 μm) ATP-dependent Ca2+ transporter that extrudes Ca2+ from the cell (Carafoli, 1991; Elwess et al. 1997). The PMCA family of proteins consist of four major isoforms, PMCA1–4, and alternative RNA splicing of these gives rise to nearly 30 PMCA proteins that differ in their tissue distribution, function and regulatory properties (Strehler & Zacharias, 2001; Thayer et al. 2002). PMCA2 (splice variants 2a and 2b) and PMCA4 (splice variant 4b) are the major PMCA isoforms expressed in the DRG (Usachev et al. 2002). PMCA4 is highly expressed in the superficial lamina of the dorsal horn, where the majority of TRPV1-positive sensory fibres terminate; PMCA2, on the other hand, is found in the deeper lamina of the dorsal horn (Tachibana et al. 2004). PMCA2 has very high affinity for Ca2+ (∼0.09 μm for PMCA2a and ∼0.06 μm for PMCA2b), suggesting that it plays an important role in maintaining low resting [Ca2+]i (Elwess et al. 1997). PMCA4b exhibits a somewhat lower apparent affinity for Ca2+ (∼0.16 μm) (Elwess et al. 1997), but its activity can be markedly increased by PKC-mediated phosphorylation (Enyedi et al. 1996). Notably, this mechanism is responsible for a marked PKC-mediated acceleration of Ca2+ efflux from the somata and axonal boutons of DRG neurons (Usachev et al. 2002).
PMCA2 and PMCA4 are also highly sensitive to stimulation by calmodulin (CaM). Ca2+-dependent binding of CaM to the PMCA autoinhibitory domain, located in its carboxy-terminus, disinhibits the pump, which results in a marked increase of both Ca2+ affinity and maximal velocity (Vmax) of PMCA (Carafoli, 1991; Strehler & Zacharias, 2001). The two major PMCA isoforms in DRG neurons, PMCA2b and PMCA4b (Usachev et al. 2002), are characterized by unusually slow dissociation of CaM (>20 min) (Caride et al. 1999, 2001). This implies that even a brief elevation in [Ca2+]i will produce a long-lasting CaM/Ca2+-dependent stimulation of PMCA. Indeed, a priming [Ca2+]i elevation has been reported to result in long-term enhancement of Ca2+ extrusion (∼1 h) in DRG neurons (Pottorf & Thayer, 2002). Thus, PMCA-mediated Ca2+ extrusion in sensory neurons is subject to modulation by both neuronal activity and PKC. In the context of spinal pain processing, the described mechanisms are predicted to accelerate presynaptic [Ca2+]i clearance during central sensitization, which is known to be associated with heightened neuronal activity in the spinal cord and enhanced release of neuromodulators coupled to PKC signalling (e.g. bradykinin and substance P). However, direct testing of this idea is hampered by a lack of isoform-specific inhibitors of PMCA and technical difficulties associated with presynaptic Ca2+ imaging in spinal cord slices or in vivo.
Ca2+ uptake by presynaptic mitochondria is another major mechanism responsible for presynaptic [Ca2+]i clearance in DRG neurons. Electron microscopy has demonstrated numerous presynaptic mitochondria in the central processes of primary sensory neurons, including TRPV1-expressing boutons (Maxwell & Rethelyi, 1987; Rethelyi et al. 1989; Hwang et al. 2003). We also found that axonal boutons are intensely labelled by the mitochondria-targeted Ca2+ indicator mtPericam (Medvedeva et al. 2008; Figs 7 and 8). Presynaptic clustering of mitochondria is consistent with the high energy demand at the synapses. Mitochondria also play important roles in shaping presynaptic [Ca2+]i signals at various synapses (Billups & Forsythe, 2002; David & Barrett, 2003; Jonas, 2004; Garcia-Chacon et al. 2006; Lee et al. 2007a), including the first sensory synapse (Medvedeva et al. 2008; Figs 5 and 6). Mitochondria buffer excessive Ca2+ via the Ca2+ uniporter mechanism, and subsequently release accumulated Ca2+ back into the cytosol via Na+/Ca2+ exchange or other mechanisms (Bernardi, 1999). Ca2+ buffering by mitochondria limits the amplitude of the cytosolic [Ca2+]i elevation, and accelerates the initial recovery of presynaptic [Ca2+]i (Medvedeva et al. 2008; Figs 5 and 6). Our data suggest that the impact of mitochondria on the amplitude of presynaptic [Ca2+]i responses is much greater than that of PMCA (FCCP or RuR induced ∼60–100% increase in the amplitude of [Ca2+]i response compared to ∼20% increase produced by pH 8.8). One possible explanation of this difference is that Ca2+ transport by the mitochondrial uniporter is much faster than that by PMCA (Miller, 1991; Elwess et al. 1997; Gunter & Sheu, 2009).
