

Abstract
Complexins (Cplxs) are small, SNARE-associated proteins believed to regulate fast, calcium-triggered exocytosis. However, studies have pointed to either an inhibitory and/or facilitatory role in exocytosis, and the role of Cplxs in synchronizing exocytosis is relatively unexplored. Here, we compare the function of two types of complexin, Cplx 1 and 2, in two model systems of calcium-dependent exocytosis. In mouse neuromuscular junctions (NMJs), we find that lack of Cplx 1 significantly reduces and desynchronizes calcium-triggered synaptic transmission; furthermore, high-frequency stimulation elicits synaptic facilitation, instead of normal synaptic depression, and the degree of facilitation is highly sensitive to the amount of cytoplasmic calcium buffering. In Cplx 2-null adrenal chromaffin cells, we also find decreased and desynchronized evoked release, and identify a significant reduction in the vesicle pool close to the calcium channels (immediately releasable pool, IRP). Viral transduction with either Cplx 1 or 2 rescues both the size of the evoked response and the synchronicity of release, and it restores the IRP size. Our findings in two model systems are mutually compatible and indicate a role of Cplx 1 and 2 in facilitating vesicle priming, and also lead to the new hypothesis that Cplxs may synchronize vesicle release by promoting coupling between secretory vesicles and calcium channels.
Key points
Knockout (KO) of complexin in mouse neuromuscular junctions (NMJs) and adrenal chromaffin cells causes a reduction and a loss in synchronicity in calcium-mediated exocytosis.
A train of high-frequency stimuli induces synaptic facilitation rather than depression in Complexin 1 KO NMJs, and the degree of facilitation is sensitive to calcium buffering.
There is a specific reduction of the vesicle pool close to the calcium channels in Complexin 2 KO chromaffin cells, which can be rescued by heterologous expression of either Complexin 1 or Complexin 2.
Complexin synchronizes vesicle release through facilitating vesicle and calcium channel coupling.
Introduction
Fast, synchronous calcium-mediated exocytosis is critical for nervous and endocrine system function. It is controlled by the SNARE complex and fine-tuned by additional associated proteins. Among the SNARE-associated proteins are complexins (Cplxs), a family of four (Cplx1–4) small (∼150-amino acids) proteins in mammals. Change in Cplx expression is found in many psychiatric and neurodegenerative disorders (Brose, 2008). It is generally accepted that Cplxs regulate exocytosis, but the detailed mechanism is not clear.
An early hypothesis for the role of complexins in regulating exocytosis is that Cplx binding to the SNARE complex ‘clamps’ it, inhibiting otherwise spontaneous membrane fusion (Ono et al. 1998; Giraudo et al. 2006; Schaub et al. 2006; Tang et al. 2006; Huntwork & Littleton, 2007). This idea was supported by early in vitro reconstitution studies showing that an increase in complexin hindered SNARE-mediated fusion (Giraudo et al. 2006; Schaub et al. 2006; Tang et al. 2006), and acute Cplx knock down in neuronal cultures and Cplx knockout in C. elegans and Drosophila led to a significant higher rate of spontaneous vesicle fusion (Huntwork & Littleton, 2007; Maximov et al. 2009; Hobson et al. 2011).
However, experiments in a number of different mammalian preparations were not entirely consistent with the clamp hypothesis. In mammals, knockout of Cplx resulted in a decrease in calcium-triggered exocytosis and no change or a decrease in spontaneous release rate (Reim et al. 2001; Cai et al. 2008; Xue et al. 2008; Strenzke et al. 2009), implying a positive regulatory role of Cplx. Hence, according to more recent hypotheses, Cplxs are capable of either inhibiting or enhancing exocytosis depending on the functional vesicle pool investigated (spontaneous, synchronous or asynchronous release) (Maximov et al. 2009; Strenzke et al. 2009; Hobson et al. 2011; Martin et al. 2011) and the species in which Cplx is studied (Xue et al. 2009; Cho et al. 2010; Hobson et al. 2011), which may be due to species-specific differences in Cplx stucture such that different domains of Cplx may dominate function (Maximov et al. 2009; Xue et al. 2009; Yang et al. 2010; Kaeser-Woo et al. 2012). The currently favoured hypothesis of Cplx function is that Cplx acts to arrrest primed vesicles from fusion but, upon calcium entry and subsequent binding to synaptotagmin, it also acts as a facilitator of vesicle fusion (Wojcik & Brose, 2007; Kaeser-Woo et al. 2012; Malsam et al. 2012).
In the study reported here, we investigated Cplx function in two complementary mouse model systems of calcium-dependent exocytosis – neuromuscular junctions (NMJs) and adrenal chromaffin cells. NMJs are a classic synaptic preparation for study of fast transmitter release. Chromaffin cells are neuroendocrine cells, in which protocols have been standardized to measure the sizes and kinetics of secretion from functional pools of vesicles, thereby making them useful for dissecting out mechanisms of proteins predicted to disrupt vesicle release. Despite the apparent morphological and functional differences between endocrine cells and synapses, the knockout of Cplx leads to parallel phenotypic changes in these two model systems. Inconsistent with the negative clamp function of Cplx, knockout of Cplx decreased rather than increased spontaneous release in mouse NMJs; on the other hand, consistent with a facilitatory function of Cplx, knockout of Cplx decreased calcium-mediated release and led to a loss in synchronicity in both mouse NMJs and chromaffin cells lacking Cplx expression. Although lack of Cplx has been shown to consistently reduce evoked vesicle fusion in multiple preparations in different species, a role for Cplx in synchronizing vesicle release has been reported previously only in murine auditory synapses (Strenzke et al. 2009), and the mechanism of how Cplx regulates the precise timing between calcium influx and vesicle release remains unknown. Here, we used chromaffin cells to assess the size of distinct, functionally defined vesicle pools, and our data suggest that Cplx not only increases the number of primed vesicles, but also facilitates the coupling between secretory vesicles and voltage-gated calcium channels (VGCCs), thereby synchronizing vesicle fusion.
Methods
Animals
Colonies of Cplx hemizygous mice (Cplx1+/− or Cplx2+/−) (Reim et al. 2001) were maintained according to NIH and USC IACUC guidelines. Mice were tailed and genotyped as described (Cai et al. 2008). Wild-type (WT) or hemizygous littermates were used as control animals.
Immunohistochemistry of NMJs
Mice were anaesthetized by intraperitoneal injection of Nembutal (sodium pentobarbital; 50 mg kg−1) or ketamine–xylazine (100 mg kg−1 ketamine–10 mg kg−1 xylazine) and transcardially perfused with normal Ringer solution followed by 4% paraformaldehyde. The extensor digitorum longus (EDL) muscle was teased into layers of five to ten fibres thick to facilitate penetration of antibodies that include: anti-complexin1/2, 3 and 4 (Synaptic systems, Germany; see also Cai et al. 2008). Muscles were incubated with the antibodies at 4°C overnight. After washing with phosphate-buffered saline (PBS), the muscles were incubated in fluorescence-conjugated secondary antibodies at 4°C overnight. Acetylcholine receptors (AChRs) were labelled by Alexa Fluor 594-conjugated α-bungarotoxin (Invitrogen, USA).
