

Abstract
Few questions on infectious disease are more important than understanding how and why avian influenza A viruses successfully emerge in mammalian populations, yet little is known about the rate and nature of the virus’ genetic adaptation in new hosts. Here, we measure, for the first time, the genomic rate of adaptive evolution of swine influenza viruses (SwIV) that originated in birds. By using a curated dataset of more than 24 000 human and swine influenza gene sequences, including 41 newly characterized genomes, we reconstructed the adaptive dynamics of three major SwIV lineages (Eurasian, EA; classical swine, CS; triple reassortant, TR). We found that, following the transfer of the EA lineage from birds to swine in the late 1970s, EA virus genes have undergone substantially faster adaptive evolution than those of the CS lineage, which had circulated among swine for decades. Further, the adaptation rates of the EA lineage antigenic haemagglutinin and neuraminidase genes were unexpectedly high and similar to those observed in human influenza A. We show that the successful establishment of avian influenza viruses in swine is associated with raised adaptive evolution across the entire genome for many years after zoonosis, reflecting the contribution of multiple mutations to the coordinated optimization of viral fitness in a new environment. This dynamics is replicated independently in the polymerase genes of the TR lineage, which established in swine following separate transmission from non-swine hosts.
Keywords: influenza, swine, avian, adaptation, genomics, zoonosis
1. Introduction
Cross-species transmission among birds, swine and humans is a key aspect of the pathogenicity and epidemiology of influenza A viruses. While wild aquatic and shore birds are the natural reservoir of the virus, transmissions to swine and humans have been repeatedly observed [1–3]. Although many such cross-species events result in isolated infections or self-limited chains of transmission in mammals, most notably for highly pathogenic H5N1 avian influenza in humans [4], some viruses have established persistent circulation in swine populations [5,6].
Of these, the Eurasian (EA) lineage of swine influenza A virus (SwIV) is particularly notable, as it represents a recent and highly successful transfer from birds to mammals. When first reported in Belgium in 1979, EA SwIV possessed a wholly avian-like genome [1], and has since become established in European swine populations [5,7]. EA viruses subsequently spread to Hong Kong and southern China around the turn of the millennium, where they largely replaced pre-existing strains [8]. By contrast, the ‘classical swine’ (CS) SwIV lineage has circulated among swine in North America for at least 80 years, and probably longer. The earliest known CS virus was isolated in 1930 and is related to the human 1918 ‘Spanish flu’ pandemic strain. CS SwIV viruses continue to circulate in North America and have, since 1974, also been detected in Asia [8,9]. The triple reassortant (TR) SwIV surfaced in North American pigs in 1998, bearing a heterogeneous genome that comprised gene segments originating from avian-, swine- and human-influenza A viruses [6,10].
For several reasons, swine influenza A viruses are important to human health. First, direct transmissions between pigs and humans are frequently observed [2,11,12]. Second, epithelial cell surfaces in the swine airway are thought to contain both α-2,3 and α-2,6 galactose sialic acid receptors, permitting entry and infection by human and avian influenza viruses (AIVs; [13]), thereby enabling the reassortment of human and avian influenza viruses when these co-infect the same pig [14]. Swine have thus long been considered an intermediate host, within which AIV might effectively adapt to human hosts [15]. Third, because of the frequent interaction between humans and pigs, human influenza pandemics can originate in swine, as demonstrated by the emergence in 2009 of pandemic H1N1 influenza (pH1N1/09), which derived from a reassortment of EA and TR swine influenza viruses [16].
