

Abstract
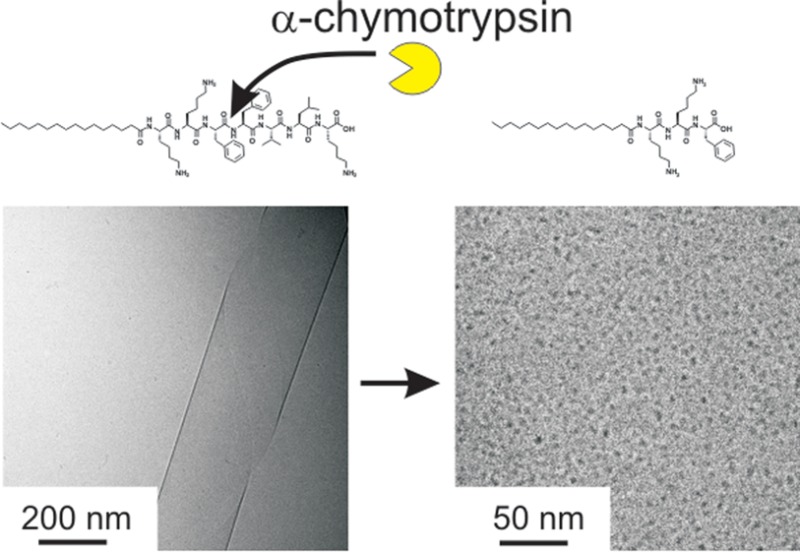
The enzymatic cleavage of a peptide amphiphile (PA) is investigated. The self-assembly of the cleaved products is distinct from that of the PA substrate. The PA C16-KKFFVLK is cleaved by α-chymotrypsin at two sites leading to products C16-KKF with FVLK and C16-KKFF with VLK. The PA C16-KKFFVLK forms nanotubes and helical ribbons at room temperature. Both PAs C16-KKF and C16-KKFF corresponding to cleavage products instead self-assemble into 5–6 nm diameter spherical micelles, while peptides FVLK and VLK do not adopt well-defined aggregate structures. The secondary structures of the PAs and peptides are examined by FTIR and circular dichroism spectroscopy and X-ray diffraction. Only C16-KKFFVLK shows substantial β-sheet secondary structure, consistent with its self-assembly into extended aggregates, based on PA layers containing hydrogen-bonded peptide headgroups. This PA also exhibits a thermoreversible transition to twisted tapes on heating.
Introduction
Peptide amphiphiles are a fascinating class of bioactive molecules in which one or more lipid chains are attached to peptides, either bioderived, bioinspired, or designed. The lipid chains drive self-assembly, leading to nanostructures in which the functional peptide unit can be presented at high density, for example, on the surface of nanofibrils.1−6
The use of enzymes to modulate the self-assembly of peptide amphiphiles (PAs) and other amphiphilic biomolecules such as amphiphilic peptides and peptide–polymer conjugates has attracted considerable attention due to potential applications in biomedicine as well as catalysis and sensing. Enzymes have been evolved by Nature to perform many essential functions in biological systems. They have excellent specificity and activity under mild environmental conditions associated with living organisms. The topic of enzyme-controlled peptide self-assembly has been the subject of a recent comprehensive review.7 Other reviews cover enzyme-driven (biocatalyzed) self-assembly of peptide derivatives,8,9 leading specifically to hydrogelation10 and the influence of enzymes on the aggregation of polymer-peptide conjugates.11,12 As well as enzyme-stimulated transitions in nanostructure, peptide systems can also undergo enzyme-mediated disassembly and enzyme-triggered self-assembly.7 By design of the peptide substrate, responsiveness to a wide variety of enzymes is possible, leading to a diversity of potential applications.
We have demonstrated a concept whereby PEGylated peptide micelles prepared from an amyloid β-peptide derived heptapeptide13,14 containing a diphenylalanine (FF) sequence attached to PEG were enzymatically degraded by α-chymotrypsin which selectively cleaves peptide bonds between aromatic residues.15 This released a hexapeptide (containing a C-terminal F) along with PEG with an N-terminal F residue. This proof-of-principle work established a possible route to create an enzyme-responsive delivery system based on PEG-coated particles which have relevance to biological applications since PEG provides a sterically stabilized corona, enabling enhanced in vivo stability.
Here, we investigate the influence of the model serine protease α-chymotrypsin on the self-assembly of the PA C16-KKFFVLK. The peptide amphiphile (PA) has been designed based on the KLVFF core motif from the amyloid beta (Aβ) peptide.16−18 This contains two F residues at the C terminus which drive self-assembly through hydrophobic and π-stacking interactions.13,14 Two additional lysine residues are incorporated between the hexadecyl lipid chain and the FFKLVFF motif to promote solubility and impart amphiphilicity. The serine protease α-chymotrypsin is expected to cleave preferentially between the two F residues, although as discussed below an additional cleavage site is also identified. The PAs corresponding to cleaved products C16-KKF and C16-KKFF are shown to self-assemble into spherical micelles, quite distinct from the nanotubes/helical ribbons formed by C16-KKFFVLK. We thus show that it is possible to develop enzyme-responsive PA systems with pronounced nanostructure transitions, leading in turn to macroscopic changes in the sample transparency.
