

Abstract
Under normal conditions histone H2AX is constitutively phosphorylated on tyrosine (Y) 142 by Williams–Beuren syndrome transcription factor kinase (WSTF). Following DNA double strand breaks (DSB), Y142 is de-phosphorylated and serine (S) 139 is phosphorylated. Here we explored DSB-dependent cross talk between H2AX residues S139 and Y142. H2axY142A mutation resulted in increased sensitivity to ionising radiation (IR), compared to H2axS139A. Interestingly, co-mutation of S139A and Y142A rescued IR sensitivity. The DSB response proteins 53Bp1 and Rad51 were recruited to IR-induced foci (IRIF) in H2axS139A, H2axY142A and H2axS139A/Y142A cells. Our results suggest that H2axY142A IR sensitivity is dependent upon the C-terminal residue, S139.
Keywords: H2AX, Y142, S139, DSB, IR, 53BP1
Abbreviations: DSB, DNA double strand break; IR, ionising radiation; WSTF, Williams–Beuren syndrome transcription factor kinase; IRIF, ionising radiation induced foci; WT, Wild type; F, phenylalanine; W, tryptophan; A, alanine; Y, tyrosine; Fig, Figure; RT-PCR, reverse transcription polymerase chain reaction; H2AX/H2ax, MDC1/Mdc1, RAD51/Rad51 and 53BP1/53Bp1, indicate human or mouse/chicken proteins respectively
Highlights
▸ H2AX-Y142A cells display a slow growth phenotype. ▸ We report H2AX-Y142A cells have increased constitutive cell death. ▸ We found that cells expressing H2AX-Y142A are IR sensitive. ▸ Combining H2AX mutations Y142A and S139A rescues the Y142A IR sensitivity. ▸ H2AX-Y142A cells can recruit the DNA repair proteins 53BP1 and Rad51.
1. Introduction
One of the earliest cellular events upon induction of DNA double strand breaks (DSB) is ATM-dependent phosphorylation of histone H2AX on Serine (S) 139 [1]. Although this modification, termed γH2AX, is the most characterised post-translational modification of H2AX, other DSB-induced modifications of H2AX have been described on lysines 5 and 36 and threonine 101 [2–4]. In higher eukaryotes the Williams–Beuren syndrome transcription factor kinase (WSTF) constitutively phosphorylates H2AX at it C-terminal residue, tyrosine 142, termed H2AXY142ph [5]. The H2AXY142ph modification is abrogated following DSB by the counteracting activity of the EYA phosphatases [5–7]. Perturbing either of these events impacts on DNA damage response signalling, through ATM, leading to increased cell death following DNA damage [5,6]. It has been proposed that persistence of Y142 phosphorylation on H2AX following DNA damage leads to apoptosis while de-phosphorylation facilitates repair [8]. Recently, a qualitative correlation between H2AX Y142 de-phosphorylation and γH2AX formation has been described, suggesting cross talk between these two modified residues [5].
Here we use the DT40 model system, a chicken B lymphocytic cell line that facilitates gene targeting, to introduce mutations into chicken H2ax that result in single and double substitutions of residues S139 and Y142 to alanines (A). This model system recapitulates many key features seen in higher vertebrate systems and Gallus gallus H2ax has high sequence homology to human H2AX [9,10]. As the mutant H2ax proteins are the sole source of H2ax in these cells we then explored the relationship between these two C-terminal DNA damage responsive sites of phosphorylation. We found that although expression of H2ax-Y142A resulted in slow growth and radio-sensitivity, co-mutation of residue S139A on the same molecule rescued the Y142A associated phenotypes. Repression of the H2ax-Y142A phenotype, by co-mutation of S139A, is likely due to loss of signalling associated with non-modifiable S139A residue.
2. Material and methods
2.1. Cell culture conditions
The DT40 chicken B lymphocyte cell lines were cultured as previously described [10]. Briefly, DT40 cell lines were cultured at 39.5 °C and 5.0% CO2 in RPMI 1640 and 10% foetal bovine serum (FBS) [Lonza (Berkshire, UK)], 1% chicken serum, 100 U/ml penicillin and 100 μg/ml streptomycin [Sigma (Dublin, Ireland)].