Our comparison of the time courses of [Ca2+]i and [Ca2+]mt changes induced by electrical stimulation demonstrated that [Ca2+]mt reaches its peak by 1–2 s later than presynaptic [Ca2+]i (Fig. 7D), indicating that mitochondrial Ca2+ uptake continues during the initial phase of [Ca2+]i recovery. This observation is consistent with the role of mitochondrial Ca2+ uptake in [Ca2+]i recovery in the axonal boutons of DRG neurons. Based on the effects of the mitochondrial Ca2+ uniporter inhibitor RuR, we estimate that mitochondria account for at least 40% of the [Ca2+]i recovery in the axonal boutons of DRG neurons. Although our analysis using the protonophore FCCP suggested that MCU has a somewhat stronger effect on the rate of presynaptic Ca2+ clearance (∼54%), this difference could be due to a partial inhibition of ATP synthesis by FCCP.
Ca2+ initially accumulated by mitochondria can be subsequently released back into the cytosol. Following intense (e.g. tetanic) stimulation, strong Ca2+ efflux from mitochondria can markedly slow recovery of presynaptic [Ca2+]i leading to a [Ca2+]i plateau lasting many minutes after stimulation is terminated (Tang & Zucker, 1997; Garcia-Chacon et al. 2006; Lee et al. 2007a). We previously reported that TRPV1 activation or intense electrical stimulation (20 Hz for 30 s) results in a long-lasting presynaptic [Ca2+]i plateau associated with enhanced glutamate release in DRG neurons (Medvedeva et al. 2008). In contrast, mild electrical stimulation such as that used in the present study (10 Hz for 2 s) was insufficient to generate a [Ca2+]i plateau in the same axonal boutons, in spite of the contribution of mitochondria to Ca2+ clearance. Thus, a specific role of presynaptic mitochondria at the sensory synapse depends on the type and intensity of the stimulus.
In addition to regulating presynaptic [Ca2+]i, mitochondrial Ca2+ uptake can stimulate the production of ATP via Ca2+-dependent dehydrogenases (Hajnoczky et al. 1995; Denton, 2009; Chouhan et al. 2012) and the generation of reactive oxygen species (ROS) (Nicholls & Budd, 2000; Hongpaisan et al. 2004). Both effects require strong neuronal activation (Hongpaisan et al. 2004; Chouhan et al. 2012), and this probably explains why mitochondrial Ca2+ uptake following the mild stimulation used in our study did not detectably impact ATP-dependent Ca2+ transport (Fig. 6D–F). In pathological pain states, neuronal activity in the spinal cord is markedly intensified, and the resulting enhancement of ROS production is thought to contribute to central sensitization (Schwartz et al. 2009). Notably, recent data suggest that mitochondrial Ca2+ uptake is essential for ROS-dependent plasticity in the spinal cord, and for the development of persistent pain (Kim et al. 2011).
In summary, this study provides the first detailed characterization of Ca2+ clearance mechanisms in presynaptic boutons of DRG neurons, and identifies PMCA and mitochondria as major regulators of Ca2+ signalling at the first sensory synapse. Development of new pharmacological and genetic tools targeting specific PMCA isoforms and mitochondrial Ca2+ transporters will be required to determine their precise contribution to the various forms of synaptic plasticity involved in spinal pain processing, such as wind-up, LTP and central sensitization.
Acknowledgments
This work was supported by National Institutes of Health grant NS072432 and the Fraternal Order of Eagles Diabetes Research Center (FOEDRC) pilot grant to Y.M.U. and by predoctoral fellowships from the American Heart Association, Midwest Affiliate, to M.S.K. and P.R.H. We thank Dr Miyawaki for his gift of the mtPericam plasmid.
Glossary
- AraC
cytosine β-d-arabinofuranoside
- [Ca2+]i
cytosolic Ca2+ concentration
- [Ca2+]mt
mitochondrial Ca2+ concentration
- CaM
calmodulin
- CPA
cyclopiazonic acid
- DRG
dorsal root ganglia
- ΔΨmt
mitochondrial membrane potential
- EPSC
excitatory postsynaptic current
- ER
endoplasmic reticulum
- FCCP
carbonyl cyanide p-trifluoromethoxyphenylhydrazone
- MCU
mitochondrial Ca2+ uniporter
- MICU1
mitochondrial calcium uptake 1
- Letm1
leucine zipper EF hand-containing transmembrane protein
- LTP
long-term potentiation
- NCX
Na+/Ca2+ exchanger
- NCKX
K+-dependent Na+/Ca2+ exchanger
- NCLX
Na+/Ca2+/Li+ exchanger
- PKA
protein kinase A
- PKC
protein kinase C
- PMCA
plasma membrane Ca2+-ATPase
- Rhod10K
tetramethylrhodamine 10K dextran
- RuR
ruthenium red
- SC
spinal cord
- SERCA
sarco-endoplasmic reticulum Ca2+-ATPase
- Tg
thapsigargin
- TRPV1
transient receptor potential vanilloid 1
Author contributions
Y.M.U. and L.P.S. designed the work. L.P.S., Y.M.U., M.S.K., P.R.H. and Y.V.M. performed the experiments. Y.M.U. wrote the paper. All authors provided critical intellectual input to the text and figures and approved the final version of the article.