There is little doubt that the cross-species transmission of influenza viruses involves adaptive evolution, particularly when viruses of avian origin emerge in mammalian hosts. Reverse genetics experiments have shown that amino acid changes in the haemagglutinin (HA) and polymerase viral proteins alter infection efficiency in mammals [17–19]. More recently, debate has followed the announcement that, through a process of experimental adaptation in the laboratory, H5N1 influenza genes of avian origin can become transmissible among ferrets [20,21]. Despite this, far less is known about the adaptive dynamics of influenza viruses to new hosts in natural populations. It is hypothesized that rates of pathogen molecular adaptation may be raised after cross-species transmission owing to the accumulation of novel host-specific adaptations [22,23], but for influenza viruses it is not known how long this process takes, and whether it is restricted to or more prominent in particular viral genes. Genes of the EA, CS and TR swine influenza lineages provide a unique system in which these questions can be studied as they originated in birds but have been established in pigs for different durations. In perhaps the only direct comparison of the molecular evolution of the CS and EA lineages, Dunham et al. [24] found that the nucleotide base composition of both changed through time since their respective cross-species transmissions, although with no apparent difference in nucleotide substitution rate. However, this study was constrained by a small sample size.
Recently, we established a framework for estimating the rate of molecular adaptation from very large sets of viral genomes and validated this through an extensive analysis of human influenza subtypes H1N1 and H3N2 [25]. This approach is different from, and complementary to, that used by methods based on dN/dS ratios. Specifically, our population genetic method directly quantifies the rate of molecular adaptation (in adaptive substitutions per site per year). It is better suited to the detection of adaptation arising from unique selective sweeps at different codons, whereas dN/dS ratio methods are most sensitive to recurrent selection at the same codon. Although the latter mode of selection prevails in the antigenic HA and NA genes of influenza, the former is likely to be prevalent in the remainder of the influenza genome. Unlike phylogenetic dN/dS methods, our approach cannot pinpoint the action of natural selection to specific amino acids, but can be practically applied to exceptionally large datasets.
Here, we use this framework to estimate, for the first time, the rate of molecular adaptation in swine influenza viruses. We reconstruct the adaptive dynamics of all gene segments of the three major SwIV lineages (EA, CS and TR). Because our method is suitable for very large datasets, we used all available SwIV gene sequences. However, despite an abundance of genomes for the CS and TR lineages, there was a paucity of EA SwIV genomes during 1990–2000, and we therefore sequenced an additional 41 EA lineage genomes from archival material. We also include two methodological improvements upon our previous work [25]. First, we undertake equivalent analyses of the HA and NA genes, making direct comparisons between them, which are reliable. Second, we use a more sophisticated model-fitting and validation procedure that allow for nonlinear rates of adaptation through time.
2. Material and methods
(a). New Eurasian lineage swine influenza genomes
We sequenced the full genomes of 41 swine influenza A isolates previously identified as subtype H1N1 by serology or HA gene sequencing. All genes of all 41 isolates belonged to the EA SwIV lineage (determined using phylogenetic methods; see below). Isolates had been collected during routine surveillance and stored at either the Animal Health & Veterinary Laboratories Agency, UK, the National Institute for Medical Research, Mill Hill, UK or the European Surveillance Network for Influenza in Pigs (ESNIP 1/2; Ghent University, Belgium). Isolates were sampled between 1992 and 2007 from UK, Italy, France, Poland, Belgium and the Czech Republic. Methods of viral extraction, amplification, sequencing, assembly and accession numbers are provided in the electronic supplementary material.
(b). Collation of datasets
We obtained all available SwIV whole genomes on 1 March 2011 from the NCBI Influenza Virus Resource. Recombinant or laboratory-generated strains, those containing segments from non-swine sources and those containing sequences with an excess (greater than 5%) of gaps/ambiguity codes, were excluded. All sequence dates, locations and labels were cross-checked with those on GenBank. Separate alignments were compiled for each virus gene (PB2, PB1, PA, HA, NP, NA, M1, M2, NS1 and NS2). Non-coding regions and overlapping reading frames were removed. Because the majority of NS2 consists of overlapping codons, this gene was excluded. We studied each swine or human influenza lineage between 1977 (or a later date of cross-species transfer) and the present day. Sequences with similar sampling dates were grouped. Time points containing less than nine sequences were not included; however, up to three contiguous years were collated into a time point to achieve adequate sample sizes. The date of each time point was taken to be the average of all the individual sequence dates comprising that time point [25]. The dates of the most ancestral sequence for each lineage were 1987 for EA, 1976 for CS, 1999 for TR and 1977 for the human H1N1 and H3N2 lineages.