Intracellular recording of the neuromuscular junction
Mice were anaesthetized by intraperitoneal injection of Nembutal (sodium pentobarbital; 50 mg kg−1) or ketamine–xylazine (100 mg kg−1 ketamine–10 mg kg−1 xylazine) The extensor digitorum longus (EDL) muscle from 1- to 4-month-old mice was dissected out with the sciatic nerve attached. Intracellular recording was performed in oxygenated normal mammalian Ringer solution (in mm: 135 NaCl, 5 KCl, 1 MgSO4, 15 NaHCO3, 1 Na2HPO4, 11 d-glucose, 2.5 calcium gluconate, pH 7.4). Muscle contraction was blocked by pre-incubating the muscle in 2–3 μmμ-conotoxin (Biomol, USA) for 30 min. The recording was then performed in toxin-free Ringer solution. Miniature endplate potentials (MEPPs) were recorded for 1–2 min, and 40–70 evoked endplate potentials (EPPs) were recorded from a given junction. EPPs were elicited by a 1 Hz train through a suction electrode, normalized to −75 mV and corrected for non-linear summation (McLachlan & Martin, 1981). The mean quantal content was calculated by the direct method (Del Castillo & Katz, 1954). Synaptic transmission was also assessed by paired-pulse stimulation (10 ms apart) and high frequency (30 Hz) stimulation. In experiments involving BAPTA-AM, the muscles were pre-incubated with normal Ringer solution containing 100 μm BAPTA-AM for 2 h before recording. Data were acquired and analysed by pClamp8 and Minialysis software.
Chromaffin cell culture and viral infection
For chromaffin cell experiments, we compared WT (+/+) and Cplx 2 KO (−/−) mice ranging from 9–13 days old from the same litter to minimize genetic variability. Mice were anaesthetized by intraperitoneal injection of Nembutal (sodium pentobarbital; 50 mg kg−1). Mouse adrenal chromaffin cells were obtained as follows: (i) adrenal glands were dissected out from 9- to 13-day-old mice; (ii) glands were digested with filtered, activated papain (Worthington, Lakewood, NJ, USA; we used ∼18 units per 1 ml of papain solution, bubbled with 95% O2–5% CO2 for >20 min by shaking in a 37°C water bath for 25–30 min); (iii) enzyme solution was replaced by chromaffin cell media and glands were triturated and plated on glass-bottomed chambers (4 per mouse). Enzyme solution contained l-cysteine-supplemented high-glucose DMEM with 1 mm CaCl2 and 0.5 mm EDTA. Chromaffin cell media contained high-glucose DMEM with 0.1% penicillin/streptomycin and 1% serum-free supplement (Insulin-Transferrin-Selenium-X). Chromaffin cells were infected with Semliki Forest virus (SFV) 1–2 days post dissection. SFV infection of chromaffin cells was previously described (Duncan et al. 1999, 2002). Here we used the DNA-based pSCA and pSCA helper vectors, generous gifts from Dr Rod Bremner of the University of Toronto (DiCiommo & Bremner, 1998). HEK293T (60–70% confluency) cells were transfected with the empty pSCA-IRES-GFP, pSCA-Cplx2-IRES-GFP, or pSCA-Cplx1-IRES-GFP vector along with a helper pSCA vector. The medium was supplemented with 10 mm sodium butyrate 1 day post transfection and replaced with normal chromaffin cell media 8 h later. Virus-containing medium was filtered (0.45 μm) and harvested the next day. Activation of SFV was done by adding 1/20th volume of α-chymotrypsin (10 mg ml−1) to viral stock and incubating for 45 min at room temperature, then adding 1/40th volume of aprotinin (10 mg ml−1) and incubating for 10 min at room temperature. Activated SFV stock (20–30 μl) containing the empty IRES-GFP, Cplx2-IRES-GFP, or Cplx1-IRES-GFP was added to plated chromaffin cells in 250 ml medium 1–2 days after dissection. Infected cells were patched ∼12–24 h post-transduction.
Electrophysiological experiments on chromaffin cells
Experiments were conducted 2–4 days post dissection. Capacitance recordings were obtained in conventional whole-cell patch clamp configuration using an Olympus IX70 inverted microscope, EPC-9 amplifier and Pulse software (HEKA Electronics), as previously described (Cai et al. 2008). The glass-bottomed chambers with adherent chromaffin cells were washed with PBS and then filled with standard extracellular solution consisting of 140 mm NaCl, 2.8 mm KCl, 10 mm Hepes, 1 mm MgCl2, 2 mm CaCl2, 10 mm glucose, pH adjusted to 7.2–7.4, and osmolarity adjusted to 290–310 mOsm/L. The pipette solutions contained 10 mm NaCl, 145 mm glutamic acid, 10 mm Hepes, 1 mm MgCl2, 0.1 mm EGTA, 2 mm ATP, 0.3 mm GTP, titrated with 5 m CsOH to pH 7.2–7.4, and osmolarity adjusted to 290–310 mOsm/L. The calcium influx was calculated by integrating the inward current trace starting from 4 ms after the stimulation onset. Because the fast sodium influx usually peaks within 2 ms, the resulting integration calculation was started at 4 ms and is attributed primarily to calcium influx.
Data analysis
Data are represented as means with the standard error of the mean (SEM) and were statistically compared using an unpaired, two-tailed Student's t test, or the Mann–Whitney non-parametric test, as indicated. For chromaffin cell experiments, we performed experiments on control cells (derived from WT and heterozygous animals) and Cplx 2 knockout cells prepared on the same day, to reduce preparation-to-preparation variability.
Results
Complexin 1 is the major isoform at the mouse neuromuscular junction
To examine which, if any, of the four Cplx isoforms is expressed at the mouse NMJ, we immunostained the Cplx proteins at the mouse NMJ, using antibodies that recognize (a) both Cplxs 1 and 2 (this antibody hereafter designated Cplx 1/2), (b) Cplx 3 only or (c) Cplx 4 only. At wild-type (WT) NMJs, we found robust Cplx 1/2 expression, which colocalized with the acetylcholine receptors (AChRs) labelled with α-bungarotoxin, whereas Cplx 3 and 4 were undetectable (Fig. 1A). Since Cplx 1/2 antibody does not differentiate between the two isoforms, we next examined NMJs in Cplx 1 KO and Cplx 2 KO mice. At Cplx 1 KO NMJs, Cplx 1/2 immunoreactivity was absent, suggesting Cplx 1 is the predominant isoform at the mouse NMJ (Fig. 1B). On the other hand, significant Cplx 1/2 immunoreactivity could be seen at Cplx 2 KO NMJs, indicating a high level of Cplx 1 (Fig. 1B). We noted that Cplx 3 and 4 immunoreactivity was absent at both Cplx 1 KO and Cplx 2 KO NMJs (data not shown). Together, these data show that Cplx 1 is the major Cplx isoform at the mouse NMJ, and knockout of Cplx 1 does not lead to upregulation of the other isoforms.
Figure 1. Complexin 1 is the major complexin isoform at the mouse NMJ.