2.2. H2AX targeting vectors and PCR primers used
The DT40 H2ax mutants were created using site directed mutagenesis of the Gallus gallus H2ax gene, sub-cloned from the previously described vector [12]. Amplification primers for the Gallus gallus H2ax open reading frame used were; forward 5′-CGCGGCAAGAGTGGCGGTAA-3′ and reverse 5′-AGCATGGGCTATTGCAGGACCC-3′.
Mutations were introduced using site directed mutagenesis (Stratagene) as per manufacturers instructions. Mutation specific primer sequences were: S139A 5′-CAGCGGGCAGCAGGCCCAGGAGTACTAGG-3′, Y142A 5′-CAGTCGCAGGAGGCCTAGCCCACCC-3′ and S139A/Y142A 5′-GCGGGCAGCAGGCCCAGGAGGCCTAGGCCTGGC-3′. All mutations introduced were verified by DNA sequencing.
The ovalbumin targeting vector was constructed as follows: ovalbumin homology arms were amplified from DT40 genomic DNA by PCR with KOD polymerase (Novagen) using the following primer pairs: 5′ ovalbumin arm: 5′- AAAAAGCGGCCGCCAGGAAGCTTTAAGGAATCATTG-3′ and 5′- CCCAGAACTAGTGGATCCTGGATGGGAGAGAAGACTGG-3′. 3′ ovalbumin arm: 5′- CATCCAGGATCCACTAGTTCTGGGACAGTTTGCTACCC-3′ and 5′- AAAAAGTCGACCTCAGTGCACAGGAATGGAG-3′. Homology arms were cloned into ploxPuro1 with a puromycin resistance cassette under the control of the CMV promoter inserted between the arms. The H2ax mutants were inserted using Nhe1 sites and all constructs verified by sequencing. All constructs were linearised by Pvu1 digestion and transfection performed as previously described [11]. Southern blot screening was carried out using a probe external to the targeting construct. The probe was amplified and labelled with α-32P using the forward primer 5′-TTTATGGGGGAAAAATGCAG-3′ and the reverse primer 5′-CAGATGAGTTGTCCCAGGTG-3′.
2.3. Proliferation and clonogenic survival analysis
For cell growth analysis, cultures were seeded in triplicate at equal cell densities (1 × 104 cells/ml) and counted using a haemocytometer every 24 h. Survival following ionising radiation treatment [137Cs irradiator (Mainance Engineering Ltd., UK), dose rate of 21.57 Gy/min] was determined by clonogenic survival assays in methylcellulose medium. Colonies formed by surviving cells were counted 7–10 days later. Results normalised to number of cells plated.
2.4. Antibodies
Western blotting was performed using the antibody recognising phosphorylated H2AX Y142 (Millipore). Immunofluorescent antibodies; 53Bp1 (Millipore) and Rad51 (ab63801, Abcam) and FITC or Texas Red-conjugated secondary antibodies (Jackson Immunoresearch).
2.5. Immunofluorescence microscopy and foci quantification
Cells were bound to poly-l-lysine coated slides prior to fixation in 4% paraformaldehyde and permeabilisation in 0.1% Triton-X-100. Cells were then blocked in 1% BSA and incubated with the indicated antibodies overnight at 4 °C.
Images acquired with an Olympus IX71 inverted microscope equipped with a Retiga-EXL Mono Lightwire 800 camera (QImaging, British Colmbia, Canada) and Image Pro-Plus software (Media Cybernetics Inc., Bethesda, USA). 0.4 μm Z-stacks were collected and brightest point projections created from stacked sections. All cell percentages were obtained from blind counting of 80–100 cells per sample. All foci percentages were determined using Cell Profiler software [13] counts of 50–100 cells per sample. Only cells with intact nuclei, no blebbing as determined by DAPI staining, were included in the counts.
2.6. Fluorescence activated flow cytometry
Cells were fixed in 70% ethanol at 4 °C overnight. Cells were then stained using propidium iodide (Sigma, Dublin, Ireland), and analysed using the FACS Calibur platform and CellQuest software (BD Biosciences, Belgium).