References
- Annunziato L, Pignataro G, Di Renzo GF. Pharmacology of brain Na+/Ca2+ exchanger: from molecular biology to therapeutic perspectives. Pharmacol Rev. 2004;56:633–654. doi: 10.1124/pr.56.4.5. [DOI] [PubMed] [Google Scholar]
- Bao J, Li JJ, Perl ER. Differences in Ca2+ channels governing generation of miniature and evoked excitatory synaptic currents in spinal laminae I and II. J Neurosci. 1998;18:8740–8750. doi: 10.1523/JNEUROSCI.18-21-08740.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baughman JM, Perocchi F, Girgis HS, Plovanich M, Belcher-Timme CA, Sancak Y, Bao XR, Strittmatter L, Goldberger O, Bogorad RL, Koteliansky V, Mootha VK. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature. 2011;476:341–345. doi: 10.1038/nature10234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benham CD, Evans ML, McBain CJ. Ca2+ efflux mechanisms following depolarization evoked calcium transients in cultured rat sensory neurones. J Physiol. 1992;455:567–583. doi: 10.1113/jphysiol.1992.sp019316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernardi P. Mitochondrial transport of cations: channels, exchangers, and permeability transition. Physiol Rev. 1999;79:1127–1155. doi: 10.1152/physrev.1999.79.4.1127. [DOI] [PubMed] [Google Scholar]
- Billups B, Forsythe ID. Presynaptic mitochondrial calcium sequestration influences transmission at mammalian central synapses. J Neurosci. 2002;22:5840–5847. doi: 10.1523/JNEUROSCI.22-14-05840.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaustein MP, Lederer WJ. Sodium/calcium exchange: its physiological implications. Physiol Rev. 1999;79:763–854. doi: 10.1152/physrev.1999.79.3.763. [DOI] [PubMed] [Google Scholar]
- Cao YQ. Voltage-gated calcium channels and pain. Pain. 2006;126:5–9. doi: 10.1016/j.pain.2006.10.019. [DOI] [PubMed] [Google Scholar]
- Carafoli E. Calcium pump of the plasma membrane. Physiol Rev. 1991;71:129–153. doi: 10.1152/physrev.1991.71.1.129. [DOI] [PubMed] [Google Scholar]
- Caride AJ, Elwess NL, Verma AK, Filoteo AG, Enyedi A, Bajzer Z, Penniston JT. The rate of activation by calmodulin of isoform 4 of the plasma membrane Ca2+ pump is slow and is changed by alternative splicing. J Biol Chem. 1999;274:35227–35232. doi: 10.1074/jbc.274.49.35227. [DOI] [PubMed] [Google Scholar]
- Caride AJ, Penheiter AR, Filoteo AG, Bajzer Z, Enyedi A, Penniston JT. The plasma membrane calcium pump displays memory of past calcium spikes. Differences between isoforms 2b and 4b. J Biol Chem. 2001;276:39797–39804. doi: 10.1074/jbc.M104380200. [DOI] [PubMed] [Google Scholar]
- Caterina MJ, Julius D. The vanilloid receptor: a molecular gateway to the pain pathway. Annu Rev Neurosci. 2001;24:487–517. doi: 10.1146/annurev.neuro.24.1.487. [DOI] [PubMed] [Google Scholar]
- Cavanaugh DJ, Chesler AT, Jackson AC, Sigal YM, Yamanaka H, Grant R, O’Donnell D, Nicoll RA, Shah NM, Julius D, Basbaum AI. Trpv1 reporter mice reveal highly restricted brain distribution and functional expression in arteriolar smooth muscle cells. J Neurosci. 2011;31:5067–5077. doi: 10.1523/JNEUROSCI.6451-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chouhan AK, Ivannikov MV, Lu Z, Sugimori M, Llinas RR, Macleod GT. Cytosolic calcium coordinates mitochondrial energy metabolism with presynaptic activity. J Neurosci. 2012;32:1233–1243. doi: 10.1523/JNEUROSCI.1301-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chouhan AK, Zhang J, Zinsmaier KE, Macleod GT. Presynaptic mitochondria in functionally different motor neurons exhibit similar affinities for Ca2+ but exert little influence as Ca2+ buffers at nerve firing rates in situ. J Neurosci. 2010;30:1869–1881. doi: 10.1523/JNEUROSCI.4701-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clayton EL, Cousin MA. The molecular physiology of activity-dependent bulk endocytosis of synaptic vesicles. J Neurochem. 2009;111:901–914. doi: 10.1111/j.1471-4159.2009.06384.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colegrove SL, Albrecht MA, Friel DD. Quantitative analysis of mitochondrial Ca2+ uptake and release pathways in sympathetic neurons – Reconstruction of the recovery after depolarization-evoked [Ca2+]i elevations. J Gen Physiol. 2000;115:371–388. doi: 10.1085/jgp.115.3.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cousin MA, Robinson PJ. The dephosphins: dephosphorylation by calcineurin triggers synaptic vesicle endocytosis. Trends Neurosci. 2001;24:659–665. doi: 10.1016/s0166-2236(00)01930-5. [DOI] [PubMed] [Google Scholar]