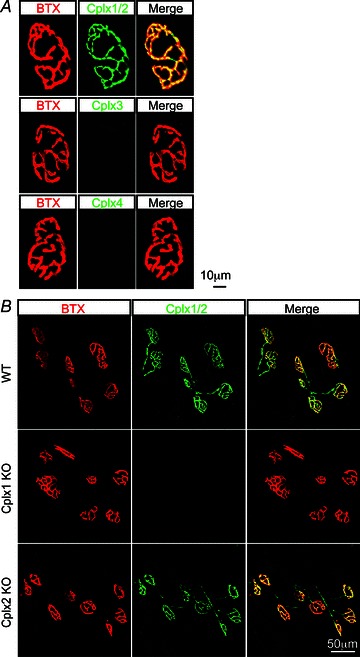
A, NMJs from adult (1–4 months old) WT mice were labelled with α-bungarotoxin for acetylcholine receptors (red) and antibodies against Cplx 1/2, 3 and 4 (green). Cplx 1/2 was found at WT NMJs but no Cplx 3 or 4 expression was detected. Since Cplx 1/2 antibody does not distinguish between the two isoforms, we next examined NMJs in complexin 1 KO and Cplx 2 KO mice. B, NMJs from adult (1–4 months old) WT, Cplx 1 KO and Cplx 2 KO mice were labelled with α-bungarotoxin for acetylcholine receptors (red) and antibodies against Cplx 1/2 (green). Cplx 2 was found to be absent at Cplx 1 KO NMJs while robust Cplx 1 expression can be detected at Cplx 2 KO NMJs.
Cplx 1 KO neuromuscular junctions show a reduced spontaneous vesicle release
To examine the function of Cplx at the mouse NMJ, we performed intracellular recordings in extensor digitorum longus (EDL) muscles of young adult (1–4 months old) Cplx 1 KO mice, using wild-type (Cplx 1+/+, WT) or hemizygous (Cplx 1+/−) littermates as controls, since these two genotypes showed no difference in NMJ synaptic transmission properties. At Cplx 1 KO NMJs, the spontaneous miniature endplate potential (MEPP) frequency was decreased by 68% (Fig. 2A, B), but the MEPP amplitude and shape (rise and decay times, Fig. 2C–E) were not changed compared to control. In contrast to Cplx 1 KO NMJs, Cplx 2 KO NMJs did not show significant differences in MEPP frequency or amplitude. The finding that there is a reduced MEPP frequency at Cplx 1 KO NMJs suggests that Cplx promotes spontaneous release at mouse NMJs.
Figure 2. Spontaneous vesicle release is reduced at Cplx 1 KO NMJs.
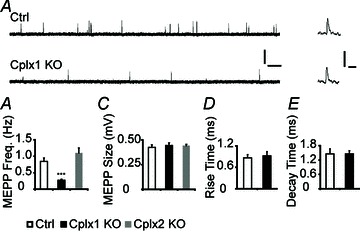
A, sample traces of MEPPs from adult (1–4 months old) control (top) and Cplx 1 KO (bottom) NMJs are shown (scale bars, 0.5 mV, 0.5 s). Sample traces of individual MEPPs from control and Cplx 1 KO NMJs are shown on the right (scale bars, 0.5 mV, 10 ms). B, MEPP frequency is significantly reduced at Cplx 1 KO but not Cplx 2 KO NMJs (Ctrl vs. Cplx 1 KO vs. Cplx 2 KO: 0.85 ± 0.11 vs. 0.27 ± 0.03 vs. 1.10 ± 0.15 Hz). C, MEPP amplitudes are similar at both Cplx 1 KO and Cplx 2 KO NMJs (Ctrl vs. Cplx 1 KO vs. Cplx 2 KO: 0.42 ± 0.03 vs. 0.44 ± 0.03 vs. 0.44 ± 0.01 mV). Rise time (D) and decay time (E) of MEPPs are similar for Cplx 1 KO and control NMJs (Ctrl vs. Cplx 1 KO rise times: 0.86 ± 0.09 vs. 0.92 ± 0.12 ms; Ctrl vs. Cplx 1 KO decay times: 1.46 ± 0.21 vs. 1.46 ± 0.13 ms). Values are means ± SEM; n= 100–130 NMJs from 5–7 mice for each genotype; ***P < 0.001, Student's t test.
Cplx 1 KO neuromuscular junctions show a reduction and loss of synchronization in evoked vesicle release
Examination of nerve-evoked release recordings revealed that NMJs lacking Cplx 1 exhibit severe defects (Fig. 3A). Cplx 1 KO NMJs showed a 70% reduction in evoked endplate potential (EPP) amplitude, and estimated quantal content (QC) was reduced by 66% (Fig. 3A–C). Moreover, the synchronicity of the fast Ca2+ triggered release was significantly disrupted in Cplx 1 KO NMJs, as evidenced by the 20–30% prolongation in the EPP rise times, decay times and half-widths (Fig. 3D–F). Whereas at control NMJs, the evoked release is so tightly synchronized that each nerve stimulation results in identical EPP peak times, at only 8% of Cplx 1 KO NMJs did the evoked release appear normally synchronized (Fig. 3G). There was a much higher incidence of asynchronous, delayed vesicle release following nerve stimulation at Cplx 1 KO NMJs (Fig. 3H). In contrast to Cplx 1 KO NMJs, Cplx 2 KO NMJs did not exhibit significant alterations in transmitter release, which corroborates the finding that Cplx 1 is the major isoform at the mouse NMJ. These data support a facilitatory role of Cplx in regulating the size of the evoked vesicle release and an additional role in synchronizing evoked release at the mouse NMJ.
Figure 3. Evoked vesicle release is reduced and desynchronized at Cplx 1 KO NMJs.
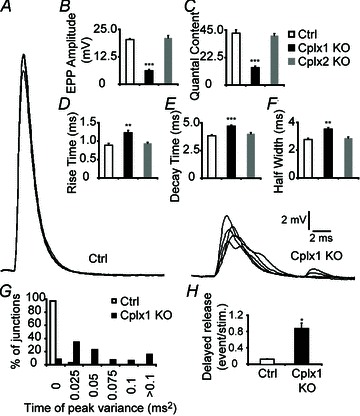
A, sample traces of EPPs from adult (1–4 months old) control (left) and Cplx 1 KO (right) NMJs are shown. B, EPP amplitude is greatly decreased at Cplx 1 KO NMJs. However, EPP amplitude is unaltered at Cplx 2 KO NMJs (Ctrl vs. Cplx 1 KO vs. Cplx 2 KO: 20.34 ± 0.46 vs. 6.20 ± 0.53 vs. 21.09 ± 1.05 mV). C, quantal content is significantly reduced at Cplx 1 KO NMJs but remained the same for Cplx 2 KO NMJs (Ctrl vs. Cplx 1 KO vs. Cplx 2 KO: 42.39 ± 2.82 vs. 14.37 ± 1.09 vs. 40.45 ± 1.74). D–F, EPP rise time (D), decay time (E) and half-width (F) were significantly increased at Cplx 1 KO NMJs (Ctrl vs. Cplx 1 KO vs. Cplx 2 KO: rise time, 0.89 ± 0.05 vs. 1.23 ± 0.06 vs. 0.94 ± 0.03 ms; decay time, 3.84 ± 0.09 vs. 4.70 ± 0.10 vs. 4.01 ± 0.11 ms; half-width, 2.77 ± 0.10 vs. 3.51 ± 0.12 vs. 2.86 ± 0.09 ms). G, there were greater variances in EPP time of peak at Cplx 1 KO NMJs. H, the delayed vesicle release was significantly increased at Cplx 1 KO NMJs (Ctrl vs. Cplx 1 KO: 0.13 ± 0.01 vs. 0.87 ± 0.13 events/stimulation). Values are means ± SEM; n= 100–130 NMJs from 5–7 mice for each genotype; *P < 0.05; **P < 0.01; ***P < 0.001, Student's t test.
