

Abstract
A series of kojic acid (5-hydroxy-2-hydroxymethyl-4H-pyran-4-one) derivatives were synthesized and tested for their ability to inhibit D-amino acid oxidase (DAAO). Various substituents were incorporated into kojic acid at its 2-hydroxymethyl group. These analogs serve as useful molecular probes to explore the secondary binding site, which can be exploited in designing more potent DAAO inhibitors.
Keywords: D-amino acid oxidase, flavoenzyme, kojic acid, schizophrenia
D-Amino acid oxidase (DAAO, EC 1.4.3.3) is a flavoenzyme that catalyzes the oxidative deamination of neutral D-amino acids and produces the corresponding α-keto acids, ammonia, and hydrogen peroxide.1, 2 In humans, DAAO is predominantly expressed in the liver, kidneys, and some regions of the brain and is primarily responsible for the metabolism of neutral D-amino acids including D-serine, an endogenous agonist at the NMDA receptor glycine modulatory site. DAAO has gained substantial interest as a therapeutic target for disorders associated with NMDA receptor hypofunction such as schizophrenia3, 4 as well as neuropathic pain in which hydrogen peroxide is believed to act as a key contributor.5 Thus, substantial efforts have been made recently to identify potent and selective DAAO inhibitors as novel therapeutic agents.
Benzoic acid6 is one of the early DAAO inhibitors and has served as a useful probe to study the mechanism and physiological role of the enzyme. Indeed, the first crystal structure of human DAAO was solved as a complex with benzoic acid,7 providing critical insights into the structural details of the DAAO active site (Figure 1A). Benzoic acid binds parallel to the flavin ring on the re face of the cofactor while it interacts with the side chain of Tyr224, which stacks against the face of the benzene ring opposite to the cofactor. The benzoate carboxylate group is coplanar with the benzene ring and forms key hydrogen bonds with Arg283 and Tyr228. Although Gly313 appears to role in the binding of benzoic acid to the DAAO active site, the carbonyl group of Gly313 participates in a critical hydrogen bond with the nitrogen group of imino-DOPA generated in situ by oxidation of D-DOPA (Figure 1B).8
Figure 1.

(A) Benzoic acid (cyan) bound to the active site of DAAO (2DU8). Key residues and FAD (flavin adenine dinucleotide) are shown in green. (B) Imino-DOPA (white) bound to the active site of DAAO (2E82). Key residues and FAD are shown in orange. Tyr224 residue of 2DU8 (green) is superimposed to highlight the repositioning (red arrow) of this residue upon binding of imino-DOPA.
In addition to benzoic acid, a variety of structurally diverse DAAO inhibitors have been discovered to date (Figure 2).4 Consistent with the crystal structure of DAAO in complex with benzoic acid, the majority of DAAO inhibitors share common structural features, namely an aromatic ring system with a carboxylic acid or its bioisostere. For instance, 6-chlorobenzo[d]isoxazol-3-ol 1 (IC50 = 188 nM),9 3-hydroxyquinolin-2(1H)-one 2a (IC50 = 4 nM),10 and 3-hydroxychromen-2-one 2b (IC50 = 440 nM)11 are considered benzoic acid derivatives in which the carboxylic acid was replaced by an isoxazol-3-ol, an α-hydroxylactam, and an α-hydroxylactone, respectively. 4,6-Difluoro-1-hydroxy-1Hbenzo[d]imidazol-2(3H)-one 3 (IC50 = 80 nM) represents a newly discovered pharmacophore based on the cyclic N-hyroxyurea scaffold.12 The relatively higher potency of compound 2a can be attributed to its ability to form a hydrogen bond with the carbonyl group of Gly313 as shown by its co-crystal structure with human DAAO.10 In these series, SAR studies indicated that sterically hindered substituents are not well tolerated on the benzene ring, which is consistent with the limited space available at the active site of DAAO.
Figure 2.

Representative inhibitors of DAAO.