- David G. Mitochondrial clearance of cytosolic Ca2+ in stimulated lizard motor nerve terminals proceeds without progressive elevation of mitochondrial matrix [Ca2+] J Neurosci. 1999;19:7495–7506. doi: 10.1523/JNEUROSCI.19-17-07495.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David G, Barrett EF. Mitochondrial Ca2+ uptake prevents desynchronization of quantal release and minimizes depletion during repetitive stimulation of mouse motor nerve terminals. J Physiol. 2003;548:425–438. doi: 10.1113/jphysiol.2002.035196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Stefani D, Raffaello A, Teardo E, Szabo I, Rizzuto R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature. 2011;476:336–340. doi: 10.1038/nature10230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denton RM. Regulation of mitochondrial dehydrogenases by calcium ions. Biochim Biophys Acta. 2009;1787:1309–1316. doi: 10.1016/j.bbabio.2009.01.005. [DOI] [PubMed] [Google Scholar]
- Elwess NL, Filoteo AG, Enyedi A, Penniston JT. Plasma membrane Ca2+ pump isoforms 2a and 2b are unusually responsive to calmodulin and Ca2+ J Biol Chem. 1997;272:17981–17986. doi: 10.1074/jbc.272.29.17981. [DOI] [PubMed] [Google Scholar]
- Empson RM, Garside ML, Knopfel T. Plasma membrane Ca2+ ATPase 2 contributes to short-term synapse plasticity at the parallel fiber to Purkinje neuron synapse. J Neurosci. 2007;27:3753–3758. doi: 10.1523/JNEUROSCI.0069-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enyedi A, Verma AK, Filoteo AG, Penniston JT. Protein kinase C activates the plasma membrane Ca2+ pump isoform 4b by phosphorylation of an inhibitory region downstream of the calmodulin-binding domain. J Biol Chem. 1996;271:32461–32467. doi: 10.1074/jbc.271.50.32461. [DOI] [PubMed] [Google Scholar]
- Garcia-Chacon LE, Nguyen KT, David G, Barrett EF. Extrusion of Ca2+ from mouse motor terminal mitochondria via a Na+-Ca2+ exchanger increases post-tetanic evoked release. J Physiol. 2006;574:663–675. doi: 10.1113/jphysiol.2006.110841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gemes G, Bangaru ML, Wu HE, Tang Q, Weihrauch D, Koopmeiners AS, Cruikshank JM, Kwok WM, Hogan QH. Store-operated Ca2+ entry in sensory neurons: functional role and the effect of painful nerve injury. J Neurosci. 2011;31:3536–3549. doi: 10.1523/JNEUROSCI.5053-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gover TD, Moreira TH, Kao JP, Weinreich D. Calcium homeostasis in trigeminal ganglion cell bodies. Cell Calcium. 2007a;41:389–396. doi: 10.1016/j.ceca.2006.08.014. [DOI] [PubMed] [Google Scholar]
- Gover TD, Moreira TH, Kao JP, Weinreich D. Calcium regulation in individual peripheral sensory nerve terminals of the rat. J Physiol. 2007b;578:481–490. doi: 10.1113/jphysiol.2006.119008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groffen AJ, Martens S, Diez Arazola R, Cornelisse LN, Lozovaya N, de Jong AP, Goriounova NA, Habets RL, Takai Y, Borst JG, Brose N, McMahon HT, Verhage M. Doc2b is a high-affinity Ca2+ sensor for spontaneous neurotransmitter release. Science. 2010;327:1614–1618. doi: 10.1126/science.1183765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruner W, Silva LR. ω-Conotoxin sensitivity and presynaptic inhibition of glutamatergic sensory neurotransmission in vitro. J Neurosci. 1994;14:2800–2808. doi: 10.1523/JNEUROSCI.14-05-02800.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Gu JGG, Macdermott AB. Activation of ATP P2x receptors elicits glutamate release from sensory neuron synapses. Nature. 1997;389:749–753. doi: 10.1038/39639. [DOI] [PubMed] [Google Scholar]
- Gunter TE, Pfeiffer DR. Mechanisms by which mitochondria transport calcium. Am J Physiol. 1990;258:C755–C786. doi: 10.1152/ajpcell.1990.258.5.C755. [DOI] [PubMed] [Google Scholar]
- Gunter TE, Sheu SS. Characteristics and possible functions of mitochondrial Ca2+ transport mechanisms. Biochim Biophys Acta. 2009;1787:1291–1308. doi: 10.1016/j.bbabio.2008.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo A, Vulchanova L, Wang J, Li X, Elde R. Immunocytochemical localization of the vanilloid receptor 1 (VR1): relationship to neuropeptides, the P2′3 purinoceptor and IB4 binding sites. Eur J Neurosci. 1999;11:946–958. doi: 10.1046/j.1460-9568.1999.00503.x. [DOI] [PubMed] [Google Scholar]
- Hajnoczky G, Robb GL, Seitz MB, Thomas AP. Decoding of cytosolic calcium oscillations in the mitochondria. Cell. 1995;82:415–424. doi: 10.1016/0092-8674(95)90430-1. [DOI] [PubMed] [Google Scholar]