Cplx 1 KO neuromuscular junctions display altered short-term synaptic plasticity
Normally, at phasic synapses such as mouse NMJs, evoked vesicle release is fast and synchronized. Because most primed vesicles are thought to be close to voltage-gated calcium channels (VGCCs), high-frequency stimulation should efficiently couple stimulation to secretion, depleting primed vesicles and leading to synaptic depression. This is indeed what we observed in control NMJs (Fig. 4A). Cplx 1 KO NMJs, however, behave more like tonic synapses, showing initially reduced evoked release, but significantly greater paired-pulse facilitation and synaptic facilitation, rather than depression, during high-frequency stimulation (Fig. 4). Asynchronous release becomes more prominent as stimulation is repeated. We hypothesize that the increase in synaptic response during consecutive stimulations in Cplx 1 KO NMJs occurs because many primed vesicles are located far from calcium channels, and successive depolarizations lead to summation and increase in the size of the high-concentration calcium cloud, which therefore reaches even those primed vesicles far from calcium channels. Because of the variable distances between calcium channels and the vesicles not associated with calcium channels, ‘jitter’ in time to vesicle fusion becomes pronounced. In support of this hypothesis, the increased facilitation in Cplx 1 KO NMJs is sensitive to cytoplasmic calcium buffering, as incubation with the calcium chelator BAPTA-AM (100 μm) caused paired-pulse facilitation to decrease significantly from 158% to 133% (Fig. 4C, D).
Figure 4. Short-term synaptic plasticity is altered at Cplx 1 KO NMJs.
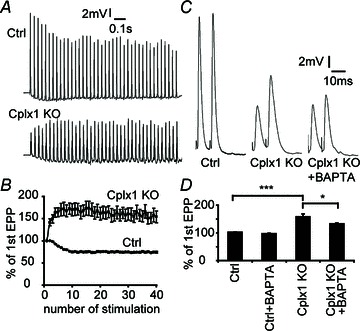
A, sample traces of EPP responses upon 30 Hz stimulation were shown. B, control NMJs displayed depression whereas Cplx 1 KO NMJs showed strong synaptic facilitation. Values are means ± SEM; n= 22–29 NMJs from 7 mice for each genotype. C, sample traces of paired-pulse (10 ms apart) responses from control, Cplx 1 KO and BAPTA-AM treated Cplx 1 KO NMJs are shown. D, stronger paired-pulse facilitation was observed at Cplx 1 KO NMJs (Ctrl vs. Cplx 1 KO, 102.1 ± 0.7 vs. 158.4 ± 10.2%; ***P < 0.0001, Student's t test; n= 16–19 NMJs from 3–5 mice for each genotype). The calcium chelator BAPTA-AM significantly reduced the paired-pulse facilitation at Cplx 1 KO NMJs (Ctrl with BAPTA-AM: 97.0 ± 1.8%; Cplx 1 KO with BAPTA-AM: 132.8 ± 2.6%; *P= 0.018, Student's t test; n= 19–20 NMJs from 3–5 mice for each group).
The immediately releasable pool size is decreased and asynchronous release is increased in Cplx 2 KO chromaffin cells
A quantitative biophysical model was proposed recently for the mechanism underlying the differences between phasic and tonic synapses (Pan & Zucker, 2009). The model tests the consequence of dividing the primed pool of vesicles into two functional subsets, one consisting of vesicles attached to voltage-gated calcium channels (VGCCs) and the other unattached. Assigning more or fewer vesicles to the pool associated with VGCCs reproduced the behaviour seen at phasic versus tonic synapses, respectively. Specifically, reducing the pool of vesicles associated with VGCCs led to the loss of synchronization of stimulated release, the lack of synaptic depression during high-frequency stimulation, and an increase in asynchronous release – all behaviour seen in tonic synapses, and behaviour that resembles what is seen at Cplx 1 KO NMJs.
We wanted to test further the idea that complexin plays a role coupling secretory vesicles to VGCCs in another model system. In adrenal chromaffin cells, the relative size of the functional vesicle pools can be readily distinguished with membrane capacitance measurements, which report the increase in plasma membrane area, as vesicle membrane is added by exocytosis. We applied a train of 10-ms depolarizing pulses in whole-cell patch clamp configuration, to elicit a highly localized calcium cloud, centred on each opened VGCC, which triggers fusion of only those vesicles in close proximity to the channels, the so-called immediately releasable pool (IRP) (Horrigan & Bookman, 1994; Voets et al. 1999). The train of brief 10-ms depolarizations was followed by a train of longer (100-ms) depolarizing pulses to trigger exocytosis of remaining vesicles from the readily releasable pool (RRP), comprising all vesicles that undergo rapid, triggered exocytosis, whether close to or far from the VGCCs. Endocytosis during capacitance measurement may cause error in measurements of exocytosis. However, previous work showed that endocytosis is essentially absent by 2 min after patch rupture in whole-cell configuration (Burgoyne, 1995). We confirmed this in our experiments, and routinely waited at least 2 min before making our recordings. It is also important to note that the presence or absence of Cplx 2 does not significantly affect endocytosis (Cai et al. 2008).
Unlike NMJs, for which Cplx 1 is the dominant isoform, chromaffin cells express only Cplx 2 (Cai et al. 2008). We have previously shown that RRP size is reduced in Cplx 2 KO (−/−) chromaffin cells compared to WT (+/+) cells (Cai et al. 2008). Here, we show that Cplx 2 KO (−/−) chromaffin cells displayed significantly reduced capacitance steps in response to short 10-ms depolarizing pulses (Fig. 5A, C, F) This suggests that the number of vesicles located near VGCCs (the immediately releasable pool or IRP) is reduced in Cplx 2 KO cells, compared to WT cells.
Figure 5. Knockout of Cplx 2 impairs secretion of vesicles in the immediately releasable pool (IRP) but enhances asynchronous secretion in mouse chromaffin cells.