Fused pyrrole carboxylic acids 4a (IC50 = 141 nM) and 4b (IC50 = 245 nM),13 pyrrole-2-carboxylic acid 5a (IC50 < 10 µM),14 and 1H-pyrazole-3-carboxylic acid 5b (IC50 < 100 µM)14 represent additional classes of DAAO inhibitors for which extensive SAR studies have been conducted. It is conceivable that these compounds bind to the DAAO active site in a manner similar to that of benzoic acid with an additional interaction between the NH (of the pyrrole or pyrazole ring) and Gly313. One unique aspect of the pyrrole and pyrazole carboxylates lies in their ability to gain affinity to DAAO by incorporating a side chain. For instance, when the 4-position of 5a was substituted by a phenethyl group, IC50 value is in the submicromolar range as exemplified by 5c (IC50 < 1 µM). Similarly substituted compounds 5d also exhibited improved potency (IC50 < 1 µM) over the parent compound 5b.14
Given the limited space available at the active site on the re face of the flavin ring, the side chains of 5c and 5d likely stretch into another hydrophobic pocket adjacent to the active site in order to gain additional binding affinity. On a related note, human DAAO in complex with imino-DOPA (2E82) shows its catechol moiety taking a position nearly perpendicular to the flavin ring (Figure 1B).8 This mode of binding was enabled by the repositioning of Tyr224, which swings away from the active site. The Tyr224 shift results in the widening of the substrate entry path of DAAO by 2–3 Å compared to the 2DU8 structure and creates space for the catechol moiety of imino-DOPA.15 We hypothesized that the side chains of compounds 5c and 5d stretched into the same hydrophobic cavity occupied by the catechol moiety of imino-DOPA. This hypothesis prompted us to design new DAAO inhibitors exploiting this secondary binding site.
Kojic acid 6a, 5-hydroxy-2-(hydroxymethyl)-4H-pyran-4-one, represents another class of DAAO inhibitors with a distinct pharmacophore. Kojic acid 6a was originally identified as an inhibitor of porcine DAAO with a Ki value of 21 µM.6 Given its structural similarity to compound 2a, we speculate that the 5-hydroxy-4H-pyran-4-one moiety acts as a carboxylic acid surrogate and binds to the active site of DAAO. Indeed, as described later, 5-methoxy derivative 6b was found to be completely devoid of any inhibitory activity. However, we speculate that the 2-hydroxymethyl group of kojic acid plays an insignificant role in binding of kojic acid to DAAO and serves as an attachment point for various substituents that may allow us to perform systematic SAR studies exploring the secondary binding site. Herein, we report the synthesis and SAR of kojic acid derivatives as molecular probes for the secondary binding site of DAAO. These new DAAO inhibitors should provide new insights into the molecular features enabling the additional interaction at the secondary binding site.
As shown in Scheme 1, 2-(hydroxymethyl)-5-((4-methoxybenzyl)oxy)-4H-pyran-4-one 7 and its derivatives 10 and 11 served as starting materials16 for the synthesis of the O-alkylated derivatives of kojic acid. Reactive organic halides can be directly coupled with compound 7 through its 2-hydroxymethyl group to give compounds 8. Subsequently, the p-methoxybenzyl group of 8 can be removed by TFA to give the desired products 9. Either mesylate 10 or bromide 11 was utilized for the reaction with alcohols to obtain compounds 8, which were subsequently converted into 9. Compounds 10 and 11 were also coupled with thiols to give 12. Deprotection with TFA provided sulfide-based analogs 13.
Scheme 1.

(a) NaH, DMF, RI or RBr, 43–75%; (b) TFA, dichloromethane, rt, 4 h, 36–96%; (c) ROH, NaH, DMF, 0 °C to rt, 4 h, 26–93%; (d) RSH, NaH, DMF, 0 °C to rt, 4 h, 16–56%; (e) TFA, dichloromethane, rt, 4 h, 30–78%.