Experimental Section
Materials
Peptide amphiphile C16-KKFFVLK was purchased from CS Bio (Menlo Park, CA). For the first batch, the molar mass by electrospray-ionization mass spectrometry was 1146.97 Da (expected 1147.57 Da). Purity was 99.73% by HPLC in a 0.1% TFA water/acetontitrile gradient. The PA was supplied as a TFA salt. For the second batch, the molar mass by ES-MSI was 1147.65 Da and the purity was 99.73% by HPLC.
Peptide amphiphile C16-KKFF was synthesized by CS Bio (Menlo Park, CA) and supplied as a TFA salt. The molecular weight was found to be 807.31 Da (expected 807.11 Da) as revealed by electrospray-ionization mass spectrometry. Purity of the sample was 99.88% by analytical HPLC in a 0.1% TFA water/acetonitirile gradient.
Peptide amphiphile C16-KKF was purchased from CS Bio (Menlo Park, CA) and supplied as a TFA salt. Electrospray-ionization mass spectrometry revealed a molecular weight of 659.73 Da (expected 659.94 Da). Purity was 99.35% by analytical HPLC in a 0.1% TFA water/acetonitirile gradient and supplied as a TFA salt.
Peptide FVLK was custom synthesized by Peptide Synthetics (Peptide Protein Research Ltd. Fareham, United Kingdom). The molecular weight was found to be 505.641 Da by electrospray-ionization mass spectrometry. The sample was purified in acetonitrile and water containing 0.1% TFA prior to lyophilization and was supplied as a TFA salt. The purity was >95% as determined by analytical HPLC.
Peptide VLK was synthesized by Peptide Synthetics (Peptide Protein Research Ltd. Fareham, United Kingdom). Molecular weight was 359.2 Da as determined by electrospray-ionization mass spectrometry. The sample was purified in acetonitrile and water containing 0.1% TFA prior to lyophilization and was supplied as a TFA salt. The purity was >95% as determined by analytical HPLC. The enzyme α-chymotrypsin, which has a molecular weight of 25 kDa, was purchased from Sigma-Aldrich (bovine pancreas extract).
To characterize self-assembly in water, solutions were made by direct dissolution in ultrafiltered water, from a Barnstead Nanopure system. The pH of a 0.5 wt % solution of C16-KKFFVLK was 4.18, and the pH was 3.84 for a 1 wt % solution.
Mass Spectrometry
Direct-infusion electrospray mass spectra (ES-MS) were obtained on a ThermoFisher Scientific Orbitrap XL instrument. The MS/MS experiments were performed with an isolated target mass of 648 Da (isolation width of 3 Da), and fragmentation was achieved in the HCD trap with a fragmentation energy of 35. A mixture of 1 wt % C16-KKFFVLK and 2 wt % α-chymotrypsin was prepared for mass spectrometry analysis. The dry powders were mixed together. Ultrapure water was then added to make up the defined concentration. A vial containing the solution was then placed in a sonicator at 50 °C for 1 h. This temperature was found to improve the activity of α-chymotrypsin (previous work showed increasing activity with temperature, but the highest temperature studied was 35 °C).19 The PA/enzyme mixture was left to age for 4 months before mass spectrometry measurements were performed.
FTIR
Spectra were recorded using a Nexus-FTIR spectrometer equipped with a DTGS detector and a multiple reflection attenuated total reflectance (ATR) system. Solutions of the PAs and peptides in D2O (0.5, 1, and 2 wt %) were sandwiched in ring spacers between two CaF2 plate windows (spacer 0.006 mm). All spectra were scanned 128 times over the range of 4000–950 cm–1.
Thioflavin T (ThT) Fluorescence Spectroscopy
Spectra were recorded with a Varian Cary Eclipse fluorescence spectrometer with samples in 4 mm inner width quartz cuvettes. The spectra were recorded from 460 to 600 nm using an excitation wavelength λex = 440 nm. ThT assays were performed using a 4.0 × 10–3 wt % ThT solution.
X-ray Diffraction (XRD)
Measurements were performed on stalks prepared by drying filaments of the PAs or peptides, prepared from 0.5, 1, or 2 wt % solutions. Solutions of the peptide were suspended between the ends of wax-coated capillaries and dried. The stalks were mounted (vertically) onto the four axis goniometer of a RAXIS IV++ X-ray diffractometer (Rigaku) equipped with a rotating anode generator and a Saturn 992 CCD camera. The sample–detector distance was 90 or 100 mm, depending on the sample. The X-ray wavelength was λ = 1.54 Å. The wavenumber scale (q = 4π sin θ/λ, where 2θ is the scattering angle) was geometrically calculated using the size of each pixel in the detector screen (0.0898 mm) and the sample–detector distance.
Small-Angle (SAXS) and Wide-Angle (WAXS) X-ray Scattering
SAXS data were collected on the BioSAXS beamline BM29 at the ESRF, Grenoble, France. Solutions containing 0.5, 1, or 2 wt % C16-KKFFVLK were loaded in PCR tubes in an automated sample changer. SAXS data was collected using a Pilatus 1 M detector. The sample–detector distance was 2.84 m. The X-ray wavelength was 0.99 Å.
Further experiments (including kinetics measurements) on a 1 wt % sample were performed on beamline ID02 at the ESRF, Grenoble, France. SAXS data were collected with a FReLoN Kodak CCD with a 1.2 m sample–detector distance, and WAXS data were measured simultaneously with an Aviex CCD. The X-ray wavelength was 0.995 Å. Solutions were injected using a syringe into ENKI KI-beam thin (0.05 mm) wall 1.85 mm diameter polycarbonate capillaries which optimize background subtraction. Measurements were performed at 25 °C. Heat/cool experiments were performed with the sample mounted in a quartz capillary instead of the ENKI polycarbonate capillary. All data were reduced to one-dimensional intensity profiles by radial integration.
