

Abstract
Mechanical insult to articular cartilage kills chondrocytes, an event that may increase the risk of post-traumatic osteoarthritis. Recent reports indicate that antioxidants decrease impact induced chondrocyte death, but the source(s) of oxidants, the time course of oxidant release, and the identity of the oxidative species generated in response to injury are unknown. A better understanding of these processes could lead to new treatments of acute joint injuries. To that end we studied the kinetics and distribution of oxidant production in osteochondral explants subjected to a single blunt impact injury. We followed superoxide production by measuring the time-dependent accumulation of chondrocyte nuclei stained with the superoxide-sensitive probe dihydroethidium. The percentage of chondrocytes that were dihydroethidium-positive was 35% above baseline 10 minutes after impact and 65% above baseline 60 minutes after impact. Most positive cells were found within and near areas contacted directly by the impact platen. Rotenone, an electron transport chain inhibitor, was used to test the hypothesis that mitochondria contribute to superoxide release. Rotenone treatment significantly reduced dihydroethidium staining, which remained steady at 15% above baseline for up to 60 minutes post-impact. Moreover, rotenone reduced chondrocyte death in impact sites by more than 40% even when administered two hours after injury (p < 0.001). These data show that much of the acute chondrocyte mortality caused by in vitro impact injuries results from superoxide release from mitochondria and suggest that brief exposure to free radical scavengers could significantly improve chondrocyte viability following joint injury.
Keywords: cartilage injury, mitochondria, viability
INTRODUCTION
Post-traumatic osteoarthritis (PTOA) is a common sequel to intra-articular fracture and other joint injuries 1,2. Despite advances in the evaluation and treatment of joint injuries over the last 20 years, the risk for PTOA following ligamentous tears and articular surface fractures has not decreased appreciably, underscoring the need for better understanding of the mechanisms responsible for joint degeneration following injury. Injurious mechanical stresses are known to play a role in the overall risk for degenerative joint disease and mechanical injury is a significant pathogenic factor in PTOA 3-5. In particular, damage to the cartilage extracellular matrix and cell death caused by high-energy impact loading of joint surfaces are believed to contribute to a degenerative cascade that progresses to end stage osteoarthritis 6.
Both necrosis and apoptosis occur in response to impact loads 6-9. Z-VAD-FMK, a cell-permeable fluoromethylketone peptide caspase inhibitor, and P188, a membrane-stabilizing surfactant, reduce impact related death 6,8,9. Caspase inhibition has also been shown to reduce the severity of osteoarthritis in vivo 6. Recent work from our laboratory showed that impact-induced chondrocyte death in a bovine explant model was blocked by treatment with the intermediate free-radical scavenger n-acetyl cysteine (NAC) 10. NAC is a powerful scavenger of highly damaging hydroxyl and hypochlorous radicals, which are formed in cells following exposure to superoxide 11. NAC applied within 2 hours of impact spared more than 75% of cells that would have otherwise died within 72 hours injury. Moreover, impact-related decline in proteoglycan content 7 or 14 days after injury was blocked by early post-impact NAC treatment. These results were consistent with the findings of Kurz et al, which showed similar effects on post-impact viability using a superoxide dismutase (SOD) mimetic 12. Taken together these studies demonstrate that the majority of acute chondrocyte death is preventable and that preserving cellularity improves matrix stability.
The beneficial effects of NAC and SOD on viability strongly implicate reactive oxygen species (ROS) in the lethal effects of impact injury. However, the sources of ROS in this context are unclear. Chondrocytes express both NADPH-oxidase (NOX) 13-15, and inducible nitric oxide synthase (iNOS) 16, enzymes that generate superoxide (O2.−) and nitric oxide (NO.) respectively in response to a variety of biochemical mediators 17-23. Increased NO and O production driven by cytokines can suppress mitochondrial respiration, promote chondrocyte apoptosis, and is associated with osteoarthritis in vivo 24-27. Mitochondria also represent a potential source of injury-related ROS in cartilage. Increased free radical production is associated with direct oxidative damage to mitochondrial electron transport complexes or to the DNA that encodes them 5,28,29. Excessive free radical production is characteristic of chondrocytes in osteoarthritic cartilage, which are also deficient in respiratory activity 28,30. A study of isolated chondrocytes has shown that mechanical deformation of the cytoplasm by pipette aspiration deforms the ER and mitochondria and triggers intracellular calcium release 31. Recent evidence from an explant injury model indicated that increased cytoplasmic calcium following blunt impact injury to cartilage induces permeability transition pore formation in the inner mitochondrial membrane. The resulting depolarization was associated with cytochrome C release, Bcl-2 degradation, and caspase-dependent apoptosis 32. Chondrocyte apoptosis was inhibited by blocking the increase in cytoplasmic calcium or by blocking permeability transition pore (PTP) formation. These results show the central role of calcium in chondrocyte responses to mechanical trauma and provide evidence that mitochondria-dependent apoptosis is a significant consequence of the disruption of calcium homeostasis.