3. Results
3.1. H2ax mutation approach and cell line construction
To examine the role of Gallus gallus H2ax residues S139 and Y142 in the DSB response we generated cell lines containing S139A, Y142A and S139A/Y142A mutations. Mutation of Y142 to phenylalanine or tryptophan, which would maintain the aromatic character of this residue, was not performed as such conservative substitutions may imitate IR induced de-phosphorylation events. This possibility is supported by the lack of disruption of either 53BP1 or MDC1 ionising radiation induced foci (IRIF) when these conservative mutations were employed [2,3]. Furthermore, the H2ax-Y142A mutation has been shown to perturb Mdc1 recruitment to IRIF in mouse cells [14]. Using a previously generated Gallus gallus DT40 H2ax−/− (null) cell line [12]. Constructs harbouring S139A, Y142A or S139A/Y142A H2ax mutations were targeted to the ovalbumin (Ova) locus. This generated three cell lines, termed H2ax-S139A, H2ax-Y142A and H2ax-S139A/Y142A, which express the indicated H2ax mutant allele as the sole source of H2ax protein in these cells. As the introduced mutations disrupt the binding of anti-H2AX antibodies from multiple sources, as previously was previously described [2], expression of each mutant was therefore confirmed by RT-PCR (Fig. 1(A)). Isogenic cells expressing wild-type H2ax (WT) were used as a control for all experiments.
Fig. 1.
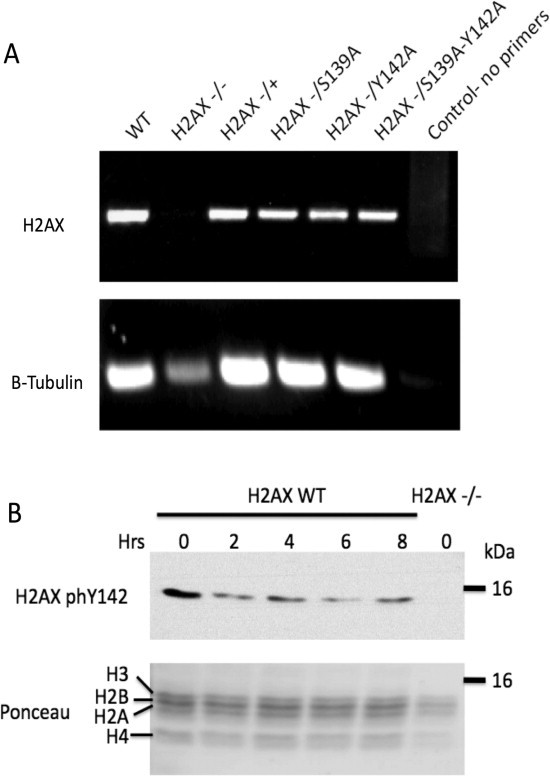
(A) Amplification of H2ax from mRNA isolated from indicated cell lines, N = 3. (B) Kinetics of H2ax phosphorylation on residue Y142 following 10 Gy IR.
It has previously been reported in human cells that phosphorylation of H2AX at its terminal Y142 residue is abrogated following DSB [5]. Following ionising radiation (IR) treatment we confirmed that de-phosphorylation of H2AX Y142 occurred in our WT DT40 cells, thereby validating our system (Fig. 1(B)). The direct effect of the H2AX mutations on the phosphorylation of Y142 could not be confirmed by western blotting, due to the disruption of H2AX antibody binding referred to above. Therefore, we examined the effect of the H2ax mutations on other cellular process.
3.2. H2ax-Y142A mutation results in slow growth and IR sensitivity
Relative to WT we found H2ax-Y142A cells exhibited a slow growth phenotype that was more pronounced than either H2ax-S139A or null cells (Fig. 2(A)). Significantly, in H2ax-S139A/Y142A cells, in which all H2ax molecules harbour both substitutions, the slow growth phenotype of H2ax-Y142A is rescued, with growth restored to levels seen in H2ax-S139A cells.
Fig. 2.
