

Abstract
Zinc, a divalent heavy metal ion and an essential mineral for life, regulates synaptic transmission and neuronal excitability via ion channels. However, its binding sites and regulatory mechanisms are poorly understood. Here, we report that Kv3 channel assembly, localization and activity are regulated by zinc through different binding sites. Local perfusion of zinc reversibly reduced spiking frequency of cultured neurons most likely by suppressing Kv3 channels. Indeed, zinc inhibited Kv3.1 channel activity and slowed activation kinetics, independent of its site in the N-terminal T1 domain. Biochemical assays surprisingly identified a novel zinc-binding site in the Kv3.1 C-terminus, critical for channel activity and axonal targeting, but not for the zinc inhibition. Finally, mutagenesis revealed an important role of the junction between the first transmembrane (TM) segment and the first extracellular loop in sensing zinc. Its mutant enabled fast spiking with relative resistance to the zinc inhibition. Therefore, our studies provide novel mechanistic insights into the multifaceted regulation of Kv3 channel activity and localization by divalent heavy metal ions.
Key points
Kv3 channels play a critical role in neuronal fast spiking and their regulation can profoundly influence the functions of various neural circuits.
Zinc reversibly inhibits fast spiking in cultured neurons by inhibiting Kv3 channels. Surprisingly, we found that the zinc inhibition is not through its known site in the N-terminal T1 domain of Kv3.1.
We have identified a new zinc-binding site in the Kv3.1 C-terminus, involved in regulating channel activity and targeting, but not the zinc inhibition.
We have also identified the junction between the first transmembrane segment and the first extracellular loop as the major zinc-sensing site in regulating Kv3 activity. This result allows us to reconstitute zinc-resistant fast spiking.
Kv3 channel assembly, localization and activity are regulated by zinc through intracellular and extracellular binding sites.
Introduction
Zinc is an essential trace element for life and is also a toxic pollutant. Zinc is widely distributed in the brain, and is usually selectively stored in presynaptic vesicles of glutamatergic neurons and co-released with glutamate (Huang, 1997; Lin et al. 2001; Smart et al. 2004; Frederickson et al. 2005). Zinc can regulate the intrinsic neuronal excitability and synaptic transmission via regulating various types of ligand-gated and voltage-gated ion channels (Frederickson et al. 2005; Mathie et al. 2006; Kay & Toth, 2008; Sensi et al. 2009). The regulations are also implicated in the pathophysiology of acute brain damage and degenerative brain diseases. For instance, ischaemic injury leads to an increase of local free zinc in some brain regions (Galasso & Dyck, 2007; Hershfinkel et al. 2009; Medvedeva et al. 2009).
Zinc stimulates the activity of the high-conductance voltage- and Ca2+-activated K+ channels, transient receptor potential cation channels A1 (TRPA1), and ATP-sensitive K+ channels (Prost et al. 2004; Hu et al. 2009; Hou et al. 2011), while it inhibits the activity of acid-sensing ion channel 1b (Jiang et al. 2011). These results raised an interesting question: does the zinc stimulation or inhibition correlate with its intracellular or extracellular binding sites? Zinc can enter the cell through some ion channels and/or via its transporters (Sensi et al. 2009). Extracellular zinc generally suppresses the activities of various isoforms of voltage-gated K+ (Kv) channels with different sensitivity (Lovinger et al. 1992; Poling et al. 1996; Zhang et al. 2001; Mathie et al. 2006; Aras et al. 2009). For cloned Kv1.5, zinc reduces ionic current, shifts the conductance–voltage (G–V) curve, and slows the activation kinetics (Zhang et al. 2001; Kehl et al. 2002). Divalent heavy metal ions, including zinc, slow the activation of the EAG family (Kv10) K+ channels (Zhang et al. 2009). For the A-type Kv channels expressed in various cells, zinc has complex effects dependent on the membrane potential (Harrison et al. 1993; Bardoni & Belluzzi, 1994; Puopolo & Belluzzi, 1998; Kuo & Chen, 1999; Horning & Trombley, 2001). Nonetheless, the exact zinc-binding site and regulatory mechanism in Kv channels remain a mystery.
The best characterized zinc-binding site in Kv channels is the conserved one (HX5CX20CC) within the cytoplasmic T1 domains of Kv2 to Kv4 channels. The N-terminal conserved T1 domains form tetramers, responsible for the proper tetramerization of Kv channels within the same subfamily (Li et al. 1992; Xu et al. 1995; Bixby et al. 1999; Jahng et al. 2002). Addition or removal of zinc can switch T1 domains between the tetramerization and monomerization states. Zinc-binding site in the T1 domain of Kv4 channels is also involved in regulating channel activity (Jahng et al. 2002; Kunjilwar et al. 2004; Wang et al. 2007), raising an intriguing possibility that free zinc can regulate Kv channel activity by controlling the assembly and disassembly of T1 tetramers. Zinc can flux into the cell through TRPA1 channels and activate TRPA1 via specific intracellular cysteine and histidine residues in the cytoplasmic domains (Hu et al. 2009). Although it is unlikely that zinc can pass through Kv3 channels, it is plausible that zinc influx via ion channels and/or transporters can bind to its binding site in Kv3 T1 domains to regulate Kv3 channel function. Furthermore, our previous studies show that ankyrin G and KIF5 play important roles in regulating polarized axon–dendrite targeting of Kv3 channels (Xu et al. 2007, 2010; Gu & Barry, 2011). While the interaction of Kv3.1 N- and C-termini is zinc dependent (Xu et al. 2007), zinc-mediated T1 tetramerization is required for the direct binding to the (kinesin superfamily protein 5 isoform B (KIF5B) tail domain, which is critical for Kv3 axonal transport (Xu et al. 2010). Taken together, Kv3 T1, containing a conserved zinc-binding site, may be essential for regulating both channel activity and trafficking.
The unique channel properties of Kv3 channels, high activation threshold and rapid activation and deactivation kinetics, are critical for fast spiking in some neurons (Rudy & McBain, 2001; Bean, 2007; Gu & Barry, 2011). In this study, we show that zinc suppresses spiking frequency through reversibly inhibiting Kv3 channel activity. By using protein biochemistry, cell biology and patch-clamp techniques, we surprisingly identified multiple zinc-binding sites in Kv3.1 channels, and further systematically analysed the role of these sites in channel activity and localization.
Methods
cDNA constructs
Abbreviations used: 31aC, the C-terminus of Kv3.1a (amino acids 442–511); 31bC, the C-terminus of Kv3.1b (amino acids 442–585); 31sC, the splice domain of Kv3.1b C-terminus (amino acids 500–585); GST, glutathione S-transferase; Kv3 channel, Shaw voltage-gated K+ channel; YFP, yellow fluorescent protein.
Kv3.1a(HA, hemagglutinin tag), Kv3.1bHA, Kv3.1bHAH77A, KIF5B-YFP, GST-31aC, GST-31bC, GST-31sC and GST-31T1 were produced as previously described (Xu et al. 2007, 2010). GST-442–452, GST-442–469, GST-442–489, GST-468–489 and GST-468–511 were made by inserting cDNA fragments encoding different regions of Kv3.1a C-terminal domain into the pGEX4T-2 vector between SalI and NheI restriction enzyme sites. GST-442–469H, GST-468–489HH and GST-468–511HH were made with Quikchange mutagenesis by mutating histidine residues to alanine (Quikchange Site Directed Mutagenesis, Stratagene). GST-31aCH, GST-31aCHH and GST-31aCHHH were made with Quickchange based on GST-31aC. YFP-31aC, YFP-31aCH, YFP-31aCHH and YFP-31aCHHH were generated by inserting Kv3.1a C-terminus and its mutants into pEYFP-C1 vector. Kv3.1bHAH459A, Kv3.1bHAHHAA and Kv3.1bHAHHH were made with Quickchange based on Kv3.1bHA. Kv3.1bHAC208A, Kv3.1bHAH212A, Kv3.1bHAC252A, Kv3.1bHAH327A, Kv3.1bHAH381A, Kv3.1bHAH383A and Kv3.1bHAH381AH383A were made with Quickchange based on Kv3.1bHA. All constructs were confirmed with sequencing.
Primary hippocampal and cerebellar neuron cultures
All animal experiments were conducted in accordance with the NIH Animal Use Guidelines and Institutional Animal Care and Use Committee (IACUC) of the Ohio State University. The rats were from Charles River Laboratories International, Inc. Pregnant rats at embryonic day 18 (E18) were killed with carbon dioxide (CO2) inhalation in a cage connected with CO2 tubing, followed by decapitation. The hippocampal neuron culture was prepared as previously described from the E18 rat hippocampi (Gu et al. 2006; Barry et al. 2010; Gu & Gu, 2010; Gu & Gu, 2011). Briefly, rats were euthanized by CO2 and followed by cervical dislocation. Hippocampi were dissected from embryonic day 18 rat embryos. For transient transfection, neurons in culture at 5–7 days in vitro (DIV) were incubated in Opti-MEM containing 0.8 μg of cDNA plasmid and 1.5 μl of Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) for 20 min at 37°C. At least three independent transfections were performed for each condition. The cerebellar neuron culture was made from the cerebella of rat pups at postnatal day 1 or 2, (Rat pups were euthanized by CO2 and followed by cervical dislocation. Hippocampi were dissected) with the same procedure as for the hippocampal neuron culture 8 rat pups were used in this experiments. The number is also indicated in Fig 1A.
Figure 1. Extracellular application of zinc reversibly reduces spiking frequency of different neurons.