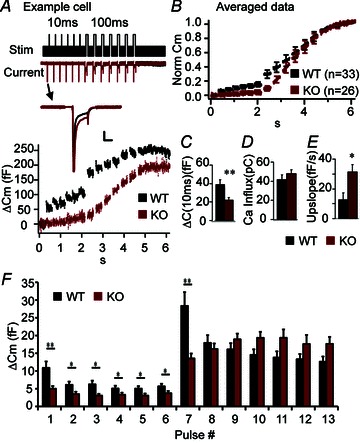
A, capacitance and current data from a WT (black) chromaffin cell and a Cplx 2 KO (red) chromaffin cell from the same litter and recorded on the same day, showing very obvious differences in secretion (ΔCm) elicited by 10-ms and 100-ms depolarizing jumps. Here, we have not normalized the traces, and it is clear that total secretion is less in the KO cells, in agreement with Cai et al. 2008. In addition, the WT cell exhibits exceptionally large amount of secretion elicited during the train of short depolarizations. The stimulation protocol used is shown at the top: a series of 6 brief 10-ms depolarization pulses (0 mV) from a resting membrane potential of −80 mV, were used to selectively elicit fusion of primed vesicles in the immediately releasable pool (IRP), followed by a series of 7 longer 100-ms depolarization pulses (0 mV) to induce fusion of remaining primed vesicles in the readily releasable pool (RRP). Inward current was not significantly different in Cplx 2 KO vs. WT cells (A, traces labelled ‘current’ on left). Inset: current traces from the first 10-ms depolarization demonstrate that the decrease in secretion is not due to a decrease in calcium influx. Scale bars, 5 ms, 0.5 nA. B, normalized and averaged capacitance recordings from 33 WT (+/+, black) and 26 KO (−/−, red) chromaffin cells. C, the IRP size (sum of non-normalized capacitance steps elicited by a train of brief 10-ms pulses) is significantly smaller in Cplx 2 KO cells (red) compared to WT cells (black) (n= 33 WT, n= 26, KO, P= 0.006 Mann–Whitney test). D, there was no significant difference in the calculated total ionic current (averaged area of the inward current, ignoring the initial large spike due to Na+ channels, summed over all depolarizing jumps) in WT (black) and KO (red) cells, as indicated. E, the capacitance upslope is used to assess the degree of asynchronous release in chromaffin cells and is calculated by measuring the slope of a fitted line to the capacitance recording between the longer 100-ms depolarizing pulses. There is significant increase in the capacitance upslope in Cplx 2 KO cells (red) compared to WT (black) (n= 33 WT, n= 26, KO, P= 0.032 Mann–Whitney test). F, averaged capacitance steps at each depolarization pulse are shown. *P < 0.05, **P < 0.01, ***P < 0.001. Error bars, SEM.
However, in contrast to WT cells, KO cells did not show depletion (large response to first depolarization followed by smaller responses) during the train of long 100-ms depolarizing pulses (Fig. 5F), so RRP size cannot be estimated in KO cells with the current protocol. This result suggests that in KO cells, the repeated stimulation results in a build-up of calcium, which reaches and triggers the exocytosis of the vesicles farther from calcium channels, a phenomenon similar to the synaptic facilitation observed in the NMJ. More importantly, in KO cells, we observed a significant increase in membrane capacitance between 100-ms depolarizations, during which time calcium channels have already closed, signifying asynchronous release (Thiagarajan, 2005). We quantified the amount of the asynchronous release by calculating the slope of the capacitance recording following each 100-ms pulse. On average, Cplx 2 KO cells showed a greater than twofold increase in the upslope of the capacitance compared to WT cells (Fig. 5B, E). The change in the capacitance steps was not a consequence of change in calcium current, as there was no significant difference in the peak current (see current trace inset in Fig. 5A), nor in the area of averaged, integrated current influx (Fig. 5D). Thus, our chromaffin cell data indicate that lack of Cplx decreases the number of vesicles close to VGCCs (IRP) and also increases asynchronous release.
Decrease of the immediately releasable pool size and asynchronous release in Cplx 2 KO chromaffin cells is rescued by expression of Cplx 2 and Cplx 1
To verify that the decrease in the IRP size and increase in the capacitance upslope is indeed due to the lack of Cplx 2 and not to other compensatory changes, we performed rescue experiments using Semliki Forest Virus (SFV), a vector known to be effective in heterologous gene expression in chromaffin cells (Duncan et al. 2002). As shown in Fig. 6, expression of Cplx 2 in Cplx 2 KO cells significantly restores the IRP size, and decreased the capacitance upslope by 71%. Furthermore, expressing Cplx 1 in Cplx 2 KO cells also rescued the IRP size and the asynchronous release, indicating that Cplx 1 and Cplx 2 not only share 86% sequence identity (Brose, 2008) but also share similar roles in regulating vesicle release.
Figure 6. Both Cplx 2 and Cplx 1 can rescue the decreased IRP secretion and the increased upslope observed in Cplx 2 knockout cells.
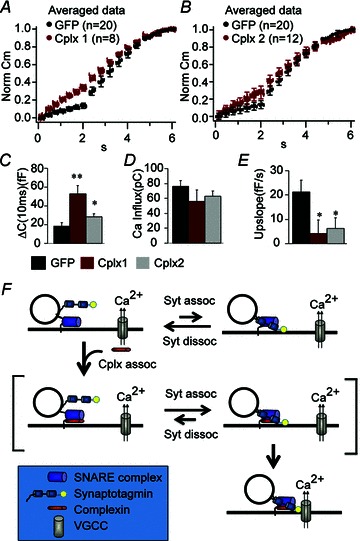
A and B, averaged and normalized capacitance traces from Cplx 2 KO chromaffin cells infected with GFP vector alone (black) or Cplx1-IRES-GFP (red; A), or Cplx2-IRES-GFP (red; B). C, there is a significant increase in IRP size in cells expressing Cplx1-IRES-GFP (red, n= 8, P= 0.001), or Cplx2-IRES-GFP (grey, n= 12, P= 0.01) compared to cells expressing GFP alone (n= 20). D there is no significant change in calcium influx. E, the increased upslope observed in Cplx 2 KO cells expressing only GFP (black, n= 20) is significantly reversed by expression of either Cplx1-IRES-GFP (red, n= 8, P= 0.04) or Cplx2-IRES-GFP (grey, n= 12, P= 0.03). P values are obtained using the Mann–Whitney non-parametric statistical test. Error bars, SEM. F, a working model for Cplx function in facilitating fast calcium-coupled secretion. When vesicles are primed, synaptotagmin (Syt) binds to the SNARE complex and can undergo calcium-triggered fusion. However, the binding of Cplx to the SNARE complex increases the probability of synaptotagmin binding to and remaining bound to the SNARE complex. The SNARE-synaptotagmin- Cplx complex, through an as-yet-undefined mechanism, favours the co-localization of the vesicle with voltage-gated calcium channels.
Discussion
We have reported here on studies concerning the role of Cplx in two distinct model systems of calcium-dependent exocytosis – one a classical synapse preparation and the other a neuroendocrine cell. As different as these two systems are, the data are mutually compatible and point to a multi-faceted role for Cplx. Our previous work showed that Cplx is involved in regulating the size of the readily releasable pool of vesicles. The present work extends that result by showing that, in addition, Cplx plays a role in increasing IRP by co-localizing the secretory vesicles and voltage-gated calcium channels.