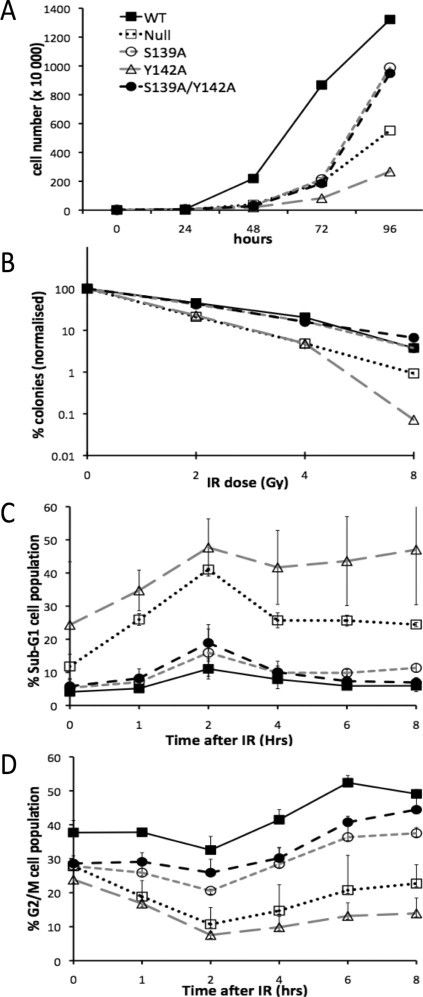
(A) Growth kinetics of indicated H2ax mutant expressing cells, N = 3. (B) Clonogenic IR survival of indicated H2ax mutant cell lines following indicated IR dose, N = 3. (C) Percent of cells in Sub G1 fraction following IR (10 Gy) treatment, N = 3. (D) Percent of cells in G2 fraction following IR (10 Gy) treatment, N = 3. Legend in (A) applies to all.
To further explore the relationship between these residues, their effect on cellular survival after ionising radiation was examined. In response to low-dose IR it was previously reported that H2ax-Y142A cells exhibited a mild sensitivity in mouse ES cells [2]. We found that our H2ax-Y142A DT40 cells displayed a higher sensitivity to IR, particularly at high dose (8 Gy) treatments not examined in previous studies [2] (Fig. 2(B)). Consistent with previous reports [12], our Null cells displayed sensitivity to IR relative to WT. Significantly, the double mutant, H2ax-S139A/Y142A, showed WT levels of IR sensitivity.
3.3. H2ax-Y142A mutations lead to increased constitutive cell death
To investigate a possible mechanism for the H2ax-Y142A mediated phenotype we performed cell cycle analyses. H2ax-Y142A cells displayed an increased apoptotic sub-G1 fraction prior to damage (Fig. 2(C)). Again, in H2ax-S139A/Y142A cells this phenotype was suppressed. Following IR treatment the same trend of apoptosis was observed in all cell lines, with H2ax-S139A/Y142A cells displaying virtually WT levels of Sub-G1 cells. However, following IR there was a significant reduction in the number of cells observed in G2/M in H2ax-Y142A cells (Fig. 2(D)). Again this was restored to almost WT levels in H2ax-S139A/Y142A cells.
3.4. H2ax-Y142A and H2ax-S139A/Y142A cells can form 53Bp1 IRIF
The constitutively high levels of sub-G1 cells observed in H2ax-Y142A cells suggested they might have intrinsic DNA damage. As previously reported [2], C-terminal mutations of tyrosine 142 of human H2AX interfere with detection of γH2AX IRIF and similarly we were unable to observe γH2ax foci in cells harbouring our H2ax mutations. Therefore, to explore the DNA damage response we examined recruitment of the DNA repair proteins 53Bp1 and Rad51 to IRIF (Fig. 3(A)). We used 53Bp1 foci formation as a surrogate for γH2AX, as 53Bp1 forms IRIF with kinetics very similar to γH2AX IRIF [16].
Fig. 3.
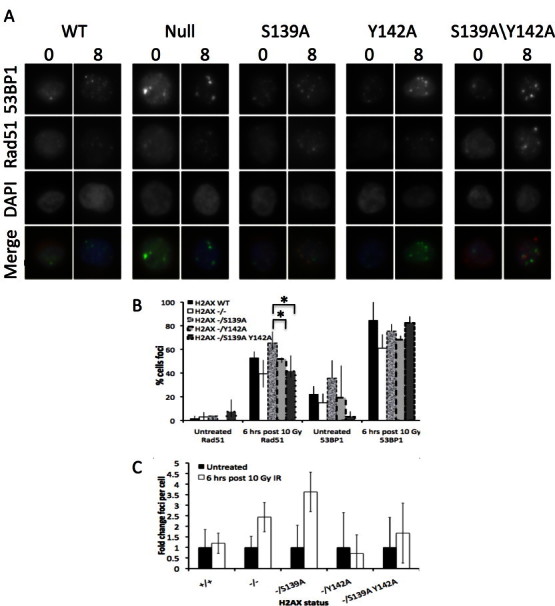
A: Localisation of 53Bp1 and Rad51 in indicated H2ax mutants following IR (8 Gy) treatment at indicated time points, N = 2. (B) Percent of cells with either 53Bp1 (>3) or Rad51 (>1) foci following IR (8 Gy) treatment, N = 2. Greater than 50 cells counted per time point per experiment. * ≥0.05 significance, students T-test. (C) Fold change in numbers of 53Bp1 IRIF per cell in indicated H2ax mutants following IR (8 Gy) treatment.