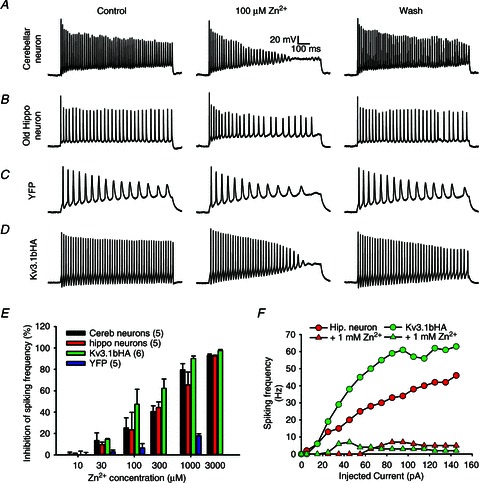
A, action potential firing of a cultured cerebellar neuron at 14 DIV was reversibly reduced by extracellular 100 μm zinc applied with a local perfusion pipette (n= 8). B, spiking frequency of a cultured hippocampal neuron at 21 DIV was reduced by 100 μm zinc. C, spiking frequency of a YFP-transfected neuron at 8 DIV was not significantly altered by 100 μm zinc. D, spiking frequency of an 8-DIV hippocampal neuron transfected with Kv3.1bHA and YFP was markedly reduced by 100 μm zinc. Square current pulses (1000 ms; 105 pA) were injected into the neuronal soma to induce action potentials. The scale bars are the same in A–D. E, inhibitory effects on spiking frequency by different concentrations of zinc. Selected concentrations of Zn2+ (10, 30, 100 and 1000 μm) were tested on YFP-expressing hippocampal neurons at 8 DIV. The number of neurons are indicated within parentheses. F, the input–output relationship of a mature hippocampal neuron (red) and a young neuron expressing Kv3.1bHA (green) in the absence and presence of 1 mm zinc.
Current-clamp recording from cultured neurons and whole-neuron perfusion
Glass pipettes with tip diameter around 1 μm for patch-clamp recording were pulled with a Model P-1000 Flaming/Brown micropipette puller (Sutter Instrument, Novato, CA, USA). Glass pipettes with diameter around 50 μm, made by adjusting the pulling parameters and filled with Hank's buffer containing different concentrations of ZnCl2, were used for perfusion of whole neurons. The procedure has been described previously (Gu et al. 2012). The perfusion lasts for approximately 10 s. The bath volume is about 2 ml, which is replaced with new Hank's buffer at the rate of 4 ml min−1. We usually record the recovery of action potentials after 5 min washing.
The internal solution and Hank's buffer have been previously described for the recording of primary cultured neurons (Gu et al. 2012). Hank's buffer: 150 mm NaCl, 4 mm KCl, 1.2 mm MgCl2, 10 mg ml−1 glucose, 1 mm CaCl2, 20 mm Hepes (pH 7.4). The internal solution of electrical pipettes: (in mm) 122 KMeSO4, 20 NaCl, 5 Mg-ATP, 0.3 GTP and 10 Hepes (pH 7.2). The current-clamp procedure has been previous described (Gu et al. 2012).
Voltage-clamp recording from transfected HEK293 cells
Biophysical properties of various Kv3 channel constructs were determined with voltage-clamp recording on transfected cells from the human embryonic kidney cell line HEK293 with the same internal solution and Hank's buffer, as previously described (Gu et al. 2012). The patch pipette resistance in the bath was about 2–2.5 MΩ. The whole-cell access resistance was between 3 and 5 MΩ. Cells with higher access resistance were discarded. The compensation for series resistance was set at >60%. In Zn2+ treatment experiments, cells whose series resistance changed by 15% were discarded. Conductance–voltage relationships (G–V curves) for Kv channel constructs were G=I/(Vm–Vrev), Vrev=−95 mV, normalized to the maximal conductance. Curves were fitted with a Boltzmann function, G/Gmax= 1/{1 + exp[–(V–V1/2)/k]}, where Gmax is the maximal conductance, V1/2 is the potential at which the value of the relative conductance is 0.5, and k is the slope factor. SigmaPlot 10.0 (Systat Software, Inc., Chicago, IL, USA) was used for fitting. To obtain the activation time constant (τon), activation curves (at +30 mV) were fitted with a single exponential function raised to the power of 4, I(t) =A(1 – exp(–t/τon))4. In studies of deactivation kinetics of Kv channels, the cells were held at −80 mV, given a 2 ms pre-pulse to +60 mV and 20 ms voltage episodes from −100 mV to −10 mV. To obtain deactivation time constant (τoff), tail currents (at −60 mV) were fitted with the equation, I(t) =Aexp(–t/τoff). Clampfit 10.0 (Molecular Devices Corporation) was used for fitting to get τon and τoff.
Expression of fusion proteins and pull down by Co2+ and Zn2+ beads
Expression of glutathione S-transferase (GST) fusion proteins was induced in BL21 E. coli cells with 1 mm isopropyl β-d-1-thiogalactopyranoside (IPTG) for 4 h at 37°C. Bacterial pellets were solubilized with sonication in the immunoprecipitation (IP) buffer at 4°C, and centrifuged at 50,000 g for 30 min at 4°C. The supernatants were incubated with Co2+ beads (Clontech Laboratories Inc., Mountain view, CA, USA) at 4°C for 3 h. After extensive washing, the beads coated with purified fusion proteins were eluted with the sample buffer containing 1 mm EDTA. Yellow fluorescent protein (YFP) fusion proteins were expressed in HEK293 cells. The supernatant of cell lysates was further incubated with Zn2+ beads. The precipitated proteins were eluted with the sample buffer.
Western blotting and Colloidal Blue staining
The precipitants were resolved by SDS-PAGE, transferred to a PVDF membrane, and subjected to Western blotting with a mouse anti-GST antibody (1:1000 dilution). Protein bands in the gels were stained with the Colloidal Blue staining kit (Invitrogen), using the procedure described by the manufacturer.
Immunostaining, imaging and quantification
The immunocytochemical procedures have been previously described (Gu et al. 2006; Xu et al. 2007). Briefly, neurons were stained under the non-permeabilized condition (without Triton X-100) to label the surface pool and under the permeabilized condition (with 0.2% Triton X-100) to label total proteins. Fluorescence images were captured with a Spot CCD camera RT slider (Diagnostic Instruments Inc., Sterling Heights, MI, USA) in a Zeiss upright microscope, Axiophot, using Plan Apo objectives ×20/0.75 and ×100/1.4 oil, saved as 16-bit TIFF files, and analysed with NIH ImageJ and Sigmaplot 10.0 for fluorescence intensity quantification. The quantification procedure has also been described previously (Xu et al. 2007, 2010).
Results
Zinc reversibly reduces the firing frequency of cultured neurons
To determine whether and how zinc regulates high-frequency firing in neurons, we applied ZnCl2 in different concentrations on cultured cerebellar neurons, mostly cerebellar granule cells, expressing Kv3.1 and Kv3.3 channels (Weiser et al. 1994; Grigg et al. 2000; Rudy & McBain, 2001). Injecting square-pulse currents into soma induced many action potentials from cultured cerebellar neurons (Fig. 1A), consistent with the presence of Kv3 channels. Extracellular 100 μm zinc significantly and reversibly suppressed the firing frequency (Fig. 1A). Next, we examined zinc's effect on hippocampal neurons. Some neurons, presumably GABAergic interneurons in mature cultured hippocampal neurons, generating relatively high firing frequency due to the presence of Kv3 channels (Gu et al. 2012), were also reversibly inhibited by extracellular zinc at 100 μm (Fig. 1B). Since 100 μm zinc is unlikely to have an inhibitory effect on voltage-gated Na+ (Nav) channels (Nigro et al. 2011), we wondered whether the zinc's reversible and inhibitory effect on spiking frequency is mainly mediated by the inhibition of Kv3 channels. The inhibitory effect of zinc on spiking frequency was indeed evident on the fast spiking of Kv3.1b-expressing young hippocampal neurons, but not YFP-expressing neurons (Fig. 1C and D). The significant inhibitory effects of zinc on Kv3.1bHA-expressing neurons, as well as cerebellar and some hippocampal neurons (at 21 DIV), started to be observed at 30 μm concentration and reached the peak around 1 mm (Fig. 1E). In contrast, the firing rate of young hippocampal neurons (at 8 DIV) expressing YFP was not significantly inhibited (Fig. 1E). Moreover, ZnCl2 (1 mm) markedly inhibited the input–output relationship both in mature hippocampal neurons and in Kv3.1b-expressing young neurons (Fig. 1F).
Zinc reversibly alters Kv3.1 biophysical properties, not through its binding site in the T1 domain
To determine whether zinc reversibly inhibits Kv3 channel activity, we performed the whole-cell voltage-clamp recording on HEK293 cells transfected with different Kv3.1 channel constructs. Kv3.1 has two alternative splicing variants, Kv3.1a and Kv3.1b. Their only difference is their C-termini. Kv3.1a is 74 residues shorter than Kv3.1b. An HA tag is inserted into their first extracellular loop (Kv3.1aHA and Kv3.1bHA) for detecting their surface expression, which does not change channel activity and localization (Xu et al. 2007). Extracellular application of 1 mm ZnCl2 markedly reduced the current amplitudes of both Kv3.1aHA (control: 8.95 ± 0.80 nA; zinc: 6.39 ± 0.40 nA) and Kv3.1bHA (control: 9.72 ± 0.75 nA; zinc: 7.29 ± 0.62 nA) (Fig. 2A, B and D). The inhibitory effect was reversed by about 5 min of wash (Fig. 2A and B). Besides current reduction, G–V curves shifted to the right (Fig. 2E). Activation time constants (τon) of both Kv3.1aHA (control: 0.38 ± 0.04 ms; zinc: 5.00 ± 0.37 ms) and Kv3.1bHA (control: 0.36 ± 0.01 ms; zinc: 4.89 ± 0.52 ms) markedly increased by more than 10-fold (Fig. 2F and Supplemental Fig. S1 in online Supplemental material). In contrast, the deactivation time constants of Kv3.1aHA (control: 0.97 ± 0.03 ms; zinc: 1.33 ± 0.05 ms) and Kv3.1bHA (control: 0.92 ± 0.03 ms; zinc: 1.08 ± 0.02 ms) only moderately increased (Fig. 2G). We also examined the effects of other divalent cations. At 1 mm concentration, Cu2+, Cd2+ and Co2+ also significantly inhibited Kv3.1bHA current in HEK293 cells, whereas Ca2+ and Mg2+ had no effect (data not shown). Therefore, Kv3.1 activity can be regulated by divalent heavy metal ions. This study mainly focuses on Zn2+.