Despite consistent observations of reduced evoked release in Cplx KO studies, much of the focus in Cplx studies has instead been on changes in spontaneous release. Some early studies showed an increase in spontaneous miniature endplate activity when Cplx expression is decreased or abolished (Huntwork & Littleton, 2007; Maximov et al. 2009; Hobson et al. 2011; Kaeser-Woo et al. 2012), apparently supporting the hypothesis that Cplx behaves as a negative clamp on vesicle exocytosis by its interaction with the SNARE proteins. It was further suggested that calcium-triggered secretion would relieve this Cplx-mediated clamp, allowing SNARE-mediated fusion to occur (Tang et al. 2006). However, Cplx studies on spontaneous release have not all been consistent. Other studies showed no change (Reim et al. 2001) or a reduction in spontaneous activity (Xue et al. 2008; Strenzke et al. 2009) when Cplx expression is removed, consistent with our observations. Furthermore, it was also reported that Cplx translocates from the cytoplasm to the plasma membrane upon antigen stimulation in rat mast cell line, rendering the negative clamp model for Cplx function unlikely, at least in this particular cell type (Tadokoro, 2005). Some of the different behaviours of Cplx may be attributable to true differences in its function in different species (BLASTp analysis revealed only 17–38% amino acid identity between mammalian and non-mammalian species), or in different cell types. Perhaps in the various species or cell types, Cplx binds to different protein partners, activating differing functions. Our data do not support the hypothesis that Cplx serves as a negative clamp for exocytosis in mouse neuromuscular junctions or in chromaffin cells.
However, in contrast to inconsistent data from studies of the role of Cplxs in spontaneous miniature endplate/exocytosis activity, other studies show that Cplx knockout has consistent effects on evoked activity across different cell types and species (Reim et al. 2001; Tadokoro, 2005; Cai et al. 2008; Xue et al. 2008; Maximov et al. 2009; Strenzke et al. 2009; Hobson et al. 2011; Martin et al. 2011; Kaeser-Woo et al. 2012). Loss of Cplx leads to a reduction in evoked transmitter release in all preparations, but also to a loss of synchronicity in evoked vesicle release in neuromuscular synapses and chromaffin cells (data presented here), auditory synapses (Strenzke et al. 2009), and most recently in Drosophila NMJs, where a reduced IRP size is also observed (Jorquera et al. 2012). The appearance of ‘jitter’ in release timing in the absence of Cplx indicates an additional regulatory function for Cplx. Here we propose the new hypothesis that Cplx is also involved in coupling the vesicle-associated SNARE complex with voltage-gated ion channels, which serves to fine-tune the precise timing of calcium-triggered fast release. It should be noted, however, that there is the alternative possibility that knocking out Cplx disrupts calcium sensing, which will also result in the lack of synaptic depression and an increase in asynchronous release. Although several studies suggested a reduced calcium sensitivity in complexin knockout neurons (Reim et al. 2001; Xue et al. 2008), our previous data suggested calcium sensitivity was not altered in Cplx KO chromaffin cells, as photolysis of caged calcium revealed no change in the kinetics of exocytosis (Cai et al. 2008).
Recent studies show that synaptotagmin is the calcium sensor in regulated exocytosis and suggest that it is a key molecule involved in synchronizing release and suppressing asynchronous release (Voets et al. 2001; Yoshihara & Littleton, 2002; Nishiki & Augustine, 2004) by positioning synaptic vesicles near calcium channels (Young & Neher, 2009). A mutation at the C2B domain of synaptotagmin disrupts its ability to synchronize release without interfering with its proper folding and calcium binding (Araçet al. 2006; Young & Neher, 2009). The similarity in vesicle release phenotypes of cells expressing this mutant synaptotagmin and those lacking Cplx indicates that Cplx and synaptotagmin may work together to facilitate the coupling of vesicles to VGCCs. Interestingly, studies have documented the existence of concurrent binding of Cplx and synaptotagmin to the SNARE complex (Chicka & Chapman, 2009; Choi et al. 2010; Vrljic et al. 2010; Kyoung et al. 2011; but cf. Tang et al. 2006), raising the possibility that the two proteins function cooperatively. There is also evidence that Cplx may interact directly with synaptotagmin (Tokumaru et al. 2008).
There are, however, important phenotype differences between synaptotagmin KOs and Cplx KOs. In caged-calcium photolysis experiments in wild-type chromaffin cells, stepping cytosolic calcium concentration up to stimulatory levels triggers a rapid-secretion exocytotic burst (comprising two fast kinetic components) that has been attributed to the rapid fusion of docked, primed vesicles. The burst phase is reduced by about two-thirds in Cplx 2 KO cells (Cai et al. 2008), whereas it is completely absent in chromaffin cells lacking synaptotagmin (Voets et al. 2001; Nagy et al. 2006). Notably, the reduction in burst amplitude seen in Cplx 2 KO cells does not involve a change in kinetics of the burst (Cai et al. 2008), indicating a reduction in the number of primed vesicles, but normal function in those vesicles that remain primed. Given that the presence of synaptotagmin is essential for the burst, we propose that synaptotagmin mediates the burst, and the action of synaptotagmin depends upon binding of synaptotagmin to the SNARE complex. Furthermore, we propose that normally synaptotagmin exists in a dynamic equilibrium between SNARE-bound and -unbound states, and Cplx binding to the SNARE complex enhances and stabilizes the SNARE-bound state of synaptotagmin, as depicted in our model (Fig. 6F). This stabilization increases the exocytotic burst, reconciling previously published studies on the role of Cplx and synaptotagmin in facilitating release (Cai et al. 2008; Young & Neher, 2009; Hobson et al. 2011).
Young and Neher previously showed that the C-terminus of synaptotagmin plays a role in coupling the vesicle-associated SNARE complex with voltage-gated calcium channels (Young & Neher, 2009). In our model, Cplx stabilizes binding of synaptotagmin to the SNARE complex, which in turn promotes the coupling of vesicles to calcium channels – a prediction that fits our observations. Note that the recruitment of calcium channels to the vesicle-associated SNARE complex may involve other proteins, and that a direct interaction between synaptotagmin or complexin and calcium channels is not needed (Zamponi, 2003).
Loss of vesicle-channel coupling has been suggested to be important in a cellular model of diabetes mellitus (Collins et al. 2010). Decreased Cplx expression has been correlated with exposure to high glucose (Dubois et al. 2006), and may contribute to the loss of vesicle-channel coupling and to the decreased insulin secretion in diabetes. Dissecting out the molecular mechanism of secretion regulation – particularly how vesicles are targeted to calcium channels – may lead to novel pharmaceutical treatments of diseases that are associated with abnormal secretory processes.
Acknowledgments
We acknowledge the support of the following: NIH GM85791 (R.H.C.), NIH K18DK091445 (R.H.C.), NIH DK60623 (R.H.C.), Human Frontiers Science Program (to T.D. Parsons, N. Brose, G. Borst and R.H. Chow), NIH NS063296 (C.-P.K.), Deutsche Forschungsgemeinschaft (N.B. and K.R.), NIH F32GM088967 (J.G.R.). We would also like to thank the following for critical feedback: Erwin Neher, Rainhard Jahn, Nils Brose, Dion Dickman, Mark Rich, Kathrin Engisch, Kassandra Kisler, Jung Hwa Cho, Reymundo Dominguez, and Robert Farley.