Cryo-Transmission Electron Microscopy (Cryo-TEM)
Cryo-TEM was carried out using a field emission cryo-electron microscope (JEOL JEM-3200FSC), operating at 200 kV voltage (except for sample VLK for which the voltage was 300 kV). Images were taken in bright field mode and using zero loss energy filtering (omega type) with slit width of 20 eV. Micrographs were recorded using a Gatan Ultrascan 4000 CCD camera. The specimen temperature was maintained at −187 °C during the imaging. Vitrified specimens were prepared using an automated FEI Vitrobot device using Quantifoil 3.5/1 holey carbon copper grids with the hole size of 3.5 μm. Just prior to use, grids were plasma cleaned using a Gatan Solarus 9500 plasma cleaner and then transferred into an environmental chamber of a FEI Vitrobot at room temperature and 100% humidity. Thereafter, 3 μL of sample solution (2 wt % concentration) was applied to the grid and it was blotted twice for 5 s and then vitrified in a 1/1 mixture of liquid ethane and propane at temperature of −180 °C. The most viscous gel (5 wt % concentration) was blotted four times for 5 s. The grid with vitrified sample solution were maintained at liquid nitrogen temperature and then cryo-transferred into the microscope.
Results and Discussion
Our objective was to examine the effect of α-chymotrypsin (αC) in cleaving the PA C16-KKFFVLK and to investigate the self-assembly of the product peptides and peptide amphiphiles. We first employed mass spectroscopy to investigate selective enzyme cleavage of the PA. Figure 1 shows an electrospray ionization mass spectrum for the PA/enzyme mixture, which reveals sharp peaks with m/z values of 660.50 and 506.33 Da. These peaks are identified as C16-KKF and FVLK, respectively, since these values correspond to the expected molar masses, listed in the Experimental Section (i.e., ions with z = 1 are produced). These two fragments are expected to be released from C16-KKFFVLK in the presence of αC as the enzyme selectively catalyzes the hydrolysis of peptide bonds on the C-terminal side of phenylalanine, which in our peptide corresponds to cleavage between the two F residues. Unexpectedly we observed an additional two peaks with high abundances with m/z values of 807.57 and 359.27 Da. The former is ascribed to C16-KKFF and the latter as the tripeptide VLK, since the m/z values correspond to the expected molar masses for these species (again z = 1). MS/MS fragmentation was employed to confirm the nature of the two unexpected fragments by sequentially knocking off amino acid residues from the C-terminus as shown in Figure 2. As the amino acids K then L and then V are removed from the C-terminus, a peak at 789.56 Da is observed, which corresponds to the C16-KKFF fragment considering loss of H2O. MS/MS fragmentation indicates that αC cleaves at an additional site between F and V residues and confirms the existence of the two fragments (C16-KKFF and VLK).
Figure 1.

ES-MS for a mixture of 1 wt % C16-KKFFVLK and 2 wt % α-chymotrypsin.
Figure 2.

MS/MS fragmentation spectrum for C16-KKFFVLK, sequentially removing lysine, leucine, and valine from the C-terminus.
To further elucidate the enzymatic degradation products of the PA by α-chymotrypsin, circular dichroism was employed. Figure 3 shows spectra for the PA and the enzyme alone, as well as the mixture. It is clear that the CD spectrum from the mixture is completely different from either of the two components and cannot be expressed as a superposition of the spectra from the species in the mixture. This is consistent with the enzymatic cleavage observed by mass spectrometry. The spectrum for αC alone is consistent with that previously reported by ourselves15 and others.20,21 The spectrum for C16-KKFFVLK is dominated by electronic transitions associated with the π–π stacking of the F residues, as discussed elsewhere.22 The expected23 CD spectrum for β-sheet structures, with a negative minimum near 216 nm and a positive maximum in the 195–202 nm range, is masked by these features. We provide evidence in a recent paper22 that this PA does form β-sheet fibrils and this is supported by further data provided herein, to be discussed shortly.
Figure 3.
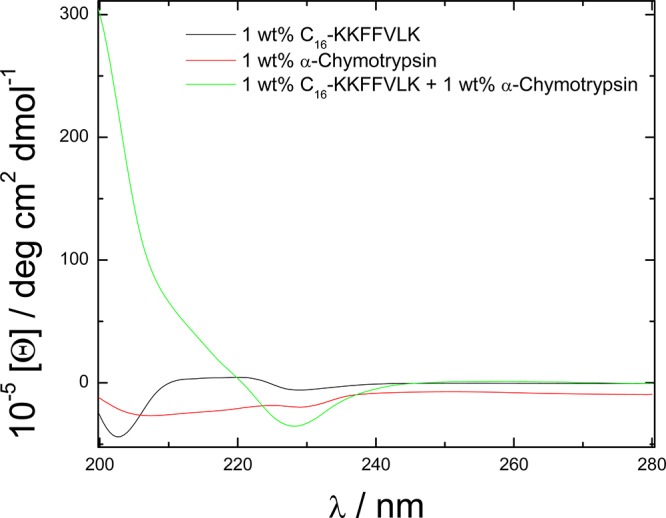
CD spectra for 1 wt % C16-KKFFVLK and 1 wt % α-chymotryspin and their mixture (1 and 1 wt %).