In vitro experiments showing that antioxidant treatment improves chondrocyte viability in impacted cartilage suggest that free radicals are released in response to such injuries. Further investigation of the mechanisms of free-radical release is needed to specifically target those pathways associated with acute trauma-related chondrocyte death. We hypothesized that superoxide released by damaged components of the mitochondrial electron transport chain causes a significant fraction of impact-induced chondrocyte death. To test this hypothesis we treated osteochondral samples before and after impact with rotenone, an agent that suppresses the release of superoxide from mitochondria by binding to the ubiquinol binding site in respiratory complex 1, thereby blocking the flow of electrons from complex I to complex III. We then measured the effects of rotenone on superoxide release and chondrocyte viability.
METHODS
Mature bovine stifle joints were obtained after slaughter from a local abattoir (Bud’s Custom Meats, Riverside, IA). Osteochondral explants were prepared by manually sawing a 25 mm by 25 mm square from the lateral tibial plateau, which included the central loaded area of the articular surface that was not covered by menisci. The explants were placed in culture medium containing 45% DMEM, 45% Ham’s F-12, and 10% fetal bovine serum (Invitrogen, Carlsbad, CA) and incubated at 37°C in an atmosphere of 5% CO2 in air.
Twenty four hours after harvest, osteochondral explants were secured in custom testing fixtures for impact loading and were kept submerged in culture medium at all times. A drop tower was used to impart loads to an indenter resting on the explant surface. The indenter was a flat-faced 5.0 mm diameter brass rod with rounded edges (r = 1 mm). Impact energy was modulated by dropping a 2 kg mass from a height of 7 cm resulting in an impact energy density of 7 J/cm2 and peak stresses in excess of 20 MPa, imposed at a rate of greater than 1000 MPa/sec. The mass was removed from the platen immediately after impact.
To study superoxide production explants were placed in phenol red-free culture medium (10% FBS, DMEM, F12) containing 5μM dihydroethidium (DHE) and 1 mM calcein AM at various time points after impact (1 hour, 3 hours, 6 hours, 24 hours, and 48 hours). The Invitrogen stained explants were imaged on a BioRad 1024 Confocal Microscope equipped with a Krypton/Argon laser (Bio-Rad Laboratories Inc., Hercules, CA). The sites were scanned to a depth of 150 μM at 20 μm intervals using wavelengths of 568 nm and 488 nm and a 10× objective with a field size of ~ 1.0 mm2. Z-axis projections of confocal images were analyzed using Image J (rsb.info.nih.gov/ij), a Java-based public domain image analysis program, to determine the average percentage of DHE-stained cells at each time point. Three sites within the impact site and 3 sites ~0.5 cm away from the impact site were imaged (Figure 1A). Four explants were used for each time point.
Figure 1.

Experimental Design. All explants (n= 44) were harvested and cultured for 2 days prior to starting the experiments. (A) The time course of oxidant production: DHE staining was measured in impact and control sites at various times after impact injury (1, 3, 6, 24, 48 hours). Four different explants were used for each time point (n =20). (B) The effects of rotenone on oxidant production: DHE staining was repeatedly measured in the same explants during the first hour after impact. The 4 groups included 2 rotenone treatments, one in which treatment was initiated before impact and carried on throughout the 60 minute imaging session (Group 1) and another in which treatment was delayed until 30 minutes post-impact (Group 2). Controls for the study included a set of explants that were impacted but not treated with rotenone (Group 3) and a set that was not impacted and not treated (Group 4). Each group consisted of 3 explants (n = 12) (C) Effects of rotenone on viability: Impact sites and control sites on the same explants were imaged in explants stained for viability (Calcein AM/ethidium homodimer) 24 hours after impact. A control group (no rotenone) and 3 rotenone treatment groups were included. The treatment groups differed with respect to the timing of rotenone dosing relative to impaction. 3 explants were used for each group.