Figure 2. Zinc markedly alters the biophysical properties of Kv3.1 channels.
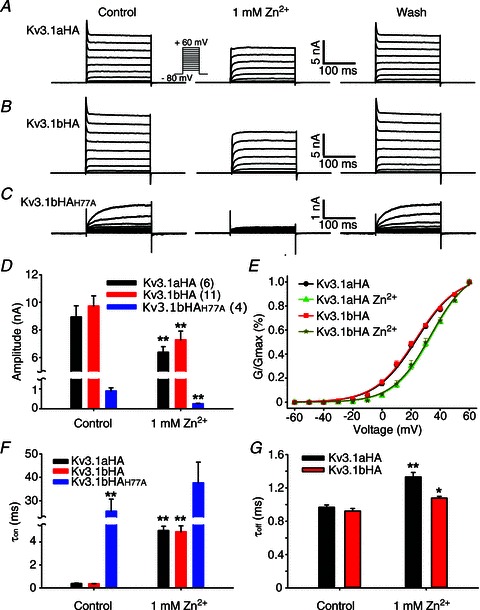
A, example traces of reversible inhibition of Kv3.1aHA currents by 1 mm zinc. Kv3.1aHA was expressed in HEK293 cells. Voltage-clamp recording was performed one day after transfection. B, reversible inhibition of Kv3.1bHA currents by 1 mm zinc. C, reversible inhibition of Kv3.1bHAH77A currents by zinc. D, inhibitory effect of Zn2+ on current amplitude. The number of neurons are indicated within parentheses, and are the same in E–G. E, effects of zinc on G–V curves of Kv3.1aHA and Kv3.1bHA. F, effects of zinc on activation time constant (τon) of three Kv3.1 constructs. G, effects of zinc on deactivation time constant (τoff) of Kv3.1aHA and Kv3.1bHA. Paired t test: **P < 0.01; *P < 0.05.
Extracellular Zn2+ can enter neuronal cytoplasm through some ion channels and its transporters. The ion channels permeable to Zn2+ include voltage-gated Ca2+ channels, Ca2+- and Zn2+-permeable GluR2-lacking AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid) receptors, and TRPA1 (Hu et al. 2009; Sensi et al. 2009). Since Kv3 channels have a zinc-binding site in the T1 domain interface, we wondered if the site mediates the Kv3 inhibition by Zn2+ entering the cell. Importantly, association and disassociation of Zn2+ may influence the dynamics of T1 tetramer assembly and disassembly, leading to altered channel biophysical properties and trafficking. Mutating any of the four residues (H77, C83, C104 and C105) in the conserved Zn2+-binding motif (HX5CX20CC) disrupts the Zn2+ binding and thereby yields monomeric T1 domains (Bixby et al. 1999; Nanao et al. 2003). Consistent with this notion, mutating H77 to alanine (A) appeared to disrupt T1 tetramerization in our previous study (Xu et al. 2010). The Kv3.1HAH77A mutant retained a significant amount of the channel current with a markedly increased activation time constant (Fig. 2C and F). However, when zinc was applied extracellularly to the mutant, its amplitude further reduced (control: 0.90 ± 0.16 nA; zinc: 0.26 ± 0.01 nA), and its activation time constant further increased (control: 25.52 ± 5.11 ms; zinc: 37.69 ± 8.78 ms). Due to reduced current amplitude, the G–V curve and deactivation time constant could not be accurately determined for Kv3.1bHAH77A. Although proper T1 domain tetramerization is certainly critical for channel current amplitude and activation kinetics, extracellular application of zinc still further inhibited channel current and increased the activation time constant (Fig. 2C and F). Therefore, the zinc inhibition must act through a different binding site.
A novel metal-binding site in the conserved C-terminal region of Kv3.1
When purifying various tagged proteins, we accidentally discovered that GST-tagged Kv3.1a C-terminus (GST-31aC) could be purified with Co2+ beads, which are usually used to purify 6×His-tagged proteins. This result suggests that there is an additional divalent heavy metal binding site(s) in the Kv3.1a C-terminus (Fig. 3A). Therefore, we made a series of GST-fusion proteins with different fragments from the Kv3.1a C-terminal domain (Fig. 3A) to examine their binding to Co2+ beads.
Figure 3. A novel binding site for divalent heavy metal ions in the Kv3.1 C-terminus.
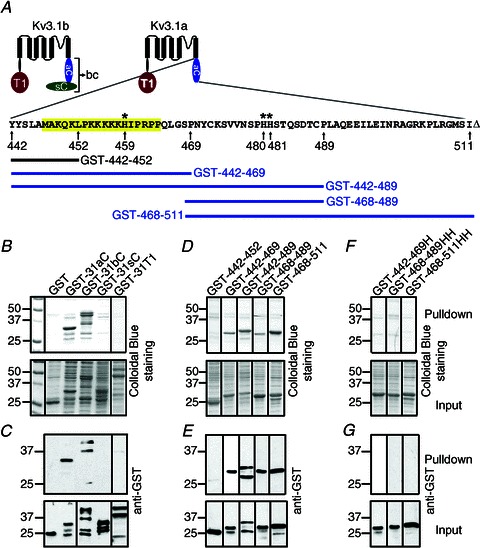
A, structural diagram of Kv3.1a and Kv3.1b. The residues of Kv3.1a cytoplasmic domain are shown (aC, amino acids (aa) 442–511 of Kv3.1a; bC, aa 442–585 of Kv3.1b; sC, aa 500–585 of Kv3.1b; T1, the N-terminal T1 domain aa 1–186). The ATM is highlighted. The numbers underneath the sequence indicate the residue numbers. Asterisks indicate the three histidine residues. ‘Δ’ shows the end of the sequence. GST-442–452 does not bind to Co2+ beads. GST-442–469, GST-442–489 and GST-468–489 bind to Co2+ beads. B, the whole C-termini of Kv3.1a and Kv3.1b, but not the splice domain or the N-terminal T1 domain, bind to Co2+ beads. Pulldown proteins (upper) and inputs (lower) are revealed by Colloidal Blue staining. C, the Western blots of B. Ten per cent of loading in B was used in Western blotting. D, the fragments in Kv3.1a C-terminus, containing at least one histidine residue, bind to Co2+ beads. E, Western blots of D. F, mutating the histidine residues in three different fragments eliminated their binding to Co2+ beads. G, Western blots of F. Each pulldown experiment was performed at least three times with consistent results.
Both the Kv3.1a and Kv3.1b C-termini fused with GST, but not GST or GST-31sC (the C-terminal splice domain of Kv3.1b, 85 residues) were pulled down by Co2+ beads (Fig. 3B and C). Surprisingly, GST-31T1, the N-terminal T1 domain containing a known zinc-binding site, was not pulled down by Co2+ beads (Fig. 3B and C). A plausible explanation is that the zinc-binding sites are buried within the T1 tetramer, which requires the proper assembly of four T1 domains.
Further analysis indicated that there are three histidine residues within the Kv3.1a C-terminus for the potential divalent heavy metal ion (Me2+) binding site (Fig. 2D–G). The first (H459) is within the axonal targeting motif (ATM) (Xu et al. 2007). A small N-terminal fragment of the Kv3.1 C-terminus, GST-442–469, but not GST-442–452, was pulled down by Co2+ beads (Fig. 3D and E). It appears that this binding requires the only histidine residue (H459), since mutating it to alanine eliminated the binding of GST-442–469 to Co2+ beads (Fig. 3F and G). This is quite surprising because this small region carries a net charge of +8, and has only one histidine and not even a single acidic residue. This region might form an oligomer upon binding to Co2+/Zn2+, or alternatively some negatively charged lipids might mediate the binding. Interestingly, our previous study showed that this region directly interacts with the N-terminal T1 domain in a zinc-dependent manner (Xu et al. 2007), which is involved in masking the ATM (Gu et al. 2012). The present study has demonstrated that both the N- and C-terminal domains of Kv3.1 channels contain zinc-binding sites, providing a structural explanation for our previous results.
The second and third histidine residues (H480H481) are located in a 22-residue region immediately downstream of the ATM (Fig. 3A). Both GST-468–489 and GST-468–511, but not GST-468–489HH or GST-468–511HH with the two histidines mutated to two alanines were pulled down by Co2+ beads (Fig. 3D–G). Therefore, it appears that there is some degree of independence of the three histidines in binding to Me2+.