- Heinke B, Balzer E, Sandkuhler J. Pre- and postsynaptic contributions of voltage-dependent Ca2+ channels to nociceptive transmission in rat spinal lamina I neurons. Eur J Neurosci. 2004;19:103–111. doi: 10.1046/j.1460-9568.2003.03083.x. [DOI] [PubMed] [Google Scholar]
- Heinke B, Gingl E, Sandkuhler J. Multiple targets of μ-opioid receptor-mediated presynaptic inhibition at primary afferent Aδ- and C-fibers. J Neurosci. 2011;31:1313–1322. doi: 10.1523/JNEUROSCI.4060-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henzi V, MacDermott AB. Characteristics and function of Ca2+- and inositol 1,4,5-trisphosphate-releasable stores of Ca2+ in neurons. Neuroscience. 1992;46:251–273. doi: 10.1016/0306-4522(92)90049-8. [DOI] [PubMed] [Google Scholar]
- Hernandez-Guijo JM, Maneu-Flores VE, Ruiz-Nuno A, Villarroya M, Garcia AG, Gandia L. Calcium-dependent inhibition of L, N, and P/Q Ca2+ channels in chromaffin cells: role of mitochondria. J Neurosci. 2001;21:2553–2560. doi: 10.1523/JNEUROSCI.21-08-02553.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollenbeck PJ. Mitochondria and neurotransmission: evacuating the synapse. Neuron. 2005;47:331–333. doi: 10.1016/j.neuron.2005.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hongpaisan J, Winters CA, Andrews SB. Strong calcium entry activates mitochondrial superoxide generation, upregulating kinase signaling in hippocampal neurons. J Neurosci. 2004;24:10878–10887. doi: 10.1523/JNEUROSCI.3278-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang SM, Bisogno T, Trevisani M, Al-Hayani A, De Petrocellis L, Fezza F, Tognetto M, Petros TJ, Krey JF, Chu CJ, Miller JD, Davies SN, Geppetti P, Walker JM, Di Marzo V. An endogenous capsaicin-like substance with high potency at recombinant and native vanilloid VR1 receptors. Proc Natl Acad Sci U S A. 2002;99:8400–8405. doi: 10.1073/pnas.122196999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang SJ, Burette A, Valtschanoff JG. VR1-positive primary afferents contact NK1-positive spinoparabrachial neurons. J Comp Neurol. 2003;460:255–265. doi: 10.1002/cne.10647. [DOI] [PubMed] [Google Scholar]
- Jeon D, Yang YM, Jeong MJ, Philipson KD, Rhim H, Shin HS. Enhanced learning and memory in mice lacking Na+/Ca2+ exchanger 2. Neuron. 2003;38:965–976. doi: 10.1016/s0896-6273(03)00334-9. [DOI] [PubMed] [Google Scholar]
- Ji RR, Kohno T, Moore KA, Woolf CJ. Central sensitization and LTP: do pain and memory share similar mechanisms. Trends Neurosci. 2003;26:696–705. doi: 10.1016/j.tins.2003.09.017. [DOI] [PubMed] [Google Scholar]
- Jiang D, Zhao L, Clapham DE. Genome-wide RNAi screen identifies Letm1 as a mitochondrial Ca2+/H+ antiporter. Science. 2009;326:144–147. doi: 10.1126/science.1175145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonas E. Regulation of synaptic transmission by mitochondrial ion channels. J Bioenerg Biomembr. 2004;36:357–361. doi: 10.1023/B:JOBB.0000041768.11006.90. [DOI] [PubMed] [Google Scholar]
- Junge HJ, Rhee JS, Jahn O, Varoqueaux F, Spiess J, Waxham MN, Rosenmund C, Brose N. Calmodulin and Munc13 form a Ca2+ sensor/effector complex that controls short-term synaptic plasticity. Cell. 2004;118:389–401. doi: 10.1016/j.cell.2004.06.029. [DOI] [PubMed] [Google Scholar]
- Kim HY, Lee KY, Lu Y, Wang J, Cui L, Kim SJ, Chung JM, Chung K. Mitochondrial Ca2+ uptake is essential for synaptic plasticity in pain. J Neurosci. 2011;31:12982–12991. doi: 10.1523/JNEUROSCI.3093-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MH, Korogod N, Schneggenburger R, Ho WK, Lee SH. Interplay between Na+/Ca2+ exchangers and mitochondria in Ca2+ clearance at the calyx of Held. J Neurosci. 2005;25:6057–6065. doi: 10.1523/JNEUROSCI.0454-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirischuk S, Veselovsky N, Grantyn R. Single-bouton-mediated synaptic transmission: postsynaptic conductance changes in their relationship with presynaptic calcium signals. Pflugers Arch. 1999;438:716–724. doi: 10.1007/s004249900075. [DOI] [PubMed] [Google Scholar]
- Kuner R. Central mechanisms of pathological pain. Nat Med. 2010;16:1258–1266. doi: 10.1038/nm.2231. [DOI] [PubMed] [Google Scholar]
- Lee D, Lee KH, Ho WK, Lee SH. Target cell-specific involvement of presynaptic mitochondria in post-tetanic potentiation at hippocampal mossy fiber synapses. J Neurosci. 2007a;27:13603–13613. doi: 10.1523/JNEUROSCI.3985-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JY, Ho WK, Lee SH. Ionic selectivity of NCKX2, NCKX3, and NCKX4 for monovalent cations at K+-binding site. Ann N Y Acad Sci. 2007b;1099:166–170. doi: 10.1196/annals.1387.012. [DOI] [PubMed] [Google Scholar]