Glossary
- Cplx
complexin
- IRP
immediately releasable pool
- NMJ
neuromuscular junction
- RRP
readily releasable pool
- SFV
Semliki Forest virus
- VGCC
voltage-gated calcium channels
- WT
wild-type
Author contributions
M.-Y.L. and J.G.R. contributed equally to the work. M.-Y.L. performed the immunostaining and electrophysiology in mouse NMJs. J.G.R. performed the electrophysiology in chromaffin cells. M.-Y.L. and J.G.R. collected and analysed the data. H.C. performed the initial preliminary experiments in chromaffin cells. K.R. provided critical input for the experiment and manuscript. R.H.C. and C.-P.K. co-supervised the experiment design and data interpretation. M.-Y.L., J.G.R., R.H.C. wrote the manuscript with inputs from K.R. and C.-P.K. All authors approved the final version of the manuscript.
References
- Araç D, Chen X, Khant HA, Ubach J, Ludtke SJ, Kikkawa M, Johnson AE, Chiu W, Südhof TC, Rizo J. Close membrane-membrane proximity induced by Ca2+-dependent multivalent binding of synaptotagmin-1 to phospholipids. Nat Struct Mol Biol. 2006;13:209–217. doi: 10.1038/nsmb1056. [DOI] [PubMed] [Google Scholar]
- Brose N. For better or for worse: Complexins regulate SNARE function and vesicle fusion. Traffic. 2008;9:1403–1413. doi: 10.1111/j.1600-0854.2008.00758.x. [DOI] [PubMed] [Google Scholar]
- Burgoyne RD. Fast exocytosis and endocytosis triggered by depolarisation in single adrenal chromaffin cells before rapid Ca2+ current run-down. Pflugers Arch. 1995;430:213–219. doi: 10.1007/BF00374652. [DOI] [PubMed] [Google Scholar]
- Cai H, Reim K, Varoqueaux F, Tapechum S, Hill K, Sorensen JB, Brose N, Chow RH. Complexin II plays a positive role in Ca2+-triggered exocytosis by facilitating vesicle priming. Proc Natl Acad Sci U S A. 2008;105:19538–19543. doi: 10.1073/pnas.0810232105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chicka MC, Chapman ER. Concurrent binding of complexin and synaptotagmin to liposome-embedded SNARE complexes. Biochemistry. 2009;48:657–659. doi: 10.1021/bi801962d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho RW, Song Y, Littleton JT. Comparative analysis of Drosophila and mammalian complexins as fusion clamps and facilitators of neurotransmitter release. Mol Cell Neurosci. 2010 doi: 10.1016/j.mcn.2010.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi UB, Strop P, Vrljic M, Chu S, Brunger AT, Weninger KR. Single-molecule FRET-derived model of the synaptotagmin 1–SNARE fusion complex. Nat Struct Mol Biol. 2010;17:318–324. doi: 10.1038/nsmb.1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins SC, Hoppa MB, Walker JN, Amisten S, Abdulkader F, Bengtsson M, Fearnside J, Ramracheya R, Toye AA, Zhang Q, Clark A, Gauguier D, Rorsman P. Progression of diet-induced diabetes in C57BL6J mice involves functional dissociation of Ca2+ channels from secretory vesicles. Diabetes. 2010;59:1192–1201. doi: 10.2337/db09-0791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Castillo J, Katz B. Quantal components of the end-plate potential. J Physiol. 1954;124:560–573. doi: 10.1113/jphysiol.1954.sp005129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiCiommo DP, Bremner R. Rapid, high level protein production using DNA-based Semliki Forest Virus vectors. J Biol Chem. 1998;273:18060–18066. doi: 10.1074/jbc.273.29.18060. [DOI] [PubMed] [Google Scholar]
- Dubois M, Vacher P, Roger B, Huyghe D, Vandewalle B, Kerr-Conte J, Pattou F, Moustaid-Moussa N, Lang J. Glucotoxicity inhibits late steps of insulin exocytosis. Endocrinology. 2006;148:1605–1614. doi: 10.1210/en.2006-1022. [DOI] [PubMed] [Google Scholar]
- Duncan RR, Don-Wauchope AC, Tapechum S, Shipston MJ, Chow RH, Estibeiro P. High-efficiency Semliki Forest virus-mediated transduction in bovine adrenal chromaffin cells. Biochem J. 1999;342:497–501. [PMC free article] [PubMed] [Google Scholar]
- Duncan RR, Greaves J, Tapechum S, Apps DK, Shipston MJ, Chow RH. Efficacy of Semliki Forest virus transduction of bovine adrenal chromaffin cells: an analysis of heterologous protein targeting and distribution. Ann N Y Acad Sci. 2002;971:641–646. doi: 10.1111/j.1749-6632.2002.tb04543.x. [DOI] [PubMed] [Google Scholar]
- Giraudo CG, Eng WS, Melia TJ, Rothman JE. A clamping mechanism involved in SNARE-dependent exocytosis. Science. 2006;313:676–680. doi: 10.1126/science.1129450. [DOI] [PubMed] [Google Scholar]
- Hobson RJ, Liu Q, Watanabe S, Jorgensen EM. Complexin maintains vesicles in the primed state in C. elegans. Curr Biol. 2011;21:106–113. doi: 10.1016/j.cub.2010.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horrigan FT, Bookman RJ. Releasable pools and the kinetics of exocytosis in adrenal chromaffin cells. Neuron. 1994;13:1119–1129. doi: 10.1016/0896-6273(94)90050-7. [DOI] [PubMed] [Google Scholar]
- Huntwork S, Littleton JT. A complexin fusion clamp regulates spontaneous neurotransmitter release and synaptic growth. Nat Neurosci. 2007;10:1235–1237. doi: 10.1038/nn1980. [DOI] [PubMed] [Google Scholar]
- Jorquera RA, Huntwork-Rodriguez S, Akbergenova Y, Cho RW, Littleton JT. Complexin controls spontaneous and evoked neurotransmitter release by regulating the timing and properties of synaptotagmin activity. J Neurosci. 2012:18234–18245. doi: 10.1523/JNEUROSCI.3212-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaeser-Woo YJ, Yang X, Sudhof TC. C-terminal complexin sequence is selectively required for clamping and priming but not for Ca2+ triggering of synaptic exocytosis. J Neurosci. 2012;32:2877–2885. doi: 10.1523/JNEUROSCI.3360-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyoung M, Srivastava A, Zhang Y, Diao J, Vrljic M, Grob P, Nogales E, Chu S, Brunger AT. In vitro system capable of differentiating fast Ca2+-triggered content mixing from lipid exchange for mechanistic studies of neurotransmitter release. Proc Natl Acad Sci U S A. 2011;108:E304–313. doi: 10.1073/pnas.1107900108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLachlan EM, Martin AR. Non-linear summation of end-plate potentials in the frog and mouse. J Physiol. 