To study the effect of rotenone on superoxide production one group of explants was treated with 2.5 μM rotenone (Sigma Aldrich, St. Louis, MO) starting 1 hour before impact and continuing for1 hour post-impact during imaging sessions. Another group of explants was dosed with rotenone 30 minutes post-impact to evaluate the effect of delaying treatment. A third group was impacted but not treated with rotenone and a fourth group was neither impacted nor treated with rotenone (Figure 1B). Impact sites and sites approximately 1 cm away (control) were imaged and analyzed as described above. Three sites within impact sites and three sites outside impact sites were imaged for each explant and 3 explants were analyzed for each group. Impact sites were imaged a final time at 70 minutes post-impact using a 4× objective to record the spatial distribution of staining on the explant surface.
Effects of impact and rotenone on chondrocyte viability were assessed 24 hours after impact, a time when impact-induced chondrocyte death was previously shown to reach a steady state 33. One group of explants was treated with 2.5 μM rotenone for 2 hours before and 2 hours after impact, a second group for 1 hour before and 1 hour after impact, a third group for 1 hour starting immediately after impact. A fourth group went untreated (Figure 1C). Calcein AM (1.0 mM) was used to stain viable cells and ethidium homdimer-2 (1.0 mM) was used to stain dead cells (Invitrogen). Explants were scanned to a depth of 200 μm at 20 μm intervals as described above and the images analyzed using Image J to determine percent viability. Three different projections were recorded within each impact or non-impact control site. Three explants were used for each treatment group.
One-way ANOVA with a post hoc Holm-Sidek correction for multiple comparisons was used to compare treatment groups. A p-value less than 0.05 was considered significant.
RESULTS
The acute physical effects of injury on cartilage were examined by histology at 24 hours after impact (Figure 2). Safranin-O/fast green-stained saggital sections through impact sites and surrounding cartilage showed surface disruption and clefts extending 100-200 μm from the surface downward through the superficial zone and into the transitional zone (Figure 2 A, B).
Figure 2.
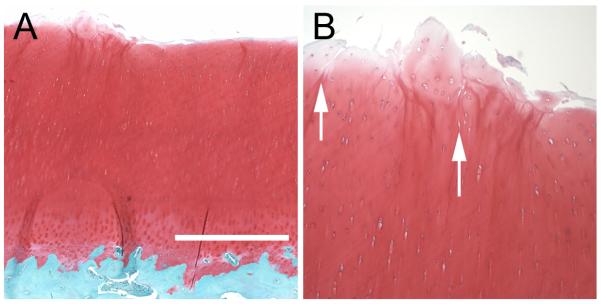
Structural Damage Caused by Impact. (A) A safranin-O and fast green stained sagittal section shows disruption of the cartilage surface near the border of an impact site. (B) Closer view of the surface of the section in A shows vertical clefts (indicated by arrows) extending through the superficial zone and into the transitional zone. The bar in A is 0.6 mm in length.
The effects of impact on superoxide production by chondrocytes were studied using confocal microscopy. Images of explant surfaces taken at low magnification (4× objective) revealed that DHE-positive chondrocytes were concentrated in a circle of ~5 mm diameter apparently corresponding to the contact area of the impact platen (Figure 3A,B). The highest density of positive cells was near the edge of injured areas, often in association with overt physical damage to the matrix. The addition of rotenone to the culture medium just after impact substantially reduced staining across such sites (Figure 3C). Higher magnification images from near the center of impact sites confirmed that rotenone treatment reduced the number of DHE-stained cells while increasing the number of calcein AM-stained (viable) cells (Figure 3D,E). These images also showed that few cells were double stained, indicating that superoxide production was associated with loss of viability. Pilot studies of “sham” impacts with the indentor in contact with the explant surface but without impact loading revealed no significant effect on DHE staining or chondrocyte viability.
Figure 3.