All cell lines were able to form 53Bp1 IRIF that persisted for at least 8 h post IR treatment (Fig. 3(A)). Quantification of the number of cells displaying 53Bp1 foci revealed that while H2ax-S139A cells have elevated levels of endogenous 53Bp1 foci, H2ax-S139A/Y142A cells have reduced levels of endogenous foci compared to the control (WT, Null) and H2ax-S139A cells (Fig. 3(B), right panel). Following IR treatment similar numbers of cells positive for 53Bp1 IRIF were observed (Fig. 3(B), far right).
Prior to treatment few cells in any line displayed Rad51, a DSB repair protein, foci (Fig. 3(C), far left panel). However, following IR treatment we found a significant increase in H2ax-S139A cells displaying Rad51 IRIF compared to WT and null cells. Furthermore, although there was an overall increase in Rad51 positive cells in all lines, when comparing between the H2ax mutants the increase in H2ax-Y142A and H2ax-S139A/Y142A cells was significantly smaller, ( p = 0.011) and ( p = 0.037) respectively, than the increase observed in H2ax-S139A cells (Fig. 3(B), centre).
Further investigation of 53Bp1 IRIF, by quantifying the number of 53Bp1 IRIF per cell revealed that null, H2ax-S139A and H2ax-S139A/Y142A cells had increased numbers of 53Bp1 IRIF per cell compared to WT (Fig. 3(C)). Significantly, the null and H2ax-S139A/Y142A cells have approximately the same increase in IRIF, while H2ax-S139A have more 53Bp1 IRIF per cell than these two lines. Although it is difficult to accurately assess 53Bp1 IRIF in H2ax-Y142A cells, due to the high level of cell death, the surviving fraction assayed displayed WT levels of 53Bp1 IRIF.
4. Discussion
In this study we demonstrate that cells expressing H2ax-Y142A, as the sole source of histone H2AX, display increased constitutive cell death and IR sensitivity. Interestingly, we found that combining the S139A and Y142A substitutions on the same H2AX molecule resulted in a suppression of the H2ax-Y142A mediated phenotypes. Furthermore, our results support the finding that DNA repair can occur, at a reduced rate, in the absence of phosphorylation at H2AX S139 [12,18].
Our analysis of cell proliferation indicates that H2ax-Y142A cells are slow growing due to an higher rate of constitutive cell death, indicated by an increased sub-G1 population (Fig. 2(A) and (C)). We propose that this is due to endogenous DNA damage which is supported by H2ax-Y142A cells displaying increased levels of constitutive 53Bp1 foci, indicative of DNA damage. As the double mutant, H2ax-S139A/Y142A, rescues this sensitivity (Figs. 2(C) and 3(C)) this suggests that the Y142A sensitivity to IR induced cell death (Fig. 2(B)) relies on signalling involving the S139 residue. H2ax-Y142A cells appear able to initiate DSB repair, as 53Bp1 is still recruited to sites of damaged DNA. However, the overall reduction in Rad51 IRIF (Fig. 3(B)) and 53Bp1 IRIF observed in H2ax-Y142A cells following DNA damage (Fig. 3(C)) is likely an under-estimation, influenced by the increased rates of cell death (see Fig. 2(C)) removing damaged cells. The observed radio-sensitivity of H2ax-Y142A cells is likely due to impairment of the homologous recombination (HR) DNA repair pathway (which has been previously reported in mouse H2ax-Y142A expressing mutant cells [2,3]) as HR is the major DNA damage repair pathway utilised by DT40 cells. In addition, it was previously demonstrated that the conservative H2AX mutations, Y142F or Y142W both of which would mimic de-phosphorylation, do not significantly affect the HR pathway [2,3]. This underscores the importance of active de-phosphorylation at H2AX Y142 for correct DNA repair and maintenance.