(c). Structural subalignments for haemagglutinin and neuraminidase
From the HA gene alignment, we created four subalignments, comprising (i) the HA1 (globular, antigenically variable) subdomain, (ii) the HA2 (proximal membrane) subdomain, (iii) all HA codons that are solvent-accessible and (iv) all HA codons that are solvent-inaccessible. For the NA gene, we created two subalignments comprising (v) all NA codons that are solvent-accessible and (vi) all NA codons that are solvent-inaccessible. To predict which residues on the surfaces of trimeric HA and NA were solvent-exposed, structure-based alignments were performed as previously reported [26]. Briefly, amino acids were aligned with MultAlin [27] using sequences from closely related HA structures as references (Protein Data Bank: 2HTY, 1RUZ, 3EMY and 1NN2). The structure-based sequence alignments were plotted with ESPRIPT [28], and the Protein Data Bank (PDB) models used to map secondary structure and calculate solvent-accessibility. Prior to the determination of solvent-accessibility, all extraneous water molecules and ligands were removed from each PDB model.
(d). Phylogenetic quality control
For each alignment and subalignment, maximum-likelihood (ML) phylogenies were estimated under a GTR + Γ nucleotide substitution model using Garli v. 2.0. These were then inspected using Path-O-Gen, which provides a regression of sampling date against root-to-tip genetic distance and can therefore identify incorrectly classified or misdated sequences. To accurately genotype all sequences, we added them to a comprehensive set of reference strains. Our study strains were classified by estimating ML phylogenies for each genome segment (see the electronic supplementary material, figure S1), within which the various SwIV lineages (EA, CS, TR and pH1N1/09) could be identified. Isolates representing sporadic swine-to-avian or swine-to-human transmissions were removed from each lineage. Following this, the sequences constituting each lineage were selected as those belonging to the largest monophyletic group descended from the founding cross-species transmission of that lineage. The temporal range of pH1N1/09 isolates was too narrow to permit analysis and these were removed. For the TR lineage, only PB2, PB1 and PA sequences were retained, as only these genes have remained monophyletic since their introduction to swine. After quality control, each EA gene alignment contained 150–216 isolates, each CS alignment contained 246–438 isolates and each TR alignment contained 130 isolates (see the electronic supplementary material). For human influenza A, we used the alignments from the study of Bhatt et al. [25], comprising 775 seasonal H1N1 and 1674 H3N2 subtype genomes (see the electronic supplementary material). All alignments are available from the authors on request.
(e). Estimating the rates of adaptive evolution
For each swine influenza lineage and gene, we estimated the number of adaptive fixations and an overall rate of adaptive substitution using the method introduced in Bhatt et al. [25]. This method, based on the work by Williamson [29], calculates the number of adaptive fixations that have occurred between an ancestral nucleotide sequence and a main alignment of nucleotide sequences (which represents a later time point). Calculation begins by classifying each nucleotide site that differs between the ancestor and the main alignment into one of the following eight categories: (i) synonymous fixation, (ii) non-synonymous fixation, (iii) synonymous high-frequency polymorphism, (iv) non-synonymous high-frequency polymorphism, (v) synonymous mid-frequency polymorphism, (vi) non-synonymous mid-frequency polymorphism, (vii) synonymous low-frequency polymorphism or (viii) non-synonymous low-frequency polymorphism. The number of sites in each category is denoted as sf, rf, sh, rh, sm, rm, sl and rl, respectively. High-, mid- and low-frequency polymorphisms are defined as having observed frequencies greater than 0.75, 0.75–0.15 and less than 0.15, respectively; these are derived states with respect to the ancestral sequence. A proportional counting method is used to account for multiple changes at the same nucleotide site and to reduce statistical error (see methods and simulations in Bhatt et al. [30]). Under the assumption that synonymous mutations and mid-frequency polymorphisms are neutral, the expected number of non-neutral substitutions in each frequency class is defined as:
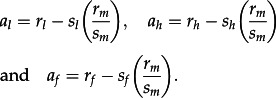 |
2.1 |
Full derivation and validation of this approach are provided in Bhatt et al. [25]. All terms in equation (2.1) are expectations [29]; hence, random error can generate negative a values when counts are small. Negative values must be set to zero, but are rare because we calculate rm and sm by combining information across time points [25]. The total number of adaptive substitutions is a = ah + af. To evaluate the rate of adaptive substitution, we calculate a for each consecutive time point, thereby generating a time series for the accumulation of adaptive substitutions through time. We also estimate the proportion of non-synonymous substitutions (or high-frequency polymorphisms) that are adaptive, as ((ah + af)/(rh + rf)). As in the study of Bhatt et al. [25], the ratio (rm/sm) for the HA and NA genes is calculated from the ‘internal’ partition to avoid bias. Computer software to calculate ah and af (called Adapt-A-Rate) is available at http://evolve.zoo.ox.ac.uk, and source code is available from the authors on request.
Previously, we estimated adaptation rates from the time series of a values using linear regression, and evaluated statistical robustness using a bootstrap approach in which codons were randomly sampled with replacement from the original alignments [25]. Here, we extend this to include fitting of orthogonal quadratic and cubic polynomials. All fitted lines or curves are constrained to pass through the origin. Because our time-series data are auto-correlated, we used a one-step-ahead cross-validation approach [31] to choose the best-fitting model. This procedure was undertaken as follows: (i) an orthogonal model (linear, quadratic or cubic) was selected. (ii) The chosen model was fitted using least-squares to a subset (containing n observations) of the full time series (comprising t observations). Given the fitted model, let 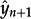 denote the forecast of the next observation (whose true value is yn+ 1). (iii) The mean square error (m.s.e.) of the predicted data point,
denote the forecast of the next observation (whose true value is yn+ 1). (iii) The mean square error (m.s.e.) of the predicted data point, 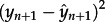 , was calculated. (iv) Steps (ii) and (iii) were repeated for n = m,…, t − 1, where m is the minimum number of observations needed to fit the model. (v) The m.s.e. for the chosen model was calculated. (vi) Steps (i)–(v) were repeated for each model. (vii) The best-fitting model was chosen as that which fits the time series with the minimum m.s.e.
, was calculated. (iv) Steps (ii) and (iii) were repeated for n = m,…, t − 1, where m is the minimum number of observations needed to fit the model. (v) The m.s.e. for the chosen model was calculated. (vi) Steps (i)–(v) were repeated for each model. (vii) The best-fitting model was chosen as that which fits the time series with the minimum m.s.e.
3. Results
ML phylogenies for the HA and NA genes of the EA lineage are shown in figure 1, in order to illustrate the phylogenetic distribution of the new EA genomes generated in this study. As expected, the EA lineage is monophyletic and descended from related AIV strains. The new EA isolates substantially increase the whole genome genetic data available for this lineage.
Figure 1.

Maximum-likelihood phylogenies illustrating the evolutionary history of the EA SwIV lineage (green) and the phylogenetic distribution of the EA genomes reported in this study (purple circles). The AIV out-group is represented by a black triangle. Scale bar is in units of estimated nucleotide substitutions per site. (a) Phylogeny estimated from HA sequences. (b) Phylogeny estimated from NA sequences. The phylogenetic position of the pH1N1/09 lineage is represented by a red triangle. Note that the AIV and pH1N1/09 isolates shown here were not included in the subsequent adaptation rate analyses.