1981;311:307–324. doi: 10.1113/jphysiol.1981.sp013586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malsam J, Parisotto D, Bharat TAM, Scheutzow A, Krause JM, Briggs JAG, Söllner TH. Complexin arrests a pool of docked vesicles for fast Ca2+-dependent release. EMBO J. 2012;31:3270–3281. doi: 10.1038/emboj.2012.164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin JA, Hu Z, Fenz KM, Fernandez J, Dittman JS. Complexin has opposite effects on two modes of synaptic vesicle fusion. Curr Biol. 2011;21:97–105. doi: 10.1016/j.cub.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maximov A, Tang J, Yang X, Pang ZP, Sudhof TC. Complexin controls the force transfer from SNARE complexes to membranes in fusion. Science. 2009;323:516–521. doi: 10.1126/science.1166505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy G, Kim JH, Pang ZP, Matti U, Rettig J, Sudhof TC, Sørensen JB. Different effects on fast exocytosis induced by Synaptotagmin 1 and 2 isoforms and abundance but not by phosphorylation. J Neurosci. 2006;26:632–643. doi: 10.1523/JNEUROSCI.2589-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishiki T, Augustine GJ. Synaptotagmin I synchronizes transmitter release in mouse hippocampal neurons. J Neurosci. 2004;24:6127–6132. doi: 10.1523/JNEUROSCI.1563-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono S, Baux G, Sekiguchi M, Fossier P, Morel NF, Nihonmatsu I, Hirata K, Awaji T, Takahashi S, Takahashi M. Regulatory roles of complexins in neurotransmitter release from mature presynaptic nerve terminals. Eur J Neurosci. 1998;10:2143–2152. doi: 10.1046/j.1460-9568.1998.00225.x. [DOI] [PubMed] [Google Scholar]
- Pan B, Zucker RS. A General model of synaptic transmission and short-term plasticity. Neuron. 2009;62:539–554. doi: 10.1016/j.neuron.2009.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reim K, Mansour M, Varoqueaux F, McMahon HT, Sudhof TC, Brose N, Rosenmund C. Complexins regulate a late step in Ca2+-dependent neurotransmitter release. Cell. 2001;104:71–81. doi: 10.1016/s0092-8674(01)00192-1. [DOI] [PubMed] [Google Scholar]
- Schaub JR, Lu X, Doneske B, Shin YK, McNew JA. Hemifusion arrest by complexin is relieved by Ca2+-synaptotagmin I. Nat Struct Mol Biol. 2006;13:748–750. doi: 10.1038/nsmb1124. [DOI] [PubMed] [Google Scholar]
- Strenzke N, Chanda S, Kopp-Scheinpflug C, Khimich D, Reim K, Bulankina AV, Neef A, Wolf F, Brose N, Xu-Friedman MA, Moser T. Complexin-I is required for high-fidelity transmission at the endbulb of held auditory synapse. J Neurosci. 2009;29:7991–8004. doi: 10.1523/JNEUROSCI.0632-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tadokoro S. Complexin II facilitates exocytotic release in mast cells by enhancing Ca2+ sensitivity of the fusion process. J Cell Sci. 2005;118:2239–2246. doi: 10.1242/jcs.02338. [DOI] [PubMed] [Google Scholar]
- Tang J, Maximov A, Shin OH, Dai H, Rizo J, Sudhof TC. A complexin/synaptotagmin 1 switch controls fast synaptic vesicle exocytosis. Cell. 2006;126:1175–1187. doi: 10.1016/j.cell.2006.08.030. [DOI] [PubMed] [Google Scholar]
- Thiagarajan R. Enhancement of asynchronous and train-evoked exocytosis in bovine adrenal chromaffin cells infected with a replication deficient adenovirus. J Neurophysiol. 2005;94:3278–3291. doi: 10.1152/jn.00336.2005. [DOI] [PubMed] [Google Scholar]
- Tokumaru H, Shimizu-Okabe C, Abe T. Direct interaction of SNARE complex binding protein synaphin/complexin with calcium sensor synaptotagmin 1. Brain Cell Biol. 2008;36:173–189. doi: 10.1007/s11068-008-9032-9. [DOI] [PubMed] [Google Scholar]
- Voets T, Moser T, Lund PE, Chow RH, Geppert M, Sudhof TC, Neher E. Intracellular calcium dependence of large dense-core vesicle exocytosis in the absence of synaptotagmin I. Proc Natl Acad Sci U S A. 2001;98:11680–11685. doi: 10.1073/pnas.201398798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voets T, Neher E, Moser T. Mechanisms underlying phasic and sustained secretion in chromaffin cells from mouse adrenal slices. Neuron. 1999;23:607–615. doi: 10.1016/s0896-6273(00)80812-0. [DOI] [PubMed] [Google Scholar]
- Vrljic M, Strop P, Ernst JA, Sutton RB, Chu S, Brunger AT. Molecular mechanism of the synaptotagmin–SNARE interaction in Ca2+-triggered vesicle fusion. Nat Struct Mol Biol. 2010;17:325–331. doi: 10.1038/nsmb.1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojcik SM, Brose N. Regulation of Membrane Fusion in Synaptic Excitation-Secretion Coupling: Speed and Accuracy Matter. Neuron. 2007;55:11–24. doi: 10.1016/j.neuron.2007.06.013. [DOI] [PubMed] [Google Scholar]
- Xue M, Lin YQ, Pan H, Reim K, Deng H, Bellen HJ, Rosenmund C. Tilting the balance between facilitatory and inhibitory functions of mammalian and Drosophila Complexins orchestrates synaptic vesicle exocytosis. Neuron. 2009;64:367–380. doi: 10.1016/j.neuron.2009.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue M, Stradomska A, Chen H, Brose N, Zhang W, Rosenmund C, Reim K. Complexins facilitate neurotransmitter release at excitatory and inhibitory synapses in mammalian central nervous system. Proc Natl Acad Sci U S A. 2008;105:7875–7880. doi: 10.1073/pnas.0803012105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Kaeser-Woo YJ, Pang ZP, Xu W, Sudhof TC. Complexin clamps asynchronous release by blocking a secondary Ca2+ sensor via its accessory alpha helix. Neuron. 2010;68:907–920. doi: 10.1016/j.neuron.2010.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshihara M, Littleton JT. Synaptotagmin I functions as a calcium sensor to synchronize neurotransmitter release. Neuron. 2002;36:897–908. doi: 10.1016/s0896-6273(02)01065-6. [DOI] [PubMed] [Google Scholar]
- Young SM, Jr, Neher E. Synaptotagmin has an essential function in synaptic vesicle positioning for synchronous release in addition to its role as a calcium sensor. Neuron. 2009;63:482–496. doi: 10.1016/j.neuron.2009.07.028. [DOI] [PubMed] [Google Scholar]
- Zamponi GW. Regulation of presynaptic calcium channels by synaptic proteins. J Pharmacol Sci. 2003;92:79–83. doi: 10.1254/jphs.92.79. [DOI] [PubMed] [Google Scholar]