CD was also used to examine the self-assembly of peptides and PAs corresponding to the cleavage products, C16-KKF and FVLK and C16-KKFF and VLK, respectively. The data are presented in Figure 4. The spectra for the two PAs (Figure 4a,b) do not show features of canonical secondary structure such as β-sheets. The spectra contain contributions arising from π–π stacking of phenylalanine residues which as shown in our papers on phenylalanine-rich peptides17,24 and peptide conjugates25 can give rise to a positive maximum in the CD spectrum at 210–220 nm. There is also a contribution from polyproline II (PPII) structure in this region. The presence of a deep minimum in a CD spectrum at around 190 nm, along with a broad positive maximum at around 210 nm, is a signature of PPII (collagen-like) secondary structure.26−28 The spectra for VLK and FVLK in Figure 4c and d show these features. The spectra for FLVK also show features in the 240–270 nm range associated with vibronic transitions of the phenylalanine chromophore.24,29,30
Figure 4.

CD spectra for solutions of 0.5, 1, and 2 wt % for (a) C16-KKFF, (b) C16-KKF, (c) VLK, and (d) FVLK.
FTIR spectroscopy was also used to investigate secondary structure formation. Spectra covering the amide I′ and amide II′ regions from 0.5, 1, and 2 wt % solutions of the PAs and peptides are shown in Figure 5. The spectra for C16-KKFFVLK show a peak at 1626 cm–1 which is due to β-sheet structure,31,32 along with a peak at 1672 cm–1 which is due to bound TFA counterions.33−35 The spectra for C16-KKFF and C16-KKF do not show peaks in the 1615–1635 cm–1 range typical of β-sheet structures but have broad peaks near 1641 cm–1. Peak positions in this range are typically ascribed to random coil structure;31 however, as discussed elsewhere, PPII structures can also give a peak in this region.36 All of the PAs also exhibit broad peaks in the amide II′ region centered on 1580 cm–1. The spectra for peptides FVLK and VLK in Figure 5d,e show broad shoulder peaks in the 1640–1650 cm–1 region due to random coil/PPII structure (along with the peak due to bound TFA counterions). This is consistent with CD. Peaks in the amide IÍ region are absent for the peptides indicating a lower extent of ordering of N–H bonds compared to the PAs.
Figure 5.

FTIR spectra for solutions of 0.5, 1, and 2 wt % for (a) C16-KKFFVLK, (b) C16-KKFF, (c) C16-KKF, (d) FVLK, and (e) VLK.
Fiber X-ray diffraction was also used to examine peptide secondary structure. Diffraction patterns from dried stalks are shown in Supporting Information Figure 1 along with meridional and equatorial intensity profiles. The peak positions were used to calculate d spacings and these are listed in Supporting Information Table 1. The pattern for C16-KKFFVLK (Supporting Information Figure 1a) shows features of a cross-β structure37 with meridional reflections from a d = 4.83 Å spacing (corresponding to the β-strand spacing) along with a series of equatorial reflections from the β-sheet stacking distance and lateral packing of the PA molecules. A series of off-axis reflections close to the equator are due to the helical twisting of nanoribbons.22 The XRD pattern shows a high degree of alignment and multiple Bragg peaks confirming the regular ordering of the PA within the nanotubes/ribbons. In contrast, the XRD patterns for C16-KKF and C16-KKFF do not show orientation, and only present a small number of reflections. The pattern for C16-KKFF indicates the lack of secondary structure for this PA since cross-β features are not observed, just sharp peaks corresponding to the packing of the lipid chains along with a broad 5.7 Å ring due to the spacing of the molecules. Unexpectedly, C16-KKF shows more order in its XRD pattern than C16-KKFF with some cross-β features (i.e., 4.79/4.65 Å spacings and a 13.4 Å peak) although the 5.7 Å ring and the 4.11 Å peak from the packing of the lipid chains predominate.
The self-assembled nanostructures of the PAs (C16-KKFFVLK and PAs corresponding to the fragments produced after enzymatic degradation) and peptide fragments were examined by cryogenic transmission electron microscopy (cryo-TEM). Representative images are shown in Figure 6. C16-KKFFVLK forms nanotubes coexisting with helical ribbons (Figure 6a and ref (22)). SAXS data (Figure 7) confirm this assignment, as discussed in detail elsewhere.22 In complete contrast, C16-KKFF and C16-KKF self-assemble into small (approximately 5 nm diameter) micelles. This was confirmed by SAXS (Figure 8) since the intensity profiles were fitted by form factors corresponding to uniform spheres,38 yielding radii of 2.5 and 2.9 nm for C16-KKF and C16-KKFF, respectively. Cryo-TEM did not reveal a significant extent of nanostructure formation for peptide fragments FVLK and VLK. There was some evidence for sparse fibril clusters for FVLK (Figure 6d), and for VLK occasional raftlike film structures were observed (Figure 6e). However, SAXS on solutions of these two peptides (data not shown) revealed no scattering above background. The lack of significant structures for these two peptides is also consistent with FTIR. The observed raftlike film structures for VLK may be due to the surface activity of this peptide.
Figure 6.

Cryo-TEM images for solutions containing 1 wt % (a) C16-KKFFVLK, (b) C16-KKFF, (c) C16-KKF, (d) FVLK, and (e) VLK.
Figure 7.