Injury and Rotenone effects on Superoxide Production. (A) The diagram represents a typical osteochondral explant (2.5 cm × 2.5 cm square) under laser illumination on the scanning confocal microscope. A 5 mm diameter impact site (70 minutes post-impact) is shown on the surface of the explant (white circle). A z-axis projection (0-200 μm-deep) of a low magnification scan (4× objective) covering the border of an impact site is shown on the right. The field subtends ~ 1/4 of the impact site area. Low magnification z-projected images show DHE (red) and calcein AM (green) staining in an impact site on an untreated explant (B) and on an explant treated with rotenone (C). The bar in B represents 1 mm. Panels D and E show higher magnification images (20× objective) from within the sites shown in B and C. The bar in E is 150 μm long.
Repeated confocal imaging sessions of DHE stained explants were performed at 5, 10, 20, 30, 45, and 60 minutes post-injury to determine the kinetics of oxidant release (Figure 4). The time of onset of DHE staining was variable, but once cells were positive they tended to remain so for the duration of the 60 minute time course. Positive cell counts increased significantly from 30% at 5 minutes post-injury to 65% at 60 minutes post-injury. Counts over the same time interval in non-impacted control cartilage were always less than 10%. The effect of immediate post-impact rotenone treatment on DHE staining kinetics was determined (Figure 5A). Staining in treated explants increased from 20% positive at 10 minutes post injury to 24% positive at 60 minutes post injury, a change that was not significant. Counts in the rotenone-treated group were significantly lower than in the untreated group at all time points (p < 0.02). Delaying rotenone treatment until 30 minutes after impact did not significantly reduce positive cells at later times. Imaging sessions conducted at later post-impact time points revealed that the percentage of DHE positives, which was near 50% at 1, 3, and 6 hours, declined significantly to 19% after 24 hours and did not change significantly from 24 to 48 hours (Figure 5B). Staining was significantly greater in impact sites than in control sites at 1, 3 and 6 hours, but not at 24 or 48 hours. The percentage of dead cells at these times post impact determined in a previous study rose steadily from 0-6 hours, but increased at a lower rate thereafter.
Figure 4.

Time Lapse Imaging of Dihydroethidium Staining at an Impact Site. Panels A-F show a single 1.2 × 1.2 mm field imaged at 5, 10, 20, 30, 45 and 60 minutes after injury. The insets in the bottom right corner are enlargements from each image showing the same small group cells, all of which remained positive until the end of the experiment.
Figure 5.

Time Course of Impact-Induced Superoxide Production. (A)The percentage of dihydroethidium-stained chondrocytes (% DHE Positive) measured repeatedly in the first 60 minutes post-impact in the same explants. Closed squares show results for impact sites in untreated explants. Closed triangles represent impact sites in explants treated with rotenone starting 1 hour before impact and during the imaging session. Open circles show data for impact sites in explants that were treated with rotenone starting at 30 minutes post-impact. Open triangles show results for untreated, non-impacted controls. Error bars indicate standard deviations based on 3 explants per group. (B) DHE positive staining at impact and control sites at the indicated times post-impact (1, 3, 6, 24, 48 hours). Asterisks indicate significant differences between impact sites and control sites. Error bars show standard deviations based on 4 explants per time point. Chondrocyte death rates over this time interval are shown in the line/symbol plot overlying the column plot.
Chondrocyte viability 24 hours after impact was measured by confocal microscopy (Figure 6). Rotenone treatment reduced chondrocyte death at impact sites by more than 40%, a highly significant effect (p < 0.001). Results were similar for explants treated 2 hours before and after impact, 1 hour before and after impact, or 2 hours after impact. In all groups (rotenone and untreated) viability in non-impacted control cartilage was near 87 +/− 8%.
Figure 6.

Rotenone Effects on Impact Site Viability. Chondrocyte viabilities are shown for explants that were treated with rotenone for varying times before and after impact. Black columns show results for impact sites and white columns for non-impact sites. Treatment times (indicated below the X axis) were 2 hours before and after impact (2/2), 1 hour before and after impact (1/1), or 2 hours after impact (0/2). Results for untreated controls (0/0) are included for reference. Columns and error bars show means and standard deviations based on 3 explants. Asterisks indicate significant differences between impact sites that were rotenone-treated and those that were not treated. (p < 0.001).
DISCUSSION
Our results show that in vitro blunt trauma to cartilage results in the acute release of superoxide radicals. Rotenone significantly inhibited superoxide at impact sites, implicating mitochondria as a major source of production. The strong positive effect of rotenone on chondrocyte viability indicates that oxidative damage from mitochondrial superoxide contributed substantially to impact-related cell death. These findings suggest that mitochondrial dysfunction could be responsible for chondrocyte mortality in joint injuries that involve blunt trauma to articular surfaces.