Point mutation mapping revealed three critical histidine residues, H459, H480 and H481, in binding to Co2+ and Zn2+ (Fig. 3). Mutating any of them appeared to reduce, but not to completely eliminate, the binding of GST-31aC to Co2+ or Zn2+ beads (Fig. 4). Only mutating all three histidine residues to alanines completely eliminated the binding (Fig. 4A). To examine whether these sites can bind to zinc and to rule out the possibility that GST contains metal binding sites, we fused 31aC to yellow fluorescence protein (YFP) C-terminus (YFP-31aC) and made three mutant constructs, YFP-31aCH, YFP-31aCHH and YFP-31aCHHH (Fig. 4B). These constructs were expressed in HEK293 cells. We used Zn2+ beads to pull down a supernatant of HEK293 cell lysates. Zinc beads pulled YFP-31aC, and YFP-31aCH and YFP-31aCHH with less efficiency, but not YFP-31aCHHH, nor YFP (Fig. 4B). Therefore, we discovered a novel zinc-binding site composed of three histidines in the C-terminal region of the Kv3.1 channel, one histidine within the ATM and the other two immediately downstream of the ATM.
Figure 4. Three histidine residues in Kv3.1a C-terminus are required for binding to Co2+ or Zn2+.
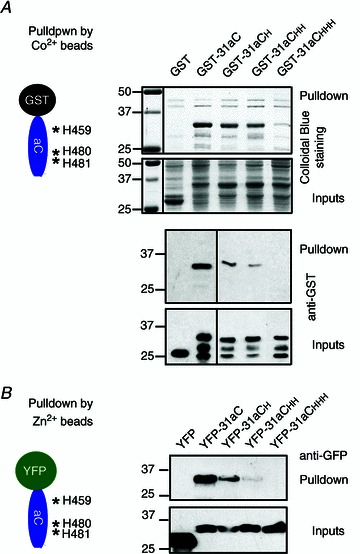
A, mutating histidine (H) residues reduced the binding of GST-31aC to Co2+ beads. Structural diagram of GST-31aC and positions of three Hs are on the upper left. Co2+ beads pulled down GST-31aC and mutants carrying single or double H mutations, but not GST and GST-31aCHHH (all three H residues mutated to A). Mutating one or two H residues reduced the pulldown. Pulldown results of Colloidal Blue staining (upper) and Western blotting with an anti-GST antibody (lower, only 10% in loading) are shown on the top, with inputs at the bottom. B, mutating H residues also reduces the binding of YFP-31aC to Zn2+ beads. 31aC (wild-type or mutants) was fused to YFP. The constructs were transfected into HEK293 cells. The supernatants of cell lysates were pulled down with Zn2+ beads. Molecular mass is indicated on the left in kDa. Each pulldown experiment was performed at least three times with consistent results.
H459 is required for axonal targeting and rapid activation of Kv3.1b channels
Kv3.1aHA and Kv3.1bHA are preferentially localized to dendritic and axonal membranes, respectively, in which the C-terminal ATM plays a critical role (Xu et al. 2007). Since one of the histidine residues (H459) is within the ATM, we examined whether and how the three histidine residues affect the channel axonal targeting. H459A single mutation (Kv3.1bHAH459A Faxon/Fdend (Faxon/Fdendrite is the ratio between the average fluorescent intensity of the axon divided by the average fluorescent intensity of the dendrites): permeabilized (perm), 0.32 ± 0.03; non-permeabilized (non-perm), 0.29 ± 0.03) and the triple mutation (Kv3.1bHAHHH Faxon/Fdend: perm, 0.30 ± 0.03; non-perm, 0.33 ± 0.04) markedly reduced the axonal level of Kv3.1bHA (Faxon/Fdend: perm, 0.45 ± 0.04; non-perm, 1.98 ± 0.11) (Fig. 5). In contrast, mutating H480 and H481 together (Kv3.1bHAHHAA Faxon/Fdend: perm, 0.46 ± 0.07; non-perm, 1.93 ± 0.13) did not affect the polarized axon–dendrite targeting of both Kv3.1aHA and Kv3.1bHA (Fig. 5). Our previous study showed that mutating H459 may also contribute to eliminating the axonal targeting of CD4, but not the binding to T1 or ankyrin G (Xu et al. 2007). The present study shows that only the histidine residue (H459) within the ATM is critical for channel polarized axon–dendrite targeting. Therefore, the newly identified zinc-binding site in Kv3.1 C-terminus is most likely to be involved in Kv3.1 axonal targeting via regulating the N-/C-terminal interaction and/or Kv3.1/ankyrin G interaction.
Figure 5. H459, but not H480 and H481, in the C-terminal domain, is required in Kv3.1b axonal targeting.
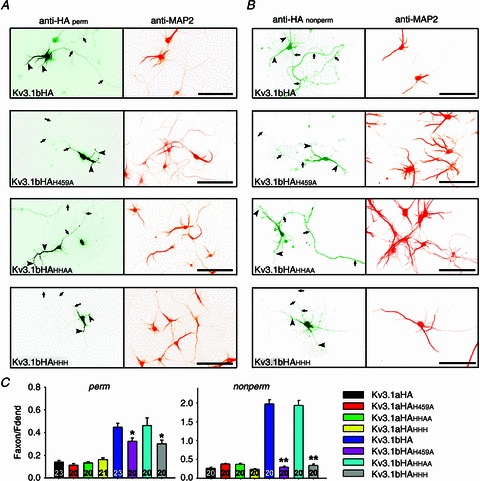
A, distribution patterns of Kv3.1bHA and three point mutants in neurons under the permeabilized condition. Anti-HA staining is on the left and anti-MAP2 (microtubule-associated protein 2) staining is on the right. Signals are inverted. B, distribution patterns of Kv3.1bHA and three point mutants in neurons under the non-permeabilized condition. Arrows, axons; arrowheads, dendrites. Scale bars, 100 μm. C, summary of the staining results. One-way analysis of variance followed by Dunn test, **P < 0.01; *P < 0.05.
To examine the biophysical properties and zinc sensitivity of these mutants, we performed whole-cell voltage-clamp recording on HEK293 cells transfected with the three mutants, Kv3.1bHAH459A, Kv3.1bHAHHAA and Kv3.1bHAHHH. All three mutants had similar current amplitude compared to the wild-type (Fig. 6A and B). The G–V curves of Kv3.1bHAH459A and Kv3.1bHAHHH shifted to the right, whereas the curve of Kv3.1bHAHHAA remained the same, compared to Kv3.1bHA (Fig. 6C). Mutating H459 within the ATM also markedly increased the activation time constant, whereas mutating the two histidines (480 and 481) outside of the ATM did not affect the τon (Fig. 6A and D). Interestingly, their currents were still markedly suppressed by 1 mm ZnCl2 (relative amplitude: Kv3.1bHAH459A, 0.80 ± 0.06; Kv3.1bHAHHAA, 0.77 ± 0.01; Kv3.1bHAHHH, 0.73 ± 0.02) (Fig. 6A and B). All the curves shifted to the right in the presence of 1 mm ZnCl2 (Fig. 6C). In the presence of 1 mm ZnCl2, the τon of all three mutants markedly increased (Kv3.1bHAH459Aτon: control 5.40 ± 0.83 ms, zinc 13.91 ± 3.33 ms; Kv3.1bHAHHAAτon: control 0.64 ± 0.26 ms, zinc 5.23 ± 0.90 ms; Kv3.1bHAHHHτon: control 2.16 ± 0.67 ms, zinc 13.17 ± 2.19 ms; Fig. 6D). Their deactivation time constants (τoff) only slightly increased (Fig. 6E). Therefore, mutating H459 within the ATM changed the channel's biophysical properties, including the G–V curve and activation time constant. However, all of these mutants were still sensitive to the zinc inhibition. Therefore, the C-terminal zinc-binding site is important for channel targeting, biophysical properties, but not the site mediating the zinc inhibition.
Figure 6. Biophysical properties of Kv3.1 channels carrying the three histidine mutations in the C-terminus and their response to extracellular zinc.
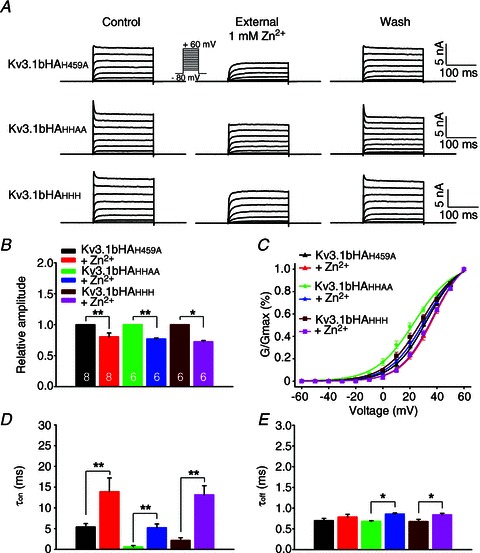
A, reversible inhibitory effects of extracellular zinc on the currents carried by channels with one to three point mutations. B, summary of effect of zinc on current amplitude of different channel constructs. The numbers of neurons are indicated within the bars, and are the same in B–E. C, summary of zinc's effects on the G–V relationship of three different channel constructs. D, zinc's effect on activation time constants (τon). E, zinc's effects on deactivation time constant (τoff). Paired t-test, **P < 0.01; *P < 0.05.