Synthesis of 5-hydroxy-2-phenethyl-4H-pyran-4-one 17 is illustrated in Scheme 2. Reaction of 2-(bromomethyl)-5-(phenylmethoxy)-4H-pyran-4-one 1416 with triethyl phosphate afforded phosphonate derivative 15. Horner-Wadsworth-Emmons reaction of 15 with benzaldehyde gave styryl derivative 16. Our attempt to remove the benzyl group and reduce the olefin in one step via catalytic hydrogenation (30 psi) resulted in the formation of multiple by-products presumably due to the concurrent reduction of the pyran ring. Thus, the desired product 17 was obtained via acid-mediated removal of the benzyl group followed by catalytic hydrogenation at atmospheric pressure. We also synthesized 3-hydroxy-1-phenethyl-4(1H)-pyridinone 19 by reacting 3-(phenylmethoxy)-4H-pyran-4-one 1817 with phenethylamine followed by catalytic hydrogenation (Scheme 2).
Scheme 2.

(a) P(OMe)3, benzene, reflux, 24h, 90%; (b) NaH, THF, 0 C, then benzaldehyde, 12h, 0 C to rt, 66%; (c) 6N HCl/HOAc, TFA, reflux, 48h then H2 (1 atm), Pd/C (5%), rt, 1h, 50%; (d) MeOH, phenethylamine, 72h, rt then H2 (1 atm), 5% Pd/C (5%), EtOAc, 1h, 50%.
Inhibitory potencies of the kojic acid derivatives were measured using recombinant human DAAO as previously reported.9 Table 1 summarizes the in vitro DAAO inhibitory data.
Table 1.
Inhibition of human DAAO by kojic acid derivatives
| Compd | Structure | IC50 (µM)a |
|---|---|---|
| 6a | 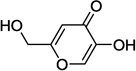 |
2 |
| 6b | 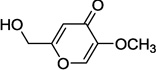 |
>100 |
| 9a | 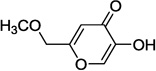 |
50 |
| 9b | 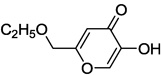 |
>100 |
| 9c | 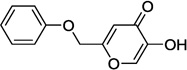 |
0.9 |
| 9d | 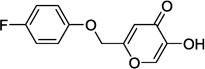 |
0.8 |
| 9e | 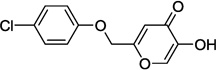 |
0.7 |
| 9f | 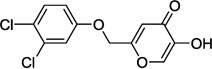 |
0.5 |
| 9g | 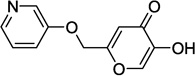 |
3 |
| 9h | 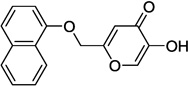 |
0.9 |
| 9i | 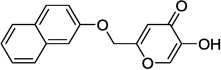 |
0.8 |
| 9j | 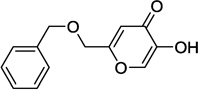 |
20 |
| 9k | 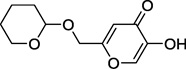 |
>100 |
Assay methods are described in the Supporting Information.
In our assay, kojic acid 6a inhibited human DAAO with an IC50 value of 2.0 µM. As mentioned earlier, methylation of the 5-hydroxy group of kojic acid resulted in complete loss of potency as in compound 6b presumably due to its inability to form hydrogen bonds with Tyr228 and Arg283. 2-Alkoxymethyl derivatives of kojic acid exhibited a wide range of DAAO inhibitory potencies (compounds 9a-9k) depending on the substituent. Saturated groups were ineffective at this position and compounds 9a, 9b, and 9k were significantly less potent than kojic acid. On the other hand, O-phenyl derivative 9c and its close analogs (compounds 9d-9i) exhibited potency comparable or superior to that of kojic acid. Insertion of one methylene unit, however, resulted in dramatic loss of potency as in compound 9j. These findings indicate that the secondary binding pocket has a preference for an aromatic side chain containing a two-atom linker to the core pyran ring.
Subsequently, we examined compounds containing other two-atom linkers (Table 2). Sulfide-based inhibitors 13a-13d were found to be slightly more potent than the corresponding ether derivatives. Among them, 13d was the most potent DAAO inhibitor within the kojic acid series with an IC50 value of 100 nM. Phenethyl derivative 17 was slightly more potent than the ether counterpart 9c. Interestingly, 4(1H)-pyridinone-based derivative 19 containing a phenethyl branch exhibited no inhibitory activity, underscoring the unique interaction formed by the 5-hydroxy-4H-pyran-4-one pharmacophore with the key residues of the DAAO active site.