Estimated rates of molecular adaptation for each gene of each SwIV lineage are shown in figure 2. Adaptation rates are expressed as average number of adaptive substitutions per codon per year, and were estimated using linear regression. Rates were estimated for all genes of the EA (green) and CS (blue) SwIV lineages, and for the polymerase genes of the TR lineage (magenta; see Material and methods). Figure 2 also includes equivalent results for two human influenza A lineages, seasonal H1N1 (orange) and H3N2 (black). The human influenza results are very similar to those in Bhatt et al. [25], except that here both the HA and NA genes are partitioned according to solvent-accessibility (see Material and methods).
Figure 2.

The estimated mean rate of adaptive substitution (per codon per year) for all genes and lineages of influenza A studied. The rate represents the gradient of a linear regression fitted to the time series shown in figures 3 and 4. Circles indicate the point estimate and the error bars show the 95% percentiles of estimate, obtained from 1000 bootstrap replicates. Lineages are colour-coded: EA, green; CS, blue; H1N1 human influenza, orange; H3N2 human influenza, black; TR swine, magenta.
The most striking result is that, in every genomic region, the estimated adaptation rates for EA genes exceed those of CS. In many genes, the 95% bootstrap percentiles of one lineage do not overlap the point estimate of the other: although this does not constitute a formal test of significance, it does indicate the observed difference is robust to sampling error. Adaptation rates are highest in the antigenic HA and NA genes, for which we report three values; the average adaptation rate across the whole gene, the rate in solvent-accessible (surface) resides and the rate in non-accessible (internal) residues. Remarkably, the HA gene adaptation rate for the EA lineage is not substantially different from that estimated for human influenza A virus (2.78×10−3 substitutions per codon per year for EA, 2.0×10−3 for human H1N1 and 3.31×10−3 for human H3N2). The high rate of EA adaptation is also observed when only the surface residues of the HA trimer are considered (HA surface). By contrast, the HA gene adaptation rate of the CS lineage (4.97×10−4) is nearly an order of magnitude lower than that of EA and human influenza A. This difference is also reflected in the percentage of non-synonymous fixations in HA that are positively selected; this value is approximately 75 per cent, approximately 76 per cent and approximately 90 per cent for the EA, human H1N1 and human H3N2 lineages, respectively, whereas the equivalent value for the CS lineage is approximately 38 per cent (see the electronic supplementary material). There is little adaptation in the internal HA residues; in this set of codons only the human H1N1 lineage has 95% bootstrap percentiles that exclude zero.
Faster adaptation of EA strains is also observed in the NA gene (as with HA, three rates are estimated; see above). The NA gene adaptation rate for the EA lineage (1.86 × 10−3) is again greater than that for CS (6.94 × 10−4) and comparable to that estimated for human influenza A. This difference remains notable (with respect to the bootstrap distributions) when only the surface residues of NA are considered, but not for the internal amino acids. Despite the lower adaptation rates seen in the NA gene of the CS lineage, the proportion of non-synonymous fixations in the NA surface residues of that lineage is high (approx. 75%) and comparable to that for the EA lineage (approx. 85%; electronic supplementary material).
For the polymerase genes (PB2, PB1 and PA), the fastest mean adaptation rates were observed in the TR lineage. The polymerase genes of the EA and TR lineages adapt faster than those of the CS lineage (none of the latter have 95% bootstrap percentiles that exclude zero). Indeed, molecular adaptation per codon in the PB2 gene of the TR lineage (9.37×10−4) is faster than that of the HA of the CS lineage. However, the absolute numbers of adaptive fixations in the polymerase genes are small (figure 4).
Figure 4.

The accumulation of adaptive substitutions through time in the PB2, PB1, PA, NP, M1, M2 and NS1 genes of avian-origin H1N1 SwIV (EA lineage, green; CS lineage, blue; TR lineage, magenta). See figure 3 legend for further details.
In all the remaining influenza A genes excluding NS1 (i.e. NP, M1 and M2), CS viruses do not exhibit adaptation rates greater than zero (assessed using the bootstrap distribution, as above). By contrast, the corresponding estimates and 95% bootstrap percentiles for the EA lineage are all greater than zero, except for the M2 gene. Although M2 gene adaptation rates appear lower in swine than human viruses, the M1 adaptation rate in EA viruses is substantially greater than that for any other lineage.