The junction of the 1st TM segment and the 1st extracellular loop in Zn2+ sensing
Given that the inhibitory effect of zinc was quite fast, usually within seconds, we hypothesized that the binding site may be in the external side or in the TM portion of the channel. Therefore, we performed the alanine scan for all six histidine and cysteine (C) residues either in the extracellular loops or TM segments of the Kv3.1 channel. We made seven mutant constructs and expressed them in HEK293 cells for voltage-clamp recording (Fig. 7A). All of them were still able to conduct currents, but with different sensitivity for zinc (Fig. 7A). The two mutations in the 1st TM segment and the 1st extracellular loop, Kv3.1bHAC208A (control: 9.24 ± 0.30 nA; zinc: 7.84 ± 0.20 nA) and Kv3.1bHAH212A (control: 4.34 ± 1.20 nA; zinc: 3.03 ± 0.78 nA), became significantly less sensitive to zinc (Fig. 7B). Although in the presence of 1 mm Zn2+ there was no significant reduction in the current amplitude of Kv3.1bHAH212A, its biophysical properties under the control condition markedly changed compared to the wild-type channel (reduced current amplitude, altered G–V curve, and dramatically increased τon; Fig. 7B–E). Therefore, Kv3.1bHAH212A is unlikely to induce fast spiking when expressed in neurons, since its activation becomes markedly slower. In contrast, Kv3.1bHAC208A was similar to the wild-type channel in biophysical properties under the control condition (Fig. 7B–E). Compared to the wild-type channel, the shifting of the G–V curve to the right in the presence of zinc was significantly less for Kv3.1bHAC208A (Fig. 7C). Its τon (control: 0.98 ± 0.46 ms; zinc: 7.78 ± 0.60 ms) still increased in the presence of 1 mm zinc (Fig. 7E), but its τoff (control: 0.99 ± 0.04 ms; zinc: 1.13 ± 0.07 ms) remained largely unchanged.
Figure 7. Mutagenesis studies searching for potential zinc-binding sites in the membrane portion of Kv3.1b.
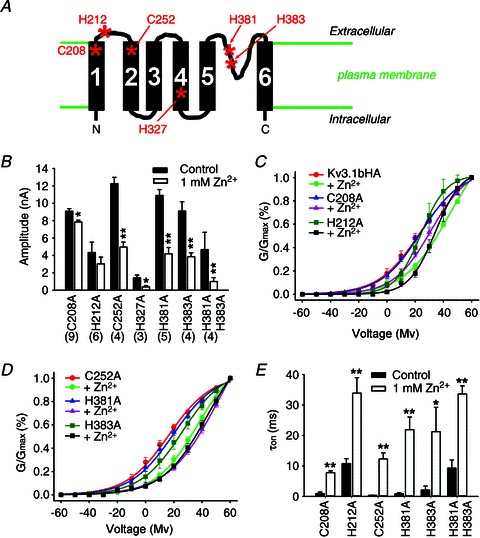
A, diagram of the membrane portion Kv3.1. All six cysteine (C) and histidine (H) residues in the transmembrane segments and extracellular loops are indicated with asterisks with residue numbers labelled. B, zinc's effects on current amplitudes of different channel mutants. The numbers of HEK293 cells are indicated in parentheses for B–E. C and D, effect of Zn2+ on the G–V relationship of the channel mutants. E, zinc's effects on activation time constant (τon). Paired t test: **P < 0.01; *P < 0.05.
Whereas the biophysical properties of Kv3.1bHAC252A (C252 is in the 2nd TM segment) remained similar to those of the wild-type, the current of Kv3.1bHAH327A (H327 is in the 4th TM segment) dramatically reduced compared to the wild-type (Fig. 7). Both mutants were still inhibited by zinc (Fig. 7).
We further examined the two histidine residues in the P loop, which are unique for Kv3 channels, not being present in other Kv channels. The location of the two histidine residues also raised an intriguing possibility that they may be involved in zinc-mediated regulation (Fig. 7A). However, mutating either one or both of them did not weaken the zinc sensitivity (Fig. 7). Mutating the histidine residues, however, did change some biophysical properties of the channel, such as reduced current amplitude for Kv3.1bHAH381AH383A, and markedly increased τon for all three mutants (Fig. 7B and E).
Among all the mutants we examined, Kv3.1bHAC208A was the one with the most dramatically reduced sensitivity to zinc. We additionally performed a detailed analysis of the zinc regulation of its channel biophysical properties at low concentrations of zinc, close to the physiological or pathological levels in vivo. We performed voltage-clamp experiments with side-by-side comparison to the wild-type channel. At 100 μm, zinc inhibited the current amplitude of Kv3.1bHA (8.1 ± 1.7% inhibition) but not Kv3.1bHAC208A (3.4 ± 1.8% inhibition) (Fig. 8A and B). Both were inhibited at 300 μm, but not affected at 30 μm (Fig. 8B). We also carefully examined the effect of zinc on the G–V relationship. The slope factor, k, was not affected by zinc (Fig. 8C). In contrast, the V1/2 of Kv3.1bHA (1.09 ± 0.02) but not Kv3.1bHAC208A (0.97 ± 0.01) increased at 30 μm zinc (Fig. 8D). At higher concentrations, Kv3.1bHA V1/2 was more sensitive to zinc than Kv3.1bHAC208A V1/2 (Fig. 8D). Importantly, at 30 μm, zinc slowed the activation kinetics of Kv3.1bHA (normalized τon: 1.18 ± 0.06) but not Kv3.1bHAC208A (normalized τon: 0.97 ± 0.02; Fig. 8E and F). In summary, our studies surprisingly identified that the junction between the 1st TM domain and the 1st extracellular loop is, at least partially, responsible for the zinc inhibition. The biophysical properties, including current amplitude and the G–V relationship of the mutant, Kv3.1bHAC208A, were similar to those of Kv3.1b, but were more resistant to the zinc inhibition compared to the wild-type channel.
Figure 8. Differential effects of Zn2+ on biophysical properties of Kv3.1bHA and Kv3.1bHAC208A.
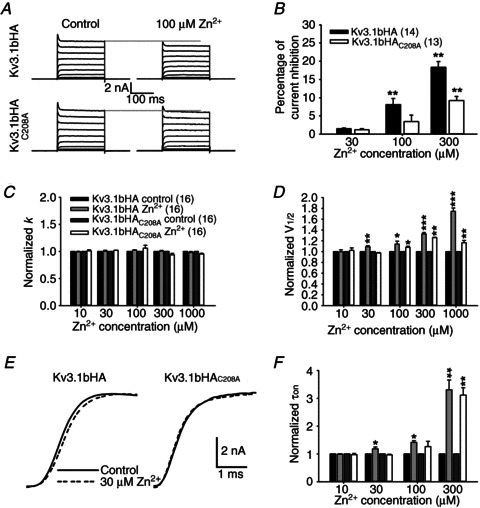
Kv3.1bHA and Kv3.1bHAC208A were transfected with YFP into HEK293 cells. Voltage-clamp recording was performed on these cells using the same protocol as in Fig. 2A. A, 100 μm Zn2+ inhibited Kv3.1bHA (top) but not Kv3.1bHAC208A (bottom). B, differential inhibitory effects of Zn2+ at low concentrations on current amplitude of Kv3.1bHA and Kv3.1bHAC208A. C, Zn2+ had no effect on the slope factor, k, in the G–V relationship of both Kv3.1bHA and Kv3.1bHAC208A. D, 30 μm Zn2+ significantly increased V1/2 of Kv3.1bHA but not Kv3.1bHAC208A. E, 30 μm Zn2+ slowed the activation of Kv3.1bHA but not Kv3.1bHAC208A at +30 mV. F, effects of Zn2+ at different concentrations on the activation time constant (τon) of Kv3.1bHA and Kv3.1bHAC208A. Paired t test: ***P < 0.001; **P < 0.01; *P < 0.05.
Construction of a Kv3-expressing and fast-spiking neuron with relative resistance to the Zn2+ inhibition
Since both channel biophysical properties and axonal localization are important for Kv3-mediated fast spiking (Gu et al. 2012), we further examined the axon–dendrite targeting pattern of Kv3.1bHAC208A on the neuronal surface. When expressed in cultured hippocampal neurons, Kv3.1bHAC208A was surprisingly restricted on somatodendritic membranes (Fig. 9A). Some puncta were often observed on the cell surface (Fig. 9A and B). In the presence of co-expressed KIF5B-YFP, Kv3.1bHAC208A, similar to Kv3.1aHA (Xu et al. 2010), was localized to axonal membranes, in both proximal and distal axons (Faxon/Fdend: +YFP 0.36 ± 0.05; +KIF5B-YFP 1.02 ± 0.08) (Fig. 9B–D). Reduced axonal localization of Kv3.1bHAC208A was unexpected, but its axonal level was restored in the presence of KIF5B-YFP, suggesting that at least its T1 domain still functions properly.
Figure 9. Constructing a fast-spiking neuron with relative resistance to the zinc inhibition.
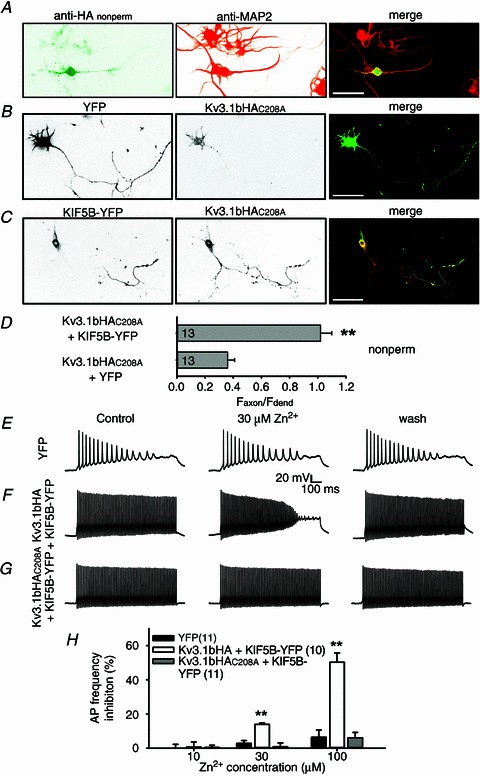
A, Kv3.1bHAC208A was restricted on somatodendritic membranes in cultured hippocampal neurons revealed by double staining of anti-HA under the non-permeabilized condition and anti-MAP2. B, Kv3.1bHAC208A enriched on somatodendritic membranes. Neurons at 5 DIV were co-transfected with Kv3.1bHAC208A and YFP, fixed 2 days later, and stained with an anti-HA antibody under the non-permeabilized condition. C, Kv3.1bAHAC208A localized to axonal membranes in the presence of KIF5B-YFP. Scale bars, 100 μm. D, summary of axonal targeting of Kv3.1bHAC208A in the absence or the presence of KIF5B-YFP. E, Zn2+ (30 μm) had no clear effect on the action potential firing rate on the neuron expressing YFP at 8 DIV. Square current pulses (1000 ms; 105 pA) were injected into the neuronal soma to induce action potentials. F, Zn2+ (30 μm) significantly reduced spiking frequency of the neuron expressing Kv3.1bHA and KIF5B-YFP. G, Zn2+ (30 μm) had no clear effect on fast spiking of the neuron expressing Kv3.1bHAC208A and KIF5B-YFP. H, summary of the effect of zinc (10, 30 and 100 μm) on spiking frequency. The number of neurons are indicated within parentheses.