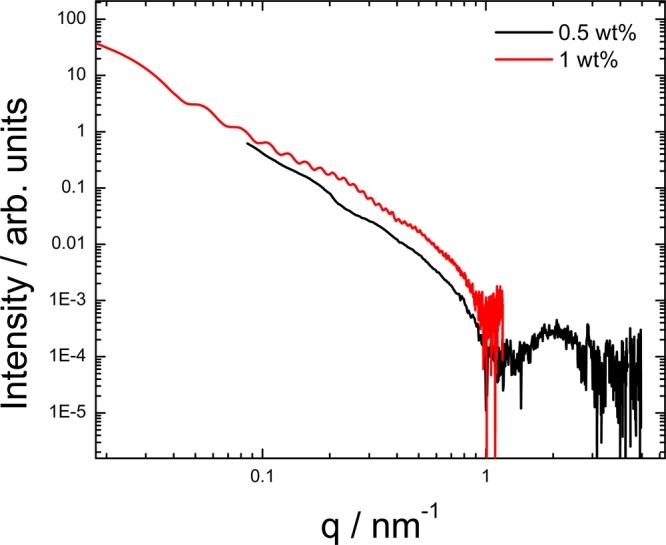
SAXS profiles for 0.5 and 1 wt % solutions of C16-KKFFVLK.
Figure 8.
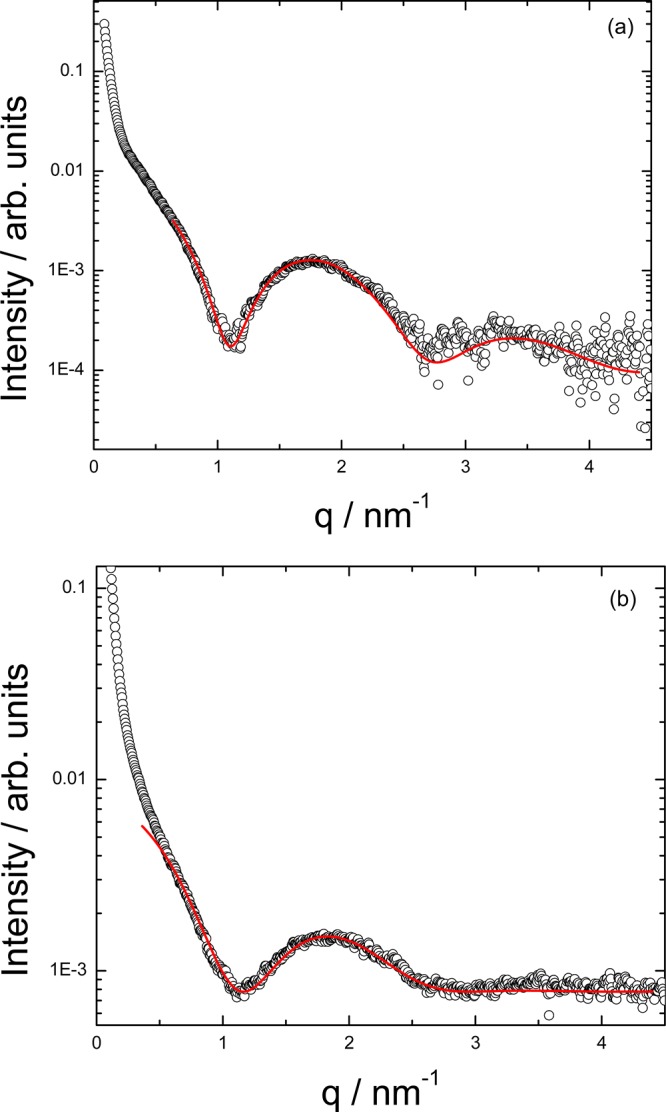
SAXS profiles fitted to a spherical shell form factor model for (a) 0.5 wt % C16-KKF and (b) 0.5 wt % C16-KKFF.
The difference in morphology of the aggregates of C16-KKFFVLK and the fragments C16-KKF and C16-KKFF leads to changes in sample transparency as shown in Figure 9. The large nanotube/ribbon structures formed by C16-KKFFVLK cause light scattering and a partly cloudy appearance, whereas the micellar structures of C16-KKF and C16-KKFF show higher transparency. Confirmation that addition of enzyme to a C16-KKFFVLK solution leads to a clear solution due to the cleavage process producing very small micelles is provided by the image shown for the mixture in Figure 9.
Figure 9.

Image of samples (1 wt %) in vials From left to right: C16 - KKFFVLK, C16-KKFF, C16-KKF, and C16-KKFFVLK with added αC (2 wt %).
As discussed in more detail elsewhere,22 C16-KKFFVLK undergoes a thermoreversible transition from nanotubes coexisting with helical ribbons (unwound nanotubes) at 20 °C to twisted tapes at higher temperature (55 °C). Figure 10 shows SAXS data obtained monitoring the kinetics of this process. The oscillations in the data at 20 °C are characteristic of a nanotube/ribbon bilayer structure as analyzed by form factor fitting. These disappear on heating, showing the break-up of the nanotubes/ribbons. However, on recooling, the oscillations develop slowly over a period of hours.
Figure 10.