Superoxide release began within 5 minutes of impact and remained limited to cartilage in and around the periphery of the contact area of the impact platen. We did not formally study the spread of distances from the indentor edge, but low magnification images taken as late as 24 hours after impact show relatively crisp borders at the impact site and little spreading of oxidant production beyond the distance observed at 1 hour post-impact. Thus, it appears that superoxide release from chondrocytes was a direct effect of impact loading which causes axial strains of up to 40% in the superficial zone 7.
Time course studies revealed complex post-impact kinetics of superoxide production. More than half the increase in DHE staining seen over the entire 60 minute follow-up was already evident at 10 minutes post-impact, the earliest time point we were able to observe. After this steep initial rise, positive cells increased at a slow linear rate from 20 to 60 minutes. Interestingly, the late addition of rotenone at 30 minutes post-impact reduced the rate of DHE positive accumulation from 40-60 minutes, indicating that delayed treatment might still have beneficial effects. Longer term follow-up showed that the percentage of positive cells remained high from 1 to 6 hours post-impact, but declined significantly by 24 hours and remained relatively low at 48 hours. These results were consistent with the kinetics of cell death in this model, which rose continuously starting immediately post-impact and peaked at 6-12 hours 33.
Some 20% of chondrocytes at impact sites produced superoxide in the presence of rotenone. Although the reason for this is unclear it is possible that NOX, a rotenone-resistant enzyme, contributed to superoxide release at impact sites. Testing the combined effects of NOX and mitochondrial electron transport inhibitors on superoxide release should help to clarify this issue. Alternatively, cells killed outright by impact injury may have released oxidants from peroxisomes and other intracellular compartments as the cells disintegrated, a process that would not be blocked by NOX or mitochondrial electron transport activity inhibitors. This would be consistent with previously published reports indicating that up to 15% of chondrocytes in impacted cartilage die by necrosis within minutes of injury 8,9. Although increased death was noted at impact sites, dead cells were not distributed evenly under the indenter and tended to concentrate at the edges, particularly in association with clefts in the matrix. It is reasonable to suppose that the clefting (typically concentrated at the edge of the indentor) represented structural failure due to excessive shear stresses. Different shaped indentors (e.g. with more rounded edges) would likely change the pattern of structural failure.
Rotenone was used to test the hypothesis that the mitochondrial electron transport chain is a source of oxidants in our model system. Although the drug had substantial positive effects on chondrocyte viability, its toxicity to neurons and other cells makes it unsuitable for in vivo use. In contrast, free radical scavengers such as n-acetyl cysteine are minimally cytotoxic and, when used to treat impacted explants, show chondrocyte-sparing effects comparable to those of rotenone 10,34. N-acetyl cysteine treatment not only improved viability, but prevented impact-related proteoglycan loss. These data, taken together with the findings of the current study, show that blocking acute oxidative damage from superoxide can promote cartilage stability.
The results of our in vitro study support the hypothesis that the timely post-injury application of free radical scavengers can prevent much of the acute cell death that may contribute to post-traumatic osteoarthritis in injured joints. However, the pathogenic significance of acute chondrocyte death is uncertain given that post-traumatic osteoarthritis is a complex, multifactorial process that occurs over time frames of up to several years. Thus, testing chondrocyte-sparing interventions in in vivo joint injury models will be required to evaluate their therapeutic potential.