Expressing Kv3.1bHA can convert a slow-spiking young hippocampal neuron to a fast-spiking one (Fig. 9E and F) (Gu et al. 2012). Here, we co-expressed Kv3.1bHAC208A and KIF5B-YFP into cultured hippocampal neurons, which also markedly increased the firing rate of transfected neurons (Fig. 9G). Here, we examined three Zn2+ concentrations, 10, 30 and 100 μm, in order to mimic its in vivo concentrations. At 30 and 100 μm, zinc markedly inhibited the firing frequency in neurons expressing Kv3.1bHA and KIF5B-YFP, but had little effect on spiking frequency in neurons expressing Kv3.1bHAC208A and KIF5B-YFP (30 μm: Kv3.1bHA + KIF5B-YFP 14.0 ± 0.8%, Kv3.1bHAC208A+ KIF5B-YFP 0.9 ± 2.3%; 100 μm: Kv3.1bHA + KIF5B-YFP 50.3 ± 5.4%, Kv3.1bHAC208A+ KIF5B-YFP 6.1 ± 3.2%; Fig. 9H). At 10 μm, zinc had no clear effect on the firing rate of either type of neurons (Fig. 9H). Therefore, based on the new mechanistic insights regarding the Kv3.1 zinc-binding site obtained in the present study, we successfully reconstituted a fast-spiking neuron with significant resistance to the zinc inhibition at concentrations close to physiological or pathological levels in vivo.
Discussion
Using mutagenesis, protein biochemistry, cell biology and electrophysiological assays, we have systematically analysed the molecular mechanism underlying the zinc-mediated regulation of Kv3.1 channels. Remarkably, we have identified multiple zinc-binding sites, involved in not only channel assembly, but also biophysical properties and localization (Fig. 10). The zinc-binding site in the T1 domain, required for channel tetramerization, is also important for channel biophysical properties and localization. The newly identified site within the C-terminus is important for channel activity and axonal targeting. The zinc inhibition is mainly mediated through a site at the junction between the 1st TM domain and the 1st extracellular loop. Taken together, this study has revealed novel mechanistic insights into the multifaceted regulation of Kv3 channel activity and localization by zinc (Fig. 10).
Figure 10. Distinct binding sites for divalent heavy metal ions in the tetrameric Kv3.1 channel complex.
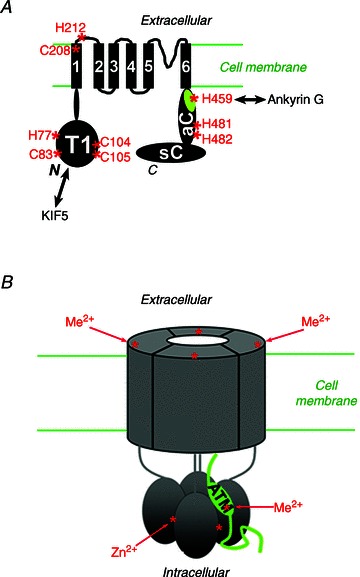
Structural diagram of Kv3.1b subunit. All the potential zinc-binding sites are indicated with asterisks and the residue numbers are labelled. B, diagram of a tetrameric Kv3 channel complex regulated by extracellular and intracellular divalent heavy metal ions.
The zinc-binding site in the N-terminal T1 domain does not mediate the zinc inhibition
Conformational rearrangements of Kv channel T1 domains are believed to be involved in gating, which is not limited to the TM core (Cushman et al. 2000; Minor et al. 2000; Wang et al. 2005; Kobrinsky et al. 2006; Lvov et al. 2009). Consistent with this notion, mutating any one of the histidine or cysteine residues in the zinc-binding site within Kv3.1 T1 not only abolishes T1 tetramerization, but also results in markedly reduced channel currents (Xu et al. 2010). Among those mutants, Kv3.1bHAH77A carries the largest current, and was therefore used in this study to further examine its sensitivity to zinc. Surprisingly, application of zinc still markedly suppressed the current of Kv3.1bHAH77A (Fig. 2C). The activation time constant of Kv3.1bHAH77A was about 100-fold bigger than that of Kv3.1b, and appeared to further increase in the presence of zinc (Fig. 2). Therefore, disassembling Kv3.1 T1 tetramers did not eliminate or even significantly reduce the zinc inhibition, indicating that the T1 domain does not mediate the zinc inhibition.
A novel zinc-binding site in the C-terminus of Kv3.1
In this study, we accidentally identified a new Me2+-binding site in the C-terminus of the Kv3.1 channel. The initial surprising result that Co2+ beads efficiently pulled down and even purified bacterially expressed GST-31aC prompted us to map critical residues that mediated the binding (Fig. 3). Co2+ beads pulled down GST-31bC but not GST-31sC, indicating that the splice domain does not contain a Me2+-binding site, although this splice domain contains 1 histidine and 4 cysteine residues (Fig. 3). Therefore, there must be a Me2+-binding site in the conserved region of C-termini of Kv3.1a and Kv3.1b. Finally, we identified three histidine residues, one (H459) within the ATM and the other two (H480H481) downstream of the ATM. All three histidine residues were required for the efficient pulldown by Zn2+ beads (Fig. 4).
The H495 residue within the ATM is conserved within the Kv3 subfamily (Kv3.1 to Kv3.4), whereas the two outside of the ATM (H480H481) are not conserved. The three histidine residues appear to play different roles in regulating channel activity and localization. Mutating H459 to alanine altered the channel's activation kinetics and voltage–conductance relationship, and decreased the axonal level of channel proteins. In sharp contrast, mutating the other two histidines did not affect either channel activity or axonal targeting (Figs 5 and 6). Furthermore, since Kv3.1 C-terminus binds to the N-terminal T1 domain in a zinc-dependent manner (Xu et al. 2007), the C-terminal zinc-binding sites are most likely involved in the N-/C-terminal binding. Overall, these binding sites will effectively increase the local Zn2+ concentration. The Kv3.1 C-terminal region is probably quite flexible, so its conformation before or after zinc binding, or during the interaction with the T1 domain, remains to be determined in future studies. Despite the novel findings in this study, how the extracellular and intracellular zinc signalling coordinate to regulate different aspects of Kv3 function remains unclear. Transient or persistent alterations of zinc homeostasis regulated by neuronal activity or during pathogenic processes might affect the intracellular trafficking and activity of Kv3 channels. These are very interesting topics for future investigation.
Interestingly, Co2+ beads pulled down the Kv3.1 C-terminus, but not its T1 domain (Fig. 3B and C). This may result from the sites being buried within T1 tetramers, which cannot be accessed by the metal ions. The Zn2+-binding site in the T1 tetramer interface is composed of residues from two T1 monomers. A single T1 monomer cannot bind to Zn2+. In the pulldown assay, bacterially expressed GST-31T1 already formed tetramers (Xu et al. 2010). Another major difference with the T1 site is that, besides Zn2+ and Co2+, the C-terminal site may bind to other divalent heavy metal ions. Nonetheless, mutating either the N- or C-terminal site failed to significantly reduce the zinc inhibition, suggesting other site(s) may be involved.
The junction between the 1st TM segment and the 1st extracellular loop in Kv3 channels
The rapid action of the zinc inhibitory effect suggests that the site might be in the extracellular side of the channel complex. We initially suspected that the two histidine residues in the P loop between the 5th and 6th TM segments might mediate the zinc inhibition. However, mutating either one or both did not significantly reduce the effect of zinc (Fig. 7). Surprisingly again, the zinc-binding site appears to be in the junction between the 1st TM segment and the 1st extracellular loop (Fig. 7). Mutating the H212 to A markedly altered channel biophysical properties, whereas mutating C208 to A did not. The zinc inhibition of both mutants was significantly reduced with respect to current amplitude and G–V curve shifting (Fig. 7). However, the slowing of activation kinetics by zinc remained prominent in Kv3.1bHAC208A (Fig. 7). Mutating either C208 or H212 still did not completely eliminate the zinc inhibition at 1 mm concentration, suggesting that another part of the channel may be involved (Fig. 7). At lower concentrations, such as 30 μm, close to its physiological level, zinc had no effect on Kv3.1bHAC208A in terms of current amplitude, G–V relationship and activation time constant (Fig. 8). In contrast, both V1/2 and τon, but not the current amplitude, of Kv3.1bHA increased at 30 μm zinc (Fig. 8). This is consistent with the result that at 30 μm, Zn2+ significantly reduced the spiking frequency of the neurons expressing Kv3.1bHA and KIF5B-YFP, but not of the neurons expressing the C208A mutant and KIF5B-YFP (Fig. 9E–H). It further indicates that alterations in V1/2 and τon of the channel are sufficient to change spiking frequency and the alteration of current amplitude is not necessary (Figs 8 and 9). Interestingly, an episodic ataxia type-1 mutation has been identified in the C-terminus of the 1st TM segment (F184C) of the human Kv1.1 channel, which is associated with a markedly higher sensitivity to zinc (Cusimano et al. 2004; Imbrici et al. 2007). This residue is in a similar position to C208 in Kv3.1b, suggesting that this junction between the 1st TM segment and 1st extracellular loop can transfer conformational changes to the channel core to alter channel biophysical properties.