Table 2.
Inhibition of human DAAO by kojic acid derivatives
| Compd | Structure | IC50 (µM)a |
|---|---|---|
| 13a | 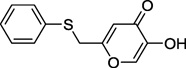 |
0.2 |
| 13b | 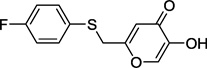 |
0.3 |
| 13c | 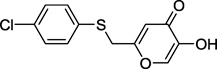 |
0.2 |
| 13d | 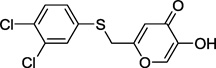 |
0.1 |
| 13e | 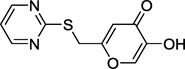 |
4 |
| 17 | 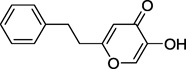 |
0.5 |
| 19 | 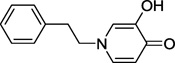 |
>100 |
Assay methods are described in the Supporting Information.
An attempt to dock compound 13d to the active site of DAAO (Figure 3) was only successful with the crystal structure containing the repositioned Tyr224 residue (2E82). The inability of compound 13d to fit the active site of 2DU8, in which Tyr224 is obstructing the secondary binding site, also supports the hypothesis that the phenylthiomethyl group of 13d is occupying the space created by the Tyr224 movement. Surprisingly, recent efforts to screen 4 million commercially available compounds in silico did not identify any DAAO inhibitors extending to the secondary binding site.11 This could be at least partially due to the concentrated use of the 2DU8 structure and/or docking grid box that failed to cover the entire secondary binding site.
Figure 3.

Proposed binding mode of 13d (white) to the active site of DAAO (2E82). Key residues and FAD are shown in green. Hydrogen-bonding interactions between 13d and the key residues are shown as gray dashed lines. Imono-DOPA (cyan) of 2E82 is superimposed for comparison.
The lack of a nitrogen atom in the core ring of kojic acid precludes hydrogen bonding with the Gly313 residue, which explains the relatively weak inhibitory potency of the kojic acid-based inhibitors compared to the series represented by compounds 2 and 3. Indeed, it was recently reported that substituted derivatives of 3-hydroxy-pyridine-2(1H)-one and 3-hydroxy-pyridazine-4(1H)-one can potently inhibit DAAO.18–20 These compounds are presumably gaining increased affinity through hydrogen bonding to the Gly313 residue in addition to the interaction with the secondary binding site.
In summary, we tested a series of kojic acid derivatives for their ability to inhibit human DAAO. These compounds likely occupy both the active site and the secondary binding site adjacent to the active site. Since the secondary binding site is a part of the funnel-shaped entrance to the active site, further structural optimization exploring a wide variety of substituents may lead to DAAO inhibitors with greater structural diversity. Furthermore, the secondary binding site can be exploited by other series of DAAO inhibitors, particularly those capable of interacting with the Gly313 residue of the active site to maximize interaction with each of the two binding sites.
Supplementary Material
Acknowledgments
This work was in part supported by National Institutes of Health (R01MH091387 to T.T.) and the Johns Hopkins Brain Science Institute Neuro Translational Drug Discovery program.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary Material
Supplementary data associated with this article can be found, in the online version, at doi####.