AKNOWLEDGMENTS
This work was supported by US DHHS, National Institutes of Health/NIAMS grant #1 P50 AR055533 and by a Merit Review Award from the Department of Veterans Affaires
REFERENCES
- 1.Buckwalter JA, Brown TD. Joint injury, repair, and remodeling: roles in post-traumatic osteoarthritis. Clin Orthop Relat Res. 2004;423:7–16. [PubMed] [Google Scholar]
- 2.Brown TD, Johnston RC, Saltzman CL, et al. Posttraumatic osteoarthritis: a first estimate of incidence, prevalence, and burden of disease. J Orthop Trauma. 2006;20:739–744. doi: 10.1097/01.bot.0000246468.80635.ef. [DOI] [PubMed] [Google Scholar]
- 3.Buckwalter JA. Articular cartilage injuries. Clin Orthop Relat Res. 2002;402:21–37. doi: 10.1097/00003086-200209000-00004. [DOI] [PubMed] [Google Scholar]
- 4.Martin JA, Buckwalter JA. Aging, articular cartilage chondrocyte senescence and osteoarthritis. Biogerontology. 2002;3:257–264. doi: 10.1023/a:1020185404126. [DOI] [PubMed] [Google Scholar]
- 5.Martin JA, Klingelhutz AJ, Moussavi-Harami F, Buckwalter JA. Effects of oxidative damage and telomerase activity on human articular cartilage chondrocyte senescence. J Gerontol A Biol Sci Med Sci. 2004;59:324–337. doi: 10.1093/gerona/59.4.b324. [DOI] [PubMed] [Google Scholar]
- 6.D’Lima D, Hermida J, Hashimoto S, et al. Caspase inhibitors reduce severity of cartilage lesions in experimental osteoarthritis. Arthritis Rheum. 2006;54:1814–1821. doi: 10.1002/art.21874. [DOI] [PubMed] [Google Scholar]
- 7.Jeffrey J, Gregory D, Aspden R. Matrix damage and chondrocyte viability following a single impact load on articular cartilage. Arch Biochem Biophys. 1995;322:87–96. doi: 10.1006/abbi.1995.1439. [DOI] [PubMed] [Google Scholar]
- 8.Phillips D, Haut R. The use of non-ionic surfactant (P188) to save chondrocytes from necrosis following impact loading of chondral explants. J Orthop Res. 2004;22:1135–1142. doi: 10.1016/j.orthres.2004.02.002. [DOI] [PubMed] [Google Scholar]
- 9.Rundell SA, Baars DC, Phillips DM, Haut RC. The limitation of acute necrosis in retro-patellar cartilage after a severe blunt impact to the in vivo rabbit patello-femoral joint. J Orthop Res. 2005;23:1363–1369. doi: 10.1016/j.orthres.2005.06.001.1100230618. [DOI] [PubMed] [Google Scholar]
- 10.Martin J, McCabe D, Walter M, et al. N-Acetylcysteine Inhibits Post-Impact Chondrocyte Death in Osteochondral Explants. J Bone Joint Surg Am. 2009 doi: 10.2106/JBJS.H.00545. Accepted. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zafarullah M, Li WQ, Sylvester J, Ahmad M. Molecular mechanisms of N-acetylcysteine actions. Cell Mol Life Sci. 2003;60:6–20. doi: 10.1007/s000180300001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kurz B, Lemke A, Kehn M, et al. Influence of tissue maturation and antioxidants on the apoptotic response of articular cartilage after injurious compression. Arthritis Rheum. 2004;50:123–130. doi: 10.1002/art.11438. [DOI] [PubMed] [Google Scholar]
- 13.Grange L, Nguyen MV, Lardy B, et al. NAD(P)H oxidase activity of Nox4 in chondrocytes is both inducible and involved in collagenase expression. Antioxid Redox Signal. 2006;8:1485–1496. doi: 10.1089/ars.2006.8.1485. [DOI] [PubMed] [Google Scholar]
- 14.Bedard K, Krause KH. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiol Rev. 2007;87:245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 15.Hiran TS, Moulton PJ, Hancock JT. In situ detection of superoxide anions within porcine articular cartilage. Br J Biomed Sci. 1998;55:199–203. [PubMed] [Google Scholar]
- 16.Maier R, Bilbe G, Rediske J, Lotz M. Inducible nitric oxide synthase from human articular chondrocytes: cDNA cloning and analysis of mRNA expression. Biochim Biophys Acta. 1994;1208:145–150. doi: 10.1016/0167-4838(94)90171-6. [DOI] [PubMed] [Google Scholar]
- 17.Hancock J, Desikan R, Neill S. Role of reactive oxygen species in cell signalling pathways. Biochem Soc Trans. 2001;29:345–350. doi: 10.1042/0300-5127:0290345. [DOI] [PubMed] [Google Scholar]