The estimated numbers of adaptive fixations through time for the swine influenza lineages (EA and CS) are shown in figures 3 and 4. The best-fitting regression model for each time series is shown (see Material and methods). Figure 3 contains the results for the antigenic HA and NA genes. In all but one case, the linear regression model provided the best-fit. The striking difference between EA and CS adaptation rates is again evident. The dataset representing surface residues of CS lineage NA genes was best-fitted by a quadratic model, suggesting a gradual reduction in the rate of adaptation after the mid 1990s.
Figure 3.

The accumulation of adaptive substitutions through time in the HA and NA genes of avian-origin H1N1 SwIV. Each point represents the estimated number of adaptive substitutions between that time point and the ancestral sequence for that lineage. The error bars show the 95% percentiles of this estimate, obtained from 1000 bootstrap replicates. The black line shows the best-fit regression model for the time series; the grey lines show the equivalent regression lines for the 1000 bootstrap replicates. All bootstrap replicates pass through the same origin, because the number of adaptive substitutions at the time of the ancestral sequence is, by definition, zero (EA lineage, green and CS lineage, blue). (a) The results for the whole HA and NA genes. (b,c) The results for the surface and internal partitions of these genes. Note that the number of adaptations here are per gene values, whereas the results in figure 2 are per codon values.
Figure 4 also highlights notable differences among SwIV lineages in the adaptive dynamics of the polymerase genes. Notably, the PB2 and PA genes of the TR lineage (which originated from AIV) both exhibit nonlinear adaptation through time and are best-fitted by cubic and quadratic models, respectively. This suggests a rapid initial burst of adaptation followed by a sharp decrease in the rate of adaptive fixation. The PA gene of the EA lineage shows a similar dynamics, but with a stronger indication that previously accrued adaptive changes were lost in recent years. The remaining time series are best-fitted by a linear model.
4. Discussion
The central result of our study is the unexpectedly high rate of molecular adaptation in the EA SwIV genome, which in all genes exceeds that of the CS lineage, and in all genes except M2 is greater than zero. By contrast, rates of adaptation of CS genes during the same time are different from zero only in HA, NA and NS1. Thus, following the cross-species transmission of EA viruses from birds to swine around 1979, the EA genome has experienced a rapid and sustained natural selection for improved replication and transmission in its new hosts. In contrast, the CS lineage had at that time already circulated among swine for greater than 60 years, and its adaptation to mammalian hosts was likely complete (as suggested by the accrual of conserved, host-specific mutations; [32]). This conclusion is independently supported by our observation of high adaptation rates in the PB2 and PA genes of the TR lineage; these genes represent a separate transmission of AIV genes to swine. Adaptive fixation was seen in almost all EA genes, indicating that the adaptation of AIV to mammals in natural populations requires a diverse set of host-specific adaptations across the whole viral genome. The accumulation of adaptive fixations in the EA lineage continues for more than a decade after cross-species transfer, suggesting that multiple mutations in all genes contribute to the progressive optimization of viral fitness in mammals; many of these may represent compensatory or epistatic changes.
Our results can help to interpret those from recent reverse genetics experiments, in which an AIV H5N1 strain [21] and an AIV/human influenza A hybrid [20] were experimentally adapted to transmissibility among ferrets. First, the small number of mutations identified in such experiments is likely to represent only the beginning of an adaptation process that may continue for years after zoonosis. Second, adaptation in influenza genes other than HA and NA will also be important for viruses of avian origin. Third, adaptive fixations in non-solvent-accessible regions of HA and NA appear to play a comparatively minor role in the adaptation of AIV to mammals (swine) in natural populations.