Furthermore, mutating C208 to A decreased its axonal targeting (Fig. 9A and B). How this mutation affects channel-polarized targeting remains an intriguing question, due to its position in the 1st TM segment. However, the mutant axonal targeting can be rescued by co-expression of KIF5B-YFP (Fig. 9), just like Kv3.1aHA (Xu et al. 2010), suggesting that the T1 tetramerization is not abolished in the mutant.
The hallmark of Kv3 biophysical properties is high activation threshold, slow activation and deactivation kinetics. The activation threshold and activation time constant appears to be highly sensitive to zinc regulation, whereas the deactivation time constant was not markedly affected. Interestingly, all three types of zinc-binding sites appear to be critical in channel biophysical properties and polarized targeting. Therefore, our study indicates that channel biophysical properties and protein trafficking are not dictated by isolated portions of channel complex, but rather rely on the conformation of whole channel protein complex.
Potential functions of the regulation under normal and pathological conditions
Free zinc ions are emerging as important signalling messengers, with dynamic fluctuations in both intracellular cytoplasm and extracellular space. In hippocampal neurons, zinc released from presynaptic vesicles can lead to synaptic concentrations of 10–30 μm (Frederickson et al. 2005; Mathie et al. 2006). Its concentration can become even higher in some pathophysiological conditions, such as Alzheimer's disease, epilepsy and ischaemia injury (Weiss et al. 2000; Galasso & Dyck, 2007; Hershfinkel et al. 2009; Medvedeva et al. 2009; Pithadia & Lim, 2012). Although GABA, glycine and NMDA receptors are well-studied targets with higher sensitivity to zinc regulation compared to Kv3 channels, the Kv3 regulation may be still important under certain physiological or pathological conditions. Zinc released by excitatory axonal terminals may act on Kv3 channels located on both presynaptic and postsynaptic membranes, to feedback-regulate the neurotransmitter release of the Kv3-expressing and fast-spiking neurons. The fast-spiking neurons are implicated not only in reflexes, learning and memory, but also in neurological disorders, such as epilepsy, schizophrenia, addiction, etc. Furthermore, this study has provided important mechanistic insights into the structure and function of Kv3 channels (Fig. 10).
Finally, in this study, we have successfully reconstituted a fast-spiking neuron with significantly reduced zinc inhibition, by using Kv3.1bHAC208A co-expressed with KIF5B-YFP (Fig. 9). Our research will contribute to the effort to manipulate the input–output relationship of neurons to modify neurophysiological functions under normal and abnormal conditions. Importantly, this study developed very sensitive protein biochemistry, cell biology and electrophysiology assays to analyse different aspects of Kv3 channel activity and trafficking, which can provide critical mechanistic insights for future studies in activity-dependent regulation of Kv3 channel functions in various disorders of the central nervous system.
Acknowledgments
This work was supported by a grant from NINDS/NIH (R01NS062720) to C.G.
Glossary
- A
alanine
- C
cysteine
- ATM
axonal targeting motif
- DIV
days in vitro
- GST
glutathione S-transferase
- H
histidine
- Kv channel
voltage-gated K+ channel
- Kv3 channel
Shaw voltage-gated K+ channel
- Me2+
divalent heavy metal ion
- Nav channel
voltage-gated Na+ channel
- TM
transmembrane
- TRPA1
transient receptor potential cation channel A1
- YFP
yellow fluorescent protein
- 31aC
the C-terminus of Kv3.1a (amino acids 442–511)
- 31bC
the C-terminus of Kv3.1b (amino acids 442–585)
- 31sC
the splice domain of Kv3.1b C-terminus (amino acids 500–585)
- HA
hemagglutinin tag
- KIF5B
kinesin superfamily protein 5 isoform B
- MAP2
microtubule-associated protein 2
Author contributions
C.G. designed and supervised the research. Y.G. and J.B. performed the experiments. Y.G., J.B. and C.G. analysed the data and wrote the paper.
Supplementary material
Supplemental Figure S1
References
- Aras MA, Saadi RA, Aizenman E. Zn2+ regulates Kv2.1 voltage-dependent gating and localization following ischemia. Eur J Neurosci. 2009;30:2250–2257. doi: 10.1111/j.1460-9568.2009.07026.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardoni R, Belluzzi O. Modifications of A-current kinetics in mammalian central neurones induced by extracellular zinc. J Physiol. 1994;479:389–400. doi: 10.1113/jphysiol.1994.sp020304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barry J, Gu Y, Gu C. Polarized targeting of L1-CAM regulates axonal and dendritic bundling in vitro. Eur J Neurosci. 2010;32:1618–1631. doi: 10.1111/j.1460-9568.2010.07447.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bean BP. The action potential in mammalian central neurons. Nat Rev Neurosci. 2007;8:451–465. doi: 10.1038/nrn2148. [DOI] [PubMed] [Google Scholar]
- Bixby KA, Nanao MH, Shen NV, Kreusch A, Bellamy H, Pfaffinger PJ, Choe S. Zn2+-binding and molecular determinants of tetramerization in voltage-gated K+ channels. Nat Struct Biol. 1999;6:38–43. doi: 10.1038/4911. [DOI] [PubMed] [Google Scholar]
- Cushman SJ, Nanao MH, Jahng AW, DeRubeis D, Choe S, Pfaffinger PJ. Voltage dependent activation of potassium channels is coupled to T1 domain structure. Nat Struct Biol. 2000;7:403–407. doi: 10.1038/75185. [DOI] [PubMed] [Google Scholar]
- Cusimano A, Cristina D’Adamo M, Pessia M. An episodic ataxia type-1 mutation in the S1 segment sensitises the hKv1.1 potassium channel to extracellular Zn2+ FEBS Lett. 2004;576:237–244. doi: 10.1016/j.febslet.2004.09.018. [DOI] [PubMed] [Google Scholar]
- Frederickson CJ, Koh JY, Bush AI. The neurobiology of zinc in health and disease. Nat Rev Neurosci. 2005;6:449–462. doi: 10.1038/nrn1671. [DOI] [PubMed] [Google Scholar]
- Galasso SL, Dyck RH. The role of zinc in cerebral ischemia. Mol Med. 2007;13:380–387. doi: 10.2119/2007-00044.Galasso. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigg JJ, Brew HM, Tempel BL. Differential expression of voltage-gated potassium channel genes in auditory nuclei of the mouse brainstem. Hear Res. 2000;140:77–90. doi: 10.1016/s0378-5955(99)00187-2. [DOI] [PubMed] [Google Scholar]
- Gu C, Barry J. Function and mechanism of axonal targeting of voltage-sensitive potassium channels. Prog Neurobiol. 2011;94:115–132. doi: 10.1016/j.pneurobio.2011.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu C, Gu Y. Clustering and activity tuning of Kv1 channels in myelinated hippocampal axons. J Biol Chem. 2011;286:25835–25847. doi: 10.1074/jbc.M111.219113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu C, Zhou W, Puthenveedu MA, Xu M, Jan YN, Jan LY. The microtubule plus-end tracking protein EB1 is required for Kv1 voltage-gated K+ channel axonal targeting. Neuron. 2006;52:803–816. doi: 10.1016/j.neuron.2006.10.022. [DOI] [PubMed] [Google Scholar]
- Gu Y, Barry J, McDougel R, Terman D, Gu C. Alternative splicing regulates Kv3.1 polarized targeting to adjust maximal spiking frequency. J Biol Chem. 2012;287:1755–1769. doi: 10.1074/jbc.M111.299305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Y, Gu C. Dynamics of Kv1 channel transport in axons. PLoS One. 2010:5. doi: 10.1371/journal.pone.0011931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrison NL, Radke HK, Talukder G, Ffrench-Mullen JM. Zinc modulates transient outward current gating in hippocampal neurons. Receptors Channels. 1993;1:153–163. [PubMed] [Google Scholar]
- Hershfinkel M, Kandler K, Knoch ME, Dagan-Rabin M, Aras MA, Abramovitch-Dahan C, Sekler I, Aizenman E. Intracellular zinc inhibits KCC2 transporter activity. Nat Neurosci. 2009;12:725–727. doi: 10.1038/nn.2316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horning MS, Trombley PQ. Zinc and copper influence excitability of rat olfactory bulb neurons by multiple mechanisms. J Neurophysiol. 2001;86:1652–1660. doi: 10.1152/jn.2001.86.4.1652. [DOI] [PubMed] [Google Scholar]