- Lee SH, Kim MH, Park KH, Earm YE, Ho WK. K+-dependent Na+/Ca2+ exchange is a major Ca2+ clearance mechanism in axon terminals of rat neurohypophysis. J Neurosci. 2002;22:6891–6899. doi: 10.1523/JNEUROSCI.22-16-06891.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li XF, Kiedrowski L, Tremblay F, Fernandez FR, Perizzolo M, Winkfein RJ, Turner RW, Bains JS, Rancourt DE, Lytton J. Importance of K+-dependent Na+/Ca2+-exchanger 2, NCKX2, in motor learning and memory. J Biol Chem. 2006;281:6273–6282. doi: 10.1074/jbc.M512137200. [DOI] [PubMed] [Google Scholar]
- Lu SG, Zhang X, Gold MS. Intracellular calcium regulation among subpopulations of rat dorsal root ganglion neurons. J Physiol. 2006;577:169–190. doi: 10.1113/jphysiol.2006.116418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ly CV, Verstreken P. Mitochondria at the synapse. Neuroscientist. 2006;12:291–299. doi: 10.1177/1073858406287661. [DOI] [PubMed] [Google Scholar]
- Lytton J. Na+/Ca2+ exchangers: three mammalian gene families control Ca2+ transport. Biochem J. 2007;406:365–382. doi: 10.1042/BJ20070619. [DOI] [PubMed] [Google Scholar]
- Mallilankaraman K, Doonan P, Cardenas C, Chandramoorthy HC, Muller M, Miller R, Hoffman NE, Gandhirajan RK, Molgo J, Birnbaum MJ, Rothberg BS, Mak DO, Foskett JK, Madesh M. MICU1 is an essential gatekeeper for MCU-mediated mitochondrial Ca2+ uptake that regulates cell survival. Cell. 2012;151:630–644. doi: 10.1016/j.cell.2012.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mata AM, Sepulveda MR. Calcium pumps in the central nervous system. Brain Res Brain Res Rev. 2005;49:398–405. doi: 10.1016/j.brainresrev.2004.11.004. [DOI] [PubMed] [Google Scholar]
- Maxwell D, Rethelyi M. Ultrastructure and synaptic connections of cutaneous afferent fibers in the spinal cord. Trends Neurosci. 1987;10:117–123. [Google Scholar]
- Mayer C, Quasthoff S, Grafe P. Confocal imaging reveals activity-dependent intracellular Ca2+ transients in nociceptive human C fibres. Pain. 1999;81:317–322. doi: 10.1016/S0304-3959(99)00015-9. [DOI] [PubMed] [Google Scholar]
- McGivern JG. Targeting N-type and T-type calcium channels for the treatment of pain. Drug Discov Today. 2006;11:245–253. doi: 10.1016/S1359-6446(05)03662-7. [DOI] [PubMed] [Google Scholar]
- Medvedeva YV, Kim MS, Schnizler K, Usachev YM. Functional tetrodotoxin-resistant Na+ channels are expressed presynaptically in rat dorsal root ganglia neurons. Neuroscience. 2009;159:559–569. doi: 10.1016/j.neuroscience.2008.12.029. [DOI] [PubMed] [Google Scholar]
- Medvedeva YV, Kim MS, Usachev YM. Mechanisms of prolonged presynaptic Ca2+ signaling and glutamate release induced by TRPV1 activation in rat sensory neurons. J Neurosci. 2008;28:5295–5311. doi: 10.1523/JNEUROSCI.4810-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer RA, Campbell JN. Myelinated nociceptive afferents account for the hyperalgesia that follows a burn to the hand. Science. 1981;213:1527–1529. doi: 10.1126/science.7280675. [DOI] [PubMed] [Google Scholar]
- Miller RJ. The control of neuronal Ca2+ homeostasis. Prog Neurobiol. 1991;37:255–285. doi: 10.1016/0301-0082(91)90028-y. [DOI] [PubMed] [Google Scholar]
- Mintz IM, Sabatini BL, Regehr WG. Calcium control of transmitter release at a cerebellar synapse. Neuron. 1995;15:675–688. doi: 10.1016/0896-6273(95)90155-8. [DOI] [PubMed] [Google Scholar]
- Nagai T, Sawano A, Park ES, Miyawaki A. Circularly permuted green fluorescent proteins engineered to sense Ca2+ Proc Natl Acad Sci U S A. 2001;98:3197–3202. doi: 10.1073/pnas.051636098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher E, Augustine GJ. Calcium gradients and buffers in bovine chromaffin cells. J Physiol. 1992;450:273–301. doi: 10.1113/jphysiol.1992.sp019127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher E, Sakaba T. Multiple roles of calcium ions in the regulation of neurotransmitter release. Neuron. 2008;59:861–872. doi: 10.1016/j.neuron.2008.08.019. [DOI] [PubMed] [Google Scholar]
- Nicholls DG, Budd SL. Mitochondria and neuronal survival. Physiol Rev. 2000;80:315–360. doi: 10.1152/physrev.2000.80.1.315. [DOI] [PubMed] [Google Scholar]
- Nicholls DG, Ward MW. Mitochondrial membrane potential and neuronal glutamate excitotoxicity: mortality and millivolts. Trends Neurosci. 2000;23:166–174. doi: 10.1016/s0166-2236(99)01534-9. [DOI] [PubMed] [Google Scholar]