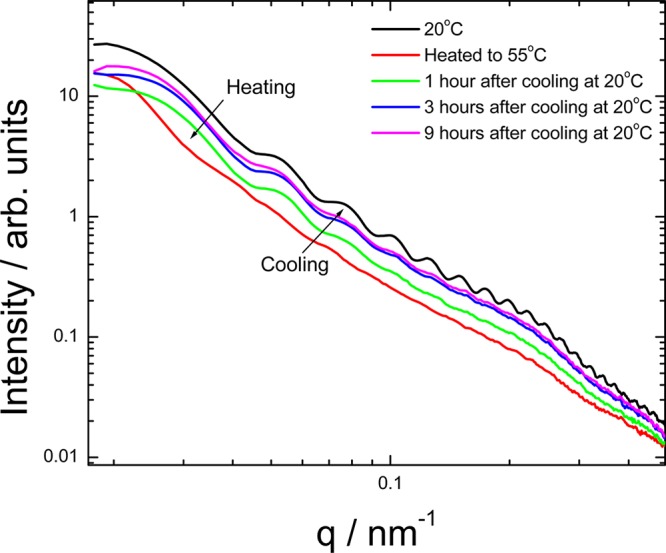
Kinetic measurements for a 1 wt % solution of C16-KKFFVLK analyzed by SAXS.
Discussion and Summary
The enzymatic cleavage of a PA using the model protease α-chymotrypsin has been demonstrated. ES-MS reveals that αC attacks two cleavage sites within the PA and preferentially cleaves between the two F residues as expected, leading to a higher abundance of both C16-KKF and FVLK. The second cleavage process occurs at the C terminus of the second F residue in C16-KKFFVLK to produce C16-KKFF and VLK.
We have shown that the PAs corresponding to the cleavage products C16-KKF and C16-KKFF assemble into very different structures than the parent PA which forms nanotubes and helical ribbons at room temperature. The C16-KKF and C16-KKFF PAs both self-assemble into spherical micelles. In contrast to C16-KKFFVLK, the two PAs with shorter peptide “headgroups” lack substantial defined secondary structure (there were some signs of β-sheet content in the XRD pattern for C16-KKF, possibly due to reduced hydrophobicity compared to C16-KKFF). This is associated with the formation of spherical micelles. Indeed, a phase diagram assembled for PAs on the basis of molecular dynamics computer simulations anticipates the formation of spherical micelles when hydrogen bonding interactions are weak.39 The near absence of intermolecular hydrogen bonded β-sheets for C16-KKF or C16-KKFF presumably reflects the insufficient length of the peptide headgroup, i.e. there are insufficient residues to participate in extensive hydrogen bonding networks. The change in nanostructure from C16-KKFFVLK to C16-KKF and C16-KKFF leads to macroscopic changes in sample appearance which might be useful for a simple enzyme sensing system, although more sensitive methods may be available.
We have recently investigated the self-assembly of the PA C16-KTTKS which forms extended β-sheet nanotapes.40 This PA has a headgroup comprising two lysine residues within the pentapeptide sequence. Considering in addition the findings reported here, our results on lysine-rich PAs suggest that a minimal sequence capable of forming β-sheets may require around five residues, provided that the peptide has both highly soluble hydrophilic residues along with hydrophobic residues. However, this is not a general rule since, for example, we have recently shown that a PA C16-βAH with only a dipeptide β-alanine-histidine headgroup can self-assemble into β-sheet fibrils in water.41
Neither of the peptide fragments VLK nor FVLK form extensive self-assembled nanostructures, although tripeptides are able to do so provided they have sufficient amphiphilicity (e.g., if they contain two hydrophobic phenylalanine residues42). Our own work also shows that a hexapeptide with one terminal phenylalanine does not self-assemble whereas a homologous heptapeptide with two C-terminal phenylalanines is capable to form β-sheet fibrils.15
One interesting application of our lysine-rich PAs may be antimicrobial agents. The antimicrobial activity of PAs C16-KxK (where x is A, G, L or K or k, i.e. d-lysine) and C16-KK and C16-K has been investigated, and this was correlated to their self-assembly properties.43 The most active PA (against microbes) C16-KKK formed very small oligomers in contrast to C16-KGK and C16-KLK which formed fibrils and C16-KAK which formed 10 nm diameter micelles.
Our results highlight the ability of enzymes to modulate the self-assembly of PA systems and the ability to tune nanostructure through use of enzymatic triggers. Our proof-of-concept work may be extended to create other enzyme-responsive assemblies or in applications where release of peptide or PA fragments is beneficial. Specifically, release of micelles in response to enzymatic degradation of extended nanostructures (which have been shown44,45 to offer enhanced circulation times in vivo) may be useful in delivery of actives.
Acknowledgments
This work was supported by EPSRC Grants EP/G026203/1 and G067538/1 to I.W.H. Beamtime at the ESRF on beamline ID02 was awarded under reference SC3468 and on BM29 under reference MX-1401, which we thank Louiza Zerrad for assistance. We thank Narayan Theyencheri (ESRF) for assistance with experiments on beamline ID02. We are grateful to Nick Spencer for assistance with XRD experiments. We thank Martin Reeves and John McKendrick for assistance with MS.