H2ax-S139A/Y142A DT40 cells display 53Bp1 IRIF suggesting that localisation of 53Bp1 at DSBs is at least partially independent of modifications to either the Y142 or S139 H2AX residues. Human 53BP1 binding is known to be mediated, at least in part, through MDC1 and as MDC1 cannot bind to H2AX without phosphorylated S139 and de-phosphorylated Y142 [15], this indicates that 53BP1 IRIF formation must not be solely dependent on H2AX mediated MDC1 localisation. Our results are consistent with previously described mouse H2AX−/− cells, which can form 53BP1 IRIF, although the number of 53BP1 foci is significantly reduced in this mouse system [14]. How the S139A and Y142A mutants effect Mdc1 binding in our system is unknown, as no working antibody exists for Gallus gallus Mdc1. However, an explanation for the observed recruitment of 53BP1 to DSB is that this localisation is mediated through the previously described interaction of MDC1 with the MRN complex [17,14].
H2ax-S139A/Y142A cells display less Rad51 IRIF compared to H2ax-S139A, suggesting that H2ax-S139A/Y142A cells can repair the damaged DNA more efficiently. Recent results support our finding that cells can actively repair DNA in the absence of γH2AX and other sites on H2AX have been suggested to play a role [18]. Our H2ax-Y142A cells also display fewer cells positive for Rad51 IRIF, which could be explained by more cells undergoing apoptosis (Fig. 3(B)).
Taken together our results indicate the H2AX mediated response to DSB is more complicated than expected. Although modification of H2AX S139 is important for the DSB response, modifications of the H2AX Y142 residue, either phosphorylation or de-phosphorylation, clearly play a significant role in determining cell fate in conjunction with phosphorylation at H2AX S139.
Statement of contribution
JALB: conception and design of the work, acquisition of all data, analysis and interpretation of data, wrote the manuscript. JKE: provided reagents. NFL: project conception, analysis and interpretation of data, edited the manuscript.
Acknowledgments
The authors wish to thank the Lowndes’ group and the CCB for stimulating discussions; Dr. Muriel Grenon and Dr. Emer Bourke for critical comments on the manuscript. We also would like to thank Prof. Takeda for his kind gift of the H2ax−/− DT40 cells and the Wild Type H2ax vector.
Contributor Information
James A.L. Brown, Email: james.brown@nuigalway.ie.
Noel F. Lowndes, Email: noel.lowndes@nuigalway.ie.
References
- 1.Rogakou E.P., Pilch D.R., Orr A.H., Ivanova V.S., Bonner W.M. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J. Biol. Chem. 1998;273(10):5858–5868. doi: 10.1074/jbc.273.10.5858. [DOI] [PubMed] [Google Scholar]
- 2.Xie A., Odate S., Chandramouly G., Scully R. H2AX post-translational modifications in the ionizing radiation response and homologous recombination. Cell Cycle. 2010;9(17):3602–3610. doi: 10.4161/cc.9.17.12884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xie A., Hartlerode A., Stucki M., Odate S., Puget N., Kwok A., Nagaraju G., Yan C., Alt F.W., Chen J., Jackson S.P., Scully R. Distinct roles of chromatin-associated proteins MDC1 and 53BP1 in mammalian double-strand break repair. Mol. Cell. 2007;28(6):1045–1057. doi: 10.1016/j.molcel.2007.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Jiang X., Xu Y., Price B.D. Acetylation of H2AX on lysine 36 plays a key role in the DNA double-strand break repair pathway. FEBS Lett. 2010;584(13):2926–2930. doi: 10.1016/j.febslet.2010.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xiao A., Li H., Shechter D., Ahn S.H., Fabrizio L.A., Erdjument-Bromage H., Ishibe-Murakami S., Wang B., Tempst P., Hofmann K., Patel D.J., Elledge S.J., Allis C.D. WSTF regulates the H2A.X DNA damage response via a novel tyrosine kinase activity. Nature. 