- Palty R, Silverman WF, Hershfinkel M, Caporale T, Sensi SL, Parnis J, Nolte C, Fishman D, Shoshan-Barmatz V, Herrmann S, Khananshvili D, Sekler I. NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc Natl Acad Sci U S A. 2010;107:436–441. doi: 10.1073/pnas.0908099107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perocchi F, Gohil VM, Girgis HS, Bao XR, McCombs JE, Palmer AE, Mootha VK. MICU1 encodes a mitochondrial EF hand protein required for Ca2+ uptake. Nature. 2010;467:291–296. doi: 10.1038/nature09358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persson AK, Black JA, Gasser A, Cheng X, Fischer TZ, Waxman SG. Sodium-calcium exchanger and multiple sodium channel isoforms in intra-epidermal nerve terminals. Mol Pain. 2010;6:84. doi: 10.1186/1744-8069-6-84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pottorf WJ, Thayer SA. Transient rise in intracellular calcium produces a long-lasting increase in plasma membrane calcium pump activity in rat sensory neurons. J Neurochem. 2002;83:1002–1008. doi: 10.1046/j.1471-4159.2002.01221.x. [DOI] [PubMed] [Google Scholar]
- Pozzan T, Rizzuto R, Volpe P, Meldolesi J. Molecular and cellular physiology of intracellular calcium stores. Physiol Rev. 1994;74:595–636. doi: 10.1152/physrev.1994.74.3.595. [DOI] [PubMed] [Google Scholar]
- Regehr WG, Atluri PP. Calcium transients in cerebellar granule cell presynaptic terminals. Biophys J. 1995;68:2156–2170. doi: 10.1016/S0006-3495(95)80398-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rethelyi M, Light AR, Perl ER. Synaptic ultrastructure of functionally and morphologically characterized neurons of the superficial spinal dorsal horn of cat. J Neurosci. 1989;9:1846–1863. doi: 10.1523/JNEUROSCI.09-06-01846.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigaud M, Gemes G, Weyker PD, Cruikshank JM, Kawano T, Wu HE, Hogan QH. Axotomy depletes intracellular calcium stores in primary sensory neurons. Anesthesiology. 2009;111:381–392. doi: 10.1097/ALN.0b013e3181ae6212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruscheweyh R, Wilder-Smith O, Drdla R, Liu XG, Sandkuhler J. Long-term potentiation in spinal nociceptive pathways as a novel target for pain therapy. Mol Pain. 2011;7:20. doi: 10.1186/1744-8069-7-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz ES, Kim HY, Wang J, Lee I, Klann E, Chung JM, Chung K. Persistent pain is dependent on spinal mitochondrial antioxidant levels. J Neurosci. 2009;29:159–168. doi: 10.1523/JNEUROSCI.3792-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwiening CJ, Kennedy HJ, Thomas RC. Calcium hydrogen exchange by the plasma membrane Ca-ATPase of voltage-clamped snail neurons. Proc R Soc Lond. 1993;253:285–289. doi: 10.1098/rspb.1993.0115. [DOI] [PubMed] [Google Scholar]
- Scott R, Rusakov DA. Main determinants of presynaptic Ca2+ dynamics at individual mossy fiber-CA3 pyramidal cell synapses. J Neurosci. 2006;26:7071–7081. doi: 10.1523/JNEUROSCI.0946-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scotti AL, Chatton JY, Reuter H. Roles of Na+-Ca2+ exchange and of mitochondria in the regulation of presynaptic Ca2+ and spontaneous glutamate release. Philos Trans R Soc Lond B Biol Sci. 1999;354:357–364. doi: 10.1098/rstb.1999.0387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shmigol A, Kostyuk P, Verkhratsky A. Role of caffeine-sensitive Ca2+ stores in Ca2+ signal termination in adult mouse DRG neurones. Neuroreport. 1994;5:2073–2076. doi: 10.1097/00001756-199410270-00021. [DOI] [PubMed] [Google Scholar]
- Sikand P, Premkumar LS. Potentiation of glutamatergic synaptic transmission by protein kinase C-mediated sensitization of TRPV1 at the first sensory synapse. J Physiol. 2007;581:631–647. doi: 10.1113/jphysiol.2006.118620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slugg RM, Meyer RA, Campbell JN. Response of cutaneous A- and C-fiber nociceptors in the monkey to controlled-force stimuli. J Neurophysiol. 2000;83:2179–2191. doi: 10.1152/jn.2000.83.4.2179. [DOI] [PubMed] [Google Scholar]
- Sørensen JB. Formation, stabilisation and fusion of the readily releasable pool of secretory vesicles. Pflugers Arch. 2004;448:347–362. doi: 10.1007/s00424-004-1247-8. [DOI] [PubMed] [Google Scholar]