Supporting Information Available
Additional figure and table as described in the text. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Supplementary Material
References
- Löwik D. W. P. M.; van Hest J. C. M. Peptide Based Amphiphiles. Chem. Soc. Rev. 2004, 33, 234. [DOI] [PubMed] [Google Scholar]
- Cavalli S.; Kros A. Scope and Applications of Amphiphilic Alkyl- and Lipopeptides. Adv. Mater. 2008, 20, 627. [Google Scholar]
- Cui H. G.; Webber M. J.; Stupp S. I. Self-Assembly of Peptide Amphiphiles: From Molecules to Nanostructures to Biomaterials. Peptide Sci. 2010, 94, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Versluis F.; Marsden H. R.; Kros A. Power Struggles in Peptide-Amphiphile Nanostructures. Chem. Soc. Rev. 2010, 39, 3434. [DOI] [PubMed] [Google Scholar]
- Hamley I. W. Self-Assembly of Amphiphilic Peptides. Soft Matter 2011, 7, 4122. [Google Scholar]
- Matson J. B.; Zha R. H.; Stupp S. I. Peptide Self-Assembly for Crafting Functional Biological Materials. Curr. Opin. Solid State Mater. Sci. 2011, 15, 225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zelzer M.; Todd S. J.; Hirst A. R.; McDonald T. O.; Ulijn R. V. Enzyme Responsive Materials: Design Strategies and Future Developments. Biomater. Sci. 2013, 1, 11–39. [DOI] [PubMed] [Google Scholar]
- Williams R. J.; Mart R. J.; Ulijn R. V. Exploiting Biocatalysis in Peptide Self-Assembly. Biopolymers 2010, 94, 107. [DOI] [PubMed] [Google Scholar]
- Hahn M. E.; Gianneschi N. C. Enzyme-Directed Assembly and Manipulation of Organic Nanomaterials. Chem. Commun. 2011, 47, 11814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z.; Liang G.; Xu B. Enzymatic Hydrogelation of Small Molecules. Acc. Chem. Res. 2008, 41, 315. [DOI] [PubMed] [Google Scholar]
- Roy D.; Cambre J. N.; Sumerlin B. S. Future Perspectives and Recent Advances in Stimuli-Responsive Materials. Prog. Polym. Sci. 2010, 35, 278. [Google Scholar]
- Börner H. G.; Kuhnle H.; Hentschel J. Making ″Smart Polymers″ Smarter: Modern Concepts to Regulate Functions in Polymer Science. J. Polym. Sci., Part A: Polym. Chem. 2010, 48, 1. [Google Scholar]
- Castelletto V.; Hamley I. W.; Hule R. A.; Pochan D. J. Helical Ribbon Formation by a β-amino Acid Modified Amyloid B Peptide Fragment. Angew. Chem., Int. Ed. 2009, 48, 2317. [DOI] [PubMed] [Google Scholar]
- Adamcik J.; Castelletto V.; Hamley I. W.; Mezzenga R. Direct Observation of Time-Resolved Polymorphic States in Self-Assembly of End-Capped Heptapeptides. Angew. Chem., Int. Ed. 2011, 50, 5495. [DOI] [PubMed] [Google Scholar]
- Castelletto V.; McKendrick J. M. E.; Hamley I. W.; Cenker C.; Olsson U. Pegylated Amyloid Peptide Nanocontainer Delivery and Release System. Langmuir 2010, 26, 11624. [DOI] [PubMed] [Google Scholar]
- Tjernberg L. O.; Naslund J.; Lindqvist F.; Johansson J.; Karlstrom A. R.; Thyberg J.; Terenius L.; Nordstedt C. Arrest of Beta-Amyloid Fibril Formation by a Pentapeptide Ligand. J. Biol. Chem. 1996, 271, 8545. [DOI] [PubMed] [Google Scholar]
- Krysmann M. J.; Castelletto V.; Kelarakis A.; Hamley I. W.; Hule R. A.; Pochan D. J. Self-Assembly and Hydrogelation of an Amyloid Peptide Fragment. Biochemistry 2008, 47, 4597. [DOI] [PubMed] [Google Scholar]
- Hamley I. W. The Amyloid Beta Peptide: A Chemist’s Perspective. Role in Alzheimer’s and Fibrillization. Chem. Rev. 2012, 112, 5147. [DOI] [PubMed] [Google Scholar]
- Asgeirsson B.; Bjarnason J. B. Structural and Kinetic-Properties of Chymotrypsin from Atlantic Cod (Gadus-Morhua) - Comparison with Bovine Chymotrypsin. Comp. Biochem. Physiol., Part B: Biochem. Mol. Biol. 1991, 99, 327. [DOI] [PubMed] [Google Scholar]
- Manavalan P.; Johnson W. C. Sensitivity of Circular Dichroism to Protein Tertiary Structure Class. Nature (London) 1983, 305, 831. [Google Scholar]
- Lahari C.; Jasti L. S.; Fadnavis N. W.; Sontakke K.; Ingavle G.; Deokar S.; Ponrathnam S. Adsorption Induced Enzyme Denaturation: The Role of Polymer Hydrophobicity in Adsorption and Denaturation of α-Chymotrypsin on Allylglycidyl Ether (Age)-Ethylene Glycol Dimethacrylate (Egdm) Copolymers. Langmuir 2010, 26, 1096. [DOI] [PubMed] [Google Scholar]
- Hamley I. W.; Dehsorkhi A.; Castelletto V.; Furzeland S.; Atkins D.; Seitsonen J.; Ruokolainen J.. Reversible Helical Ribbon Unwinding Transition of a Self-Assembling Peptide Amphiphile. Submitted.