- 18.Henrotin Y, Kurz B, Aigner T. Oxygen and reactive oxygen species in cartilage degradation: friends or foes? Osteoarthritis Cartilage. 2005;13:643–654. doi: 10.1016/j.joca.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 19.Henrotin YE, Bruckner P, Pujol JP. The role of reactive oxygen species in homeostasis and degradation of cartilage. Osteoarthritis Cartilage. 2003;11:747–755. doi: 10.1016/s1063-4584(03)00150-x. [DOI] [PubMed] [Google Scholar]
- 20.Krenn V, Molitoris R, Berek C, et al. A novel monospecific IgG2/lambda-autoantibody with specificity for a mitochondrial antigen: evidence for an antigen-driven pathogenetic B-cell response in rheumatoid synovial tissue, induced by tissue alteration. Lab Invest. 1998;78:485–496. [PubMed] [Google Scholar]
- 21.Lo Y, Conquer J, Grinstein S, Cruz T. Interleukin-1 beta induction of c-fos and collagenase expression in articular chondrocyte involvement of reactive oxygen species. J Cell Biochem. 1998;69:19–29. doi: 10.1002/(sici)1097-4644(19980401)69:1<19::aid-jcb3>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 22.Lo Y, Wong J, Cruz T. Reactive oxygen species mediate cytokine activation of c-Jun NH2-terminal kinases. J BIol Chem. 1996;271:15703–15707. doi: 10.1074/jbc.271.26.15703. [DOI] [PubMed] [Google Scholar]
- 23.Oh M, Fukuda K, Asada S, et al. Concurrent generation of nitric oxide and superoxide inhibits proteoglycan synthesis in bovine articular chondrocytes: involvement of peroxynitrite. J Rheumatol. 1998;25:2169–2174. [PubMed] [Google Scholar]
- 24.Lotz M. The role of nitric oxide in articular cartilage damage. Rheum Dis Clin North Am. 1999;25:269–282. doi: 10.1016/s0889-857x(05)70067-3. [DOI] [PubMed] [Google Scholar]
- 25.Pelletier J, Lascau-Coman V, Jovanovic D, et al. Selective inhibition of inducible nitric oxide synthase in experimental osteoarthritis is associated with reduction in tissue levels of catabolic factors. J Rheumatol. 1999;26:2002–2014. [PubMed] [Google Scholar]
- 26.Tomita M, Sato E, Nishikawa M, et al. Nitric oxide regulates mitochondrial respiration and functions of articular chondrocytes. Arthiritis Rheum. 2001;44:96–104. doi: 10.1002/1529-0131(200101)44:1<96::AID-ANR13>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 27.Golden T, Melov S. Mitochondrial DNA mutations, oxidative stress, and aging. Mech Ageing Dev. 2001;122:1577–1589. doi: 10.1016/s0047-6374(01)00288-3. [DOI] [PubMed] [Google Scholar]
- 28.Terkeltaub R, Johnson K, Murphy A, Ghosh S. Invited review: the mitochondrion in osteoarthritis. Mitochondrion. 2002;1:301–319. doi: 10.1016/s1567-7249(01)00037-x. [DOI] [PubMed] [Google Scholar]
- 29.Vincent F, Corral-Debrinski M, Adolphe M. Transient mitochondrial transcript level decay in oxidative stressed chondrocytes. J Cell Physiol. 1994;158:128–132. doi: 10.1002/jcp.1041580116. [DOI] [PubMed] [Google Scholar]
- 30.Milner PI, Wilkins RJ, Gibson JS. The role of mitochondrial reactive oxygen species in pH regulation in articular chondrocytes. Osteoarthritis Cartilage. 2007;15:735–742. doi: 10.1016/j.joca.2007.01.008. [DOI] [PubMed] [Google Scholar]
- 31.Ohashi T, Hagiwara M, Bader DL, Knight MM. Intracellular mechanics and mechanotransduction associated with chondrocyte deformation during pipette aspiration. Biorheology. 2006;43:201–214. [PubMed] [Google Scholar]
- 32.Huser CA, Davies ME. Calcium signaling leads to mitochondrial depolarization in impact-induced chondrocyte death in equine articular cartilage explants. Arthritis Rheum. 2007;56:2322–2334. doi: 10.1002/art.22717. [DOI] [PubMed] [Google Scholar]
- 33.Martin JA, McCabe D, Walter M, et al. N-acetylcysteine inhibits post-impact chondrocyte death in osteochondral explants. J Bone Joint Surg Am. 2009;91:1890–1897. doi: 10.2106/JBJS.H.00545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Beecher BR, Martin JA, Pedersen DR, et al. Antioxidants block cyclic loading induced chondrocyte death. Iowa Orthop J. 2007;27:1–8. [PMC free article] [PubMed] [Google Scholar]