2009;457(7225):57–62. doi: 10.1038/nature07668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cook P.J., Ju B.G., Telese F., Wang X., Glass C.K., Rosenfeld M.G. Tyrosine dephosphorylation of H2AX modulates apoptosis and survival decisions. Nature. 2009;458(7238):591–596. doi: 10.1038/nature07849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Krishnan N., Jeong D.G., Jung S.K., Ryu S.E., Xiao A., Allis C.D., Kim S.J., Tonks N.K. Dephosphorylation of the C-terminal tyrosyl residue of the DNA damage-related histone H2A.X is mediated by the protein phosphatase eyes absent. J. Biol. Chem. 2009;284(24):16066–16070. doi: 10.1074/jbc.C900032200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Banerjee T., Chakravarti D. A peek into the complex realm of histone phosphorylation. Mol. Cell Biol. 2011;31(24):4858–4873. doi: 10.1128/MCB.05631-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li A., Yu Y., Lee S.C., Ishibashi T., Lees-Miller S.P., Ausió J. Phosphorylation of histone H2A.X by DNA-dependent protein kinase is not affected by core histone acetylation, but it alters nucleosome stability and histone H1 binding. J. Biol. Chem. 2010;285(23):17778–17788. doi: 10.1074/jbc.M110.116426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Caldwell R.B., Fiedler P., Schoetz U., Buerstedde J.M. Gene function analysis using the chicken B-cell line DT40. Methods Mol. Biol. 2007;408:193–210. doi: 10.1007/978-1-59745-547-3_11. [DOI] [PubMed] [Google Scholar]
- 11.FitzGerald J., Moureau S., Drogaris P., O’Connell E., Abshiru N., Verreault A., Thibault P., Grenon M., Lowndes N.F. Regulation of the DNA damage response and gene expression by the Dot1L histone methyltransferase and the 53Bp1 tumour suppressor. PLoS ONE. 2011;6(2):e14714. doi: 10.1371/journal.pone.0014714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sonoda E., Zhao G.Y., Kohzaki M., Dhar P.K., Kikuchi K., Redon C., Pilch D.R., Bonner W.M., Nakano A., Watanabe M., Nakayama T., Takeda S., Takami Y. Collaborative roles of gammaH2AX and the Rad51 paralog Xrcc3 in homologous recombinational repair. DNA Repair (Amst) 2007;6(3):280–292. doi: 10.1016/j.dnarep.2006.10.025. [DOI] [PubMed] [Google Scholar]
- 13.Kamentsky L., Jones T.R., Fraser A., Bray M., Logan D., Madden K., Ljosa V., Rueden C., Harris G.B., Eliceiri K., Carpenter A.E. Improved structure, function, and compatibility for CellProfiler: modular high-throughput image analysis software. Bioinformatics. 2011;27(8):1179–1180. doi: 10.1093/bioinformatics/btr095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stucki M., Clapperton J.A., Mohammad D., Yaffe M.B., Smerdon S.J., Jackson S.P. MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell. 2005;123(7):1213–1226. doi: 10.1016/j.cell.2005.09.038. [DOI] [PubMed] [Google Scholar]
- 15.Lee M.S., Edwards R.A., Thede G.L., Glover J.N. Structure of the BRCT repeat domain of MDC1 and its specificity for the free COOH-terminal end of the {gamma}-H2AX histone tail. J. Biol. Chem. 2005;280:32053–32056. doi: 10.1074/jbc.C500273200. [DOI] [PubMed] [Google Scholar]
- 16.Xu X., Stern D.F. NFBD1/MDC1 regulates ionizing radiation-induced focus formation by DNA checkpoint signaling and repair factors. FASEB J. 2003;17:1842–1848. doi: 10.1096/fj.03-0310com. [DOI] [PubMed] [Google Scholar]
- 17.Schultz L.B., Chehab N.H., Malikzay A., Halazonetis T.D. p53 binding protein 1 (53BP1) is an early participant in the cellular response to DNA double-strand breaks. J. Cell Biol. 2000;151(7):1381–1390. doi: 10.1083/jcb.151.7.1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Revet I., Feeney L., Bruguera S., Wilson W., Dong T.K., Oh D.H., Dankort D., Cleaver J.E. Functional relevance of the histone gammaH2Ax in the response to DNA damaging agents. Proc. Natl. Acad. Sci. USA. 2011;108(21):8663–8667. doi: 10.1073/pnas.1105866108. [DOI] [PMC free article] [PubMed] [Google Scholar]