- Nordén B.; Rodger A.; Dafforn T. R.. Linear Dichroism and Circular Dichroism: A Textbook on Polarized-Light Spectroscopy; RSC: Cambridge, 2010. [Google Scholar]
- Hamley I. W.; Castelletto V.; Moulton C. M.; Rodriguez-Perez J.; Squires A. M.; Eralp T.; Held G.; Hicks M.; Rodger A. Alignment of a Model Amyloid Peptide Fragment in Bulk and at a Solid Surface. J. Phys. Chem. B 2010, 114, 8244. [DOI] [PubMed] [Google Scholar]
- Castelletto V.; Hamley I. W. Self Assembly of a Model Amphiphilic Phenylalanine Peptide/ Polyethylene Glycol Block Copolymer in Aqueous Solution. Biophys. Chem. 2009, 141, 169. [DOI] [PubMed] [Google Scholar]
- Woody R. W. Circular Dichroism. Methods Enzymol. 1995, 246, 34. [DOI] [PubMed] [Google Scholar]
- Shi Z. S.; Woody R. W.; Kallenbach N. R.. Is Polyproline Ii a Major Backbone Conformation in Unfolded Proteins? In Unfolded Proteins, Rose G. D., Ed.; Academic Press Inc: San Diego, 2002; Vol. 62, p 163. [DOI] [PubMed] [Google Scholar]
- Woody R. W. Circular Dichroism Spectrum of Peptides in the Poly(Pro)Ii Conformation. J. Am. Chem. Soc. 2009, 131, 8234. [DOI] [PubMed] [Google Scholar]
- Du H.; Fuh R. C. A.; Li J. Z.; Corkan L. A.; Lindsey J. S. Photochemcad: A Computer-Aided Design and Research Tool in Photochemistry. Photochem. Photobiol. 1998, 68, 141. [Google Scholar]
- Perkampus H.-H.UV–Vis Atlas of Organic Compounds; VCH: Weinheim, 1992; Vol. 2. [Google Scholar]
- Stuart B.Biological Applications of Infrared Spectroscopy; Wiley: Chichester, 1997. [Google Scholar]
- Barth A.; Zscherp C. What Vibrations Tell Us About Proteins. Q. Rev. Biophys. 2002, 35, 369. [DOI] [PubMed] [Google Scholar]
- Haris P.; Chapman D. The Conformational Analysis of Peptide Using Fourier Transform Ir Spectroscopy. Biopolymers 1995, 37, 251. [DOI] [PubMed] [Google Scholar]
- Gaussier H.; Morency H.; Lavoie M. C.; Subirade M. Replacement of Trifluoroacetic Acid with HCl in the Hydrophobic Purification Steps of Pediocin Pa-1: A Structural Effect. Appl. Environ. Microbiol. 2002, 68, 4803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelton J. T.; McLean L. R. Spectroscopic Methods for Analysis of Protein Secondary Structure. Anal. Biochem. 2000, 277, 167. [DOI] [PubMed] [Google Scholar]
- Keiderling T. A.; Xu Q., Unfolded Peptides and Proteins Studied with Infrared Absorption and Vibrational Circular Dichroism Spectra; Academic Press: San Diego, 2002; Vol. 62, p 111. [DOI] [PubMed] [Google Scholar]
- Serpell L. C. Alzheimer’s Amyloid Fibrils: Structure and Assembly. Biochim. Biophys. Acta 2000, 1502, 16. [DOI] [PubMed] [Google Scholar]
- Pedersen J. S. Analysis of Small-Angle Scattering Data from Colloids and Polymer Solutions: Modeling and Least-Squares Fitting. Adv. Colloid Interface Sci. 1997, 70, 171. [Google Scholar]
- Velichko Y. S.; Stupp S. I.; Olvera de la Cruz M. Molecular Simulation Study of Peptide Amphiphile Self-Assembly. J. Phys. Chem. B 2008, 112, 2326. [DOI] [PubMed] [Google Scholar]
- Castelletto V.; Hamley I. W.; Perez J.; Abezgauz L.; Danino D. Fibrillar Superstructure from Extended Nanotapes Formed by a Collagen-Stimulating Peptide. Chem. Commun. 2010, 46, 9185. [DOI] [PubMed] [Google Scholar]
- Castelletto V.; Cheng G.; Stain C.; Connon C. J.; Hamley I. W. Self-Assembly of a Peptide Amphiphile Containing L-Carnosine and Its Mixtures with a Multilamellar Vesicle Forming Lipid. Langmuir 2012, 28, 11599. [DOI] [PubMed] [Google Scholar]
- Marchesan S.; Easton C. D.; Kushkaki F.; Waddington L.; Hartley P. G. Tripeptide Self-Assembled Hydrogels: Unexpected Twists of Chirality. Chem. Commun. 2012, 48, 2195. [DOI] [PubMed] [Google Scholar]
- Makovitzki A.; Baram J.; Shai Y. Antimicrobial Lipopolypeptides Composed of Palmitoyl Di- and Tricationic Peptides: In Vitro and in Vivo Activities, Self-Assembly to Nanostructures, and a Plausible Mode of Action. Biochemistry 2008, 47, 10630. [DOI] [PubMed] [Google Scholar]
- Geng Y.; Dalhaimer P.; Cai S. S.; Tsai R.; Tewari M.; Minko T.; Discher D. E. Shape Effects of Filaments Versus Spherical Particles in Flow and Drug Delivery. Nat. Nanotechnol. 2007, 2, 249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christian D. A.; Cai S.; Garbuzenko O. B.; Harada T.; Zajac A. L.; Minko T.; Discher D. E. Flexible Filaments for in Vivo Imaging and Delivery: Persistent Circulation of Filomicelles Opens the Dosage Window for Sustained Tumor Shrinkage. Mol. Pharmaceutics 2009, 6, 1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.