References and notes
- 1.Dixon M, Kleppe K. Biochim. Biophys. Acta. 1965;96:357. doi: 10.1016/0005-2787(65)90556-3. [DOI] [PubMed] [Google Scholar]
- 2.Dixon M, Kleppe K. Biochim. Biophys. Acta. 1965;96:368. doi: 10.1016/0005-2787(65)90558-7. [DOI] [PubMed] [Google Scholar]
- 3.Smith SM, Uslaner JM, Hutson PH. Open Med. Chem. J. 2010;4:3. doi: 10.2174/1874104501004020003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ferraris DV, Tsukamoto T. Curr. Pharm. Des. 2011;17:103. doi: 10.2174/138161211795049633. [DOI] [PubMed] [Google Scholar]
- 5.Lu JM, Gong N, Wang YC, Wang YX. Br. J. Pharmacol. 2011;165:1941. doi: 10.1111/j.1476-5381.2011.01680.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Klein JR. J. Biol. Chem. 1953;205:725. [PubMed] [Google Scholar]
- 7.Kawazoe T, Tsuge H, Pilone MS, Fukui K. Protein Sci. 2006;15:2708. doi: 10.1110/ps.062421606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kawazoe T, Tsuge H, Imagawa T, Aki K, Kuramitsu S, Fukui K. Biochem. Biophys. Res. Commun. 2007;355:385. doi: 10.1016/j.bbrc.2007.01.181. [DOI] [PubMed] [Google Scholar]
- 9.Ferraris D, Duvall B, Ko YS, Thomas AG, Rojas C, Majer P, Hashimoto K, Tsukamoto T. J. Med. Chem. 2008;51:3357. doi: 10.1021/jm800200u. [DOI] [PubMed] [Google Scholar]
- 10.Duplantier AJ, Becker SL, Bohanon MJ, Borzilleri KA, Chrunyk BA, Downs JT, Hu LY, El-Kattan A, James LC, Liu S, Lu J, Maklad N, Mansour MN, Mente S, Piotrowski MA, Sakya SM, Sheehan S, Steyn SJ, Strick CA, Williams VA, Zhang L. J. Med. Chem. 2009;52:3576. doi: 10.1021/jm900128w. [DOI] [PubMed] [Google Scholar]
- 11.Katane M, Osaka N, Matsuda S, Maeda K, Kawata T, Saitoh Y, Sekine M, Furuchi T, Doi I, Hirono S, Homma H. J. Med. Chem. 2013;56:1894. doi: 10.1021/jm3017865. [DOI] [PubMed] [Google Scholar]
- 12.Berry JF, Ferraris DV, Duvall B, Hin N, Rais R, Alt J, Thomas AG, Rojas C, Hashimoto K, Slusher BS, Tsukamoto T. ACS Med. Chem. Lett. 2012;3:839. doi: 10.1021/ml300212a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sparey T, Abeywickrema P, Almond S, Brandon N, Byrne N, Campbell A, Hutson PH, Jacobson M, Jones B, Munshi S, Pascarella D, Pike A, Prasad GS, Sachs N, Sakatis M, Sardana V, Venkatraman S, Young MB. Bioorg. Med. Chem. Lett. 2008;18:3386. doi: 10.1016/j.bmcl.2008.04.020. [DOI] [PubMed] [Google Scholar]
- 14.Fang KQ, Hopkins S, Heffernan M, Chytil M. 7,488,747 U.S. Patent. 2009
- 15. The repositioning of Tyr224 was also observed with other types of DAAO inhibitors (see ref 18).
- 16.Chen Y-H, Lu P-J, Hulme C, Shaw A. Med. Chem. Res. 2013;22:995. [Google Scholar]
- 17.Ozturk G, Erol DD, Uzbay T, Aytemir MD. Farmaco. 2001;56:251. doi: 10.1016/s0014-827x(01)01083-7. [DOI] [PubMed] [Google Scholar]
- 18.Hondo T, Warizaya M, Niimi T, Namatame I, Yamaguchi T, Nakanishi K, Hamajima T, Harada K, Sakashita H, Matsumoto Y, Orita M, Watanabe T, Takeuchi M. MEDI 98; Abstracts of Papers, 244th National Meeting of the American Chemical Society; Philadelphia, PA. Washington, DC: American Chemical Society; 2012. [Google Scholar]
- 19.Farnaby W, Fieldhouse C, Kerr C, Kinsella N, Livermore D, Merchant K, Miller D, Hazel K. PCT Int. Appl. WO 2013004996
- 20.Farnaby W, Fieldhouse C, Hazel K, Kerr C, Kinsella N, Livermore D, Merchant K, Miller D. PCT Int. Appl. WO 2013027000
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.