- Spat A, Szanda G, Csordas G, Hajnoczky G. High- and low-calcium-dependent mechanisms of mitochondrial calcium signalling. Cell Calcium. 2008;44:51–63. doi: 10.1016/j.ceca.2007.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strehler EE, Zacharias DA. Role of alternative splicing in generating isoform diversity among plasma membrane calcium pumps. Physiol Rev. 2001;81:21–50. doi: 10.1152/physrev.2001.81.1.21. [DOI] [PubMed] [Google Scholar]
- Sugita S, Shin OH, Han W, Lao Y, Sudhof TC. Synaptotagmins form a hierarchy of exocytotic Ca2+ sensors with distinct Ca2+ affinities. EMBO J. 2002;21:270–280. doi: 10.1093/emboj/21.3.270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szallasi A, Cortright DN, Blum CA, Eid SR. The vanilloid receptor TRPV1: 10 years from channel cloning to antagonist proof-of-concept. Nat Rev Drug Discov. 2007;6:357–372. doi: 10.1038/nrd2280. [DOI] [PubMed] [Google Scholar]
- Tachibana T, Ogura H, Tokunaga A, Dai Y, Yamanaka H, Seino D, Noguchi K. Plasma membrane calcium ATPase expression in the rat spinal cord. Brain Res Mol Brain Res. 2004;131:26–32. doi: 10.1016/j.molbrainres.2004.08.001. [DOI] [PubMed] [Google Scholar]
- Tang YG, Zucker RS. Mitochondrial involvement in post-tetanic potentiation of synaptic transmission. Neuron. 1997;18:483–491. doi: 10.1016/s0896-6273(00)81248-9. [DOI] [PubMed] [Google Scholar]
- Thayer SA, Usachev YM, Pottorf WJ. Modulating Ca2+ clearance from neurons. Front Biosci. 2002;7:D1255–1279. doi: 10.2741/A838. [DOI] [PubMed] [Google Scholar]
- Thomas D, Hanley MR. Pharmacological tools for perturbing intracellular calcium storage. Methods Cell Biol. 1994;40:65–89. doi: 10.1016/s0091-679x(08)61110-3. [DOI] [PubMed] [Google Scholar]
- Toescu EC, Verkhratsky A. Neuronal ageing from an intraneuronal perspective: roles of endoplasmic reticulum and mitochondria. Cell Calcium. 2003;34:311–323. doi: 10.1016/s0143-4160(03)00142-8. [DOI] [PubMed] [Google Scholar]
- Tsuzuki K, Xing H, Ling J, Gu JG. Menthol-induced Ca2+ release from presynaptic Ca2+ stores potentiates sensory synaptic transmission. J Neurosci. 2004;24:762–771. doi: 10.1523/JNEUROSCI.4658-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner KM, Burgoyne RD, Morgan A. Protein phosphorylation and the regulation of synaptic membrane traffic. Trends Neurosci. 1999;22:459–464. doi: 10.1016/s0166-2236(99)01436-8. [DOI] [PubMed] [Google Scholar]
- Usachev YM, DeMarco SJ, Campbell C, Strehler EE, Thayer SA. Bradykinin and ATP accelerate Ca2+ efflux from rat sensory neurons via protein kinase C and the plasma membrane Ca2+ pump isoform 4. Neuron. 2002;33:113–122. doi: 10.1016/s0896-6273(01)00557-8. [DOI] [PubMed] [Google Scholar]
- Usachev YM, Thayer SA. Ca2+ influx in resting rat sensory neurones that regulates and is regulated by ryanodine-sensitive Ca2+ stores. J Physiol. 1999;519:115–130. doi: 10.1111/j.1469-7793.1999.0115o.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vangheluwe P, Sepulveda MR, Missiaen L, Raeymaekers L, Wuytack F, Vanoevelen J. Intracellular Ca2+- and Mn2+-transport ATPases. Chem Rev. 2009;109:4733–4759. doi: 10.1021/cr900013m. [DOI] [PubMed] [Google Scholar]
- Vikman KS, Kristensson K, Hill RH. Sensitization of dorsal horn neurons in a two-compartment cell culture model: wind-up and long-term potentiation-like responses. J Neurosci. 2001;21:RC169. doi: 10.1523/JNEUROSCI.21-19-j0004.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wanaverbecq N, Marsh SJ, Al-Qatari M, Brown DA. The plasma membrane calcium-ATPase as a major mechanism for intracellular calcium regulation in neurones from the rat superior cervical ganglion. J Physiol. 2003;550:83–101. doi: 10.1113/jphysiol.2002.035782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolf CJ, Salter MW. Plasticity and pain: role of the dorsal horn. In: McMahon SB, Koltzenburg M, editors. Textbook of Pain. Amsterdam: Elsevier; 2006. pp. 91–105. [Google Scholar]
- Xu J, Mashimo T, Sudhof TC. Synaptotagmin-1, -2, and -9: Ca2+ sensors for fast release that specify distinct presynaptic properties in subsets of neurons. Neuron. 2007;54:567–581. doi: 10.1016/j.neuron.2007.05.004. [DOI] [PubMed] [Google Scholar]
- Yang K, Kumamoto E, Furue H, Yoshimura M. Capsaicin facilitates excitatory but not inhibitory synaptic transmission in substantia gelatinosa of the rat spinal cord. Neurosci Lett. 1998;255:135–138. doi: 10.1016/s0304-3940(98)00730-7. [DOI] [PubMed] [Google Scholar]
- Yao J, Gaffaney JD, Kwon SE, Chapman ER. Doc2 is a Ca2+ sensor required for asynchronous neurotransmitter release. Cell. 2011;147:666–677. doi: 10.1016/j.cell.2011.09.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]