- Hou S, Vigeland LE, Zhang G, Xu R, Li M, Heinemann SH, Hoshi T. Zn2+ activates large conductance Ca2+-activated K+ channel via an intracellular domain. J Biol Chem. 2011;285:6434–6442. doi: 10.1074/jbc.M109.069211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu H, Bandell M, Petrus MJ, Zhu MX, Patapoutian A. Zinc activates damage-sensing TRPA1 ion channels. Nat Chem Biol. 2009;5:183–190. doi: 10.1038/nchembio.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang EP. Metal ions and synaptic transmission: think zinc. Proc Natl Acad Sci U S A. 1997;94:13386–13387. doi: 10.1073/pnas.94.25.13386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imbrici P, D’Adamo MC, Cusimano A, Pessia M. Episodic ataxia type 1 mutation F184C alters Zn2+-induced modulation of the human K+ channel Kv1.4-Kv1.1/Kvβ1.1. Am J Physiol Cell Physiol. 2007;292:C778–C787. doi: 10.1152/ajpcell.00259.2006. [DOI] [PubMed] [Google Scholar]
- Jahng AW, Strang C, Kaiser D, Pollard T, Pfaffinger P, Choe S. Zinc mediates assembly of the T1 domain of the voltage-gated K channel 4.2. J Biol Chem. 2002;277:47885–47890. doi: 10.1074/jbc.M208416200. [DOI] [PubMed] [Google Scholar]
- Jiang Q, Inoue K, Wu X, Papasian CJ, Wang JQ, Xiong ZG, Chu XP. Cysteine 149 in the extracellular finger domain of acid-sensing ion channel 1b subunit is critical for zinc-mediated inhibition. Neuroscience. 2011;193:89–99. doi: 10.1016/j.neuroscience.2011.07.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kay AR, Toth K. Is zinc a neuromodulator. Sci Signal. 2008;1:re3. doi: 10.1126/stke.119re3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kehl SJ, Eduljee C, Kwan DC, Zhang S, Fedida D. Molecular determinants of the inhibition of human Kv1.5 potassium currents by external protons and Zn2+ J Physiol. 2002;541:9–24. doi: 10.1113/jphysiol.2001.014456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobrinsky E, Stevens L, Kazmi Y, Wray D, Soldatov NM. Molecular rearrangements of the Kv2.1 potassium channel termini associated with voltage gating. J Biol Chem. 2006;281:19233–19240. doi: 10.1074/jbc.M601231200. [DOI] [PubMed] [Google Scholar]
- Kunjilwar K, Strang C, DeRubeis D, Pfaffinger PJ. KChIP3 rescues the functional expression of Shal channel tetramerization mutants. J Biol Chem. 2004;279:54542–54551. doi: 10.1074/jbc.M409721200. [DOI] [PubMed] [Google Scholar]
- Kuo CC, Chen FP. Zn2+ modulation of neuronal transient K+ current: fast and selective binding to the deactivated channels. Biophys J. 1999;77:2552–2562. doi: 10.1016/s0006-3495(99)77090-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Jan YN, Jan LY. Specification of subunit assembly by the hydrophilic amino-terminal domain of the Shaker potassium channel. Science. 1992;257:1225–1230. doi: 10.1126/science.1519059. [DOI] [PubMed] [Google Scholar]
- Lin DD, Cohen AS, Coulter DA. Zinc-induced augmentation of excitatory synaptic currents and glutamate receptor responses in hippocampal CA3 neurons. J Neurophysiol. 2001;85:1185–1196. doi: 10.1152/jn.2001.85.3.1185. [DOI] [PubMed] [Google Scholar]
- Lovinger DM, Harrison NL, Lambert NA. The actions of 3-aminopropanephosphinic acid at GABAB receptors in rat hippocampus. Eur J Pharmacol. 1992;211:337–341. doi: 10.1016/0014-2999(92)90390-p. [DOI] [PubMed] [Google Scholar]
- Lvov A, Greitzer D, Berlin S, Chikvashvili D, Tsuk S, Lotan I, Michaelevski I. Rearrangements in the relative orientation of cytoplasmic domains induced by a membrane-anchored protein mediate modulations in Kv channel gating. J Biol Chem. 2009;284:28276–28291. doi: 10.1074/jbc.M109.028761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathie A, Sutton GL, Clarke CE, Veale EL. Zinc and copper: pharmacological probes and endogenous modulators of neuronal excitability. Pharmacol Ther. 2006;111:567–583. doi: 10.1016/j.pharmthera.2005.11.004. [DOI] [PubMed] [Google Scholar]
- Medvedeva YV, Lin B, Shuttleworth CW, Weiss JH. Intracellular Zn2+ accumulation contributes to synaptic failure, mitochondrial depolarization, and cell death in an acute slice oxygen-glucose deprivation model of ischemia. J Neurosci. 2009;29:1105–1114. doi: 10.1523/JNEUROSCI.4604-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minor DL, Lin YF, Mobley BC, Avelar A, Jan YN, Jan LY, Berger JM. The polar T1 interface is linked to conformational changes that open the voltage-gated potassium channel. Cell. 2000;102:657–670. doi: 10.1016/s0092-8674(00)00088-x. [DOI] [PubMed] [Google Scholar]
- Nanao MH, Zhou W, Pfaffinger PJ, Choe S. Determining the basis of channel-tetramerization specificity by x-ray crystallography and a sequence-comparison algorithm: Family Values (FamVal) Proc Natl Acad Sci U S A. 2003;100:8670–8675. doi: 10.1073/pnas.1432840100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nigro MJ, Perin P, Magistretti J. Differential effects of Zn2+ on activation, deactivation, and inactivation kinetics in neuronal voltage-gated Na+ channels. Pflugers Arch. 2011;462:331–347. doi: 10.1007/s00424-011-0972-z. [DOI] [PubMed] [Google Scholar]
- Pithadia AS, Lim MH. Metal-associated amyloid-β species in Alzheimer's disease. Curr Opin Chem Biol. 2012;16:67–73. doi: 10.1016/j.cbpa.2012.01.016. [DOI] [PubMed] [Google Scholar]
- Poling JS, Vicini S, Rogawski MA, Salem N., Jr Docosahexaenoic acid block of neuronal voltage-gated K+ channels: subunit selective antagonism by zinc. Neuropharmacology. 1996;35:969–982. doi: 10.1016/0028-3908(96)00127-x. [DOI] [PubMed] [Google Scholar]
- Prost AL, Bloc A, Hussy N, Derand R, Vivaudou M. Zinc is both an intracellular and extracellular regulator of KATP channel function. J Physiol. 2004;559:157–167. doi: 10.1113/jphysiol.2004.065094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puopolo M, Belluzzi O. Functional heterogeneity of periglomerular cells in the rat olfactory bulb. Eur J Neurosci. 1998;10:1073–1083. doi: 10.1046/j.1460-9568.1998.00115.x. [DOI] [PubMed] [Google Scholar]
- Rudy B, McBain CJ. Kv3 channels: voltage-gated K+ channels designed for high-frequency repetitive firing. Trends Neurosci. 2001;24:517–526. doi: 10.1016/s0166-2236(00)01892-0. [DOI] [PubMed] [Google Scholar]
- Sensi SL, Paoletti P, Bush AI, Sekler I. Zinc in the physiology and pathology of the CNS. Nat Rev Neurosci. 2009;10:780–791. doi: 10.1038/nrn2734. [DOI] [PubMed] [Google Scholar]
- Smart TG, Hosie AM, Miller PS. Zn2+ ions: modulators of excitatory and inhibitory synaptic activity. Neuroscientist. 2004;10:432–442. doi: 10.1177/1073858404263463. [DOI] [PubMed] [Google Scholar]
- Wang G, Shahidullah M, Rocha CA, Strang C, Pfaffinger PJ, Covarrubias M. Functionally active T1–T1 interfaces revealed by the accessibility of intracellular thiolate groups in Kv4 channels. J Gen Physiol. 2005;126:55–69. doi: 10.1085/jgp.200509288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Strang C, Pfaffinger PJ, Covarrubias M. Zn2+-dependent redox switch in the intracellular T1–T1 interface of a Kv channel. J Biol Chem. 2007;282:13637–13647. doi: 10.1074/jbc.M609182200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiser M, Vega-Saenz de Miera E, Kentros C, Moreno H, Franzen L, Hillman D, Baker H, Rudy B. Differential expression of Shaw-related K+ channels in the rat central nervous system. J Neurosci. 1994;14:949–972. doi: 10.1523/JNEUROSCI.14-03-00949.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiss JH, Sensi SL, Koh JY. Zn2+: a novel ionic mediator of neural injury in brain disease. Trends Pharmacol Sci. 2000;21:395–401. doi: 10.1016/s0165-6147(00)01541-8. [DOI] [PubMed] [Google Scholar]
- Xu J, Yu W, Jan YN, Jan LY, Li M. Assembly of voltage-gated potassium channels. Conserved hydrophilic motifs determine subfamily-specific interactions between the α-subunits. J Biol Chem. 1995;270:24761–24768. doi: 10.1074/jbc.270.42.24761. [DOI] [PubMed] [Google Scholar]
- Xu M, Cao R, Xiao R, Zhu MX, Gu C. The axon-dendrite targeting of Kv3 (Shaw) channels is determined by a targeting motif that associates with the T1 domain and ankyrin G. J Neurosci. 2007;27:14158–14170. doi: 10.1523/JNEUROSCI.3675-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu M, Gu Y, Barry J, Gu C. Kinesin I transports tetramerized Kv3 channels through the axon initial segment via direct binding. J Neurosci. 2010;30:15987–16001. doi: 10.1523/JNEUROSCI.3565-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S, Kehl SJ, Fedida D. Modulation of Kv1.5 potassium channel gating by extracellular zinc. Biophys J. 2001;81:125–136. doi: 10.1016/S0006-3495(01)75686-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Bursulaya B, Lee CC, Chen B, Pivaroff K, Jegla T. Divalent cations slow activation of EAG family K+ channels through direct binding to S4. Biophys J. 2009;97:110–120. doi: 10.1016/j.bpj.2009.04.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.