The high adaptation rates we estimate for residues on the surface of the HA and NA genes of the EA lineage raise the possibility that at least some of this adaptation is caused by natural selection for escape from antibody responses. It is commonly thought that SwIV undergoes less antigenic change than human influenza viruses [33,34], possibly because the short lifespan of farmed swine (approx. 150 days) lowers the chance of re-infection. However, several studies have concluded that the EA lineage HA gene undergoes more antigenic drift than the CS lineage HA gene [8,24,35], and it has been hypothesized that this provided EA viruses with a selective advantage, contributing to it replacing CS and TR strains in Asia [8]. Antibodies arising from CS and TR infection cross-react with many (but not all) EA viruses, which could explain some adaptation observed in the EA lineage HA and NA genes. However, we urge caution in these interpretations, as our understanding of the selective forces on HA and NA is still limited. While much attention has focused on the selection of mutations in known HA antibody epitopes, other regions of HA and NA may contribute to viral fitness through glycosylation [36], compensatory or epistatic interactions [37] or increased avidity for host cell receptors [38]. Further, adaptive evolution, even when not directly driven by humoural immunity, may be more commonly tolerated on macromolecular surfaces owing to stronger structural constraints on internal residues [39].
Mutations in the polymerase genes are considered to be important for the mammalian adaptation of AIV [17]. Our results are consistent with this as we observe rapid adaptation in the polymerase genes of the EA and TR lineages (although the absolute number of adaptations is small). In the TR lineage, PB2 and PA were reassorted from North American AIV and PB1 from human H3N2 influenza viruses. In the EA lineage, all three polymerase genes were derived from European AIV. Notably, in PB2 and PA of the TR lineage (and in PA of the EA lineage), we see a rapid burst of adaptation (approx. five fixations) soon after cross-species transfer followed by a decrease in the rate of molecular adaptation (figure 4). Visual inspection of the time series for the PB2 and PB1 genes of the EA lineage suggests a similar nonlinear behaviour, but for these the model-fitting results indicate the nonlinear trend is non-significant.
Population genetic theory highlights the factors determining rates of molecular adaptation: (i) per capita mutation rate, (ii) virus generation time, (iii) effective population size and (iv) selective pressures acting on the population. We posit that the latter provides the most probable explanation for the differences in EA and CS lineage adaptation rates. Changes in mutation rate or generation time should affect all gene segments equally, whereas we observe that ratios of EA/CS adaptation rates vary among genes (adaptation rates of M1, PB1 and PB2 are at least 10 times greater in EA viruses than in CS viruses, whereas those for NS1 or M2 are not significantly different). This heterogeneity points to selective pressures as the drivers of differences between EA and CS adaptation. Although it has been suggested that the mutation rate of the EA lineage may be higher than that of other lineages [40], subsequent experiments failed to observe a significant difference between the mutation rate of EA and that of AIV subtype H2N2 (thought to be slowly evolving; [41]).
The new methodological improvements implemented here demonstrate the utility of our framework [25,29,30]. Its simplicity means that it will remain computationally tractable as the datasets generated by next-generation sequencing technologies continue to increase greatly in size, length and temporal density. We have also illustrated the use of estimated three-dimensional protein structures to partition genomic sites into structural categories; further development and use of structural data will improve the study of adaptation in these and other viral genes. In future studies, we aim to further improve our framework by implementing a more formal approach to statistical significance testing.
Acknowledgements
Combating Swine Influenza Consortium (COSI) members are listed in the electronic supplementary material. We thank the institutes named in Material and methods for providing isolates, contributors to ESNIP for depositing samples and Dr K. van Reeth for facilitating access. This work was supported by UK COSI (BB/H014306/1), co-funded by BBSRC, MRC, the Wellcome Trust and Defra. Surveillance in GB pigs was supported by Defra (ED1204). O.G.P. was supported by the Royal Society and ESNIP3; S.J.L. by the BBSRC, the Wellcome Trust and COSI; T.A.B. by the Wellcome Trust; J.L.N.W. by the Alborada Trust and the RAPIDD programme of the Science and Technology Directorate, Department of Homeland Security and Fogarty International Center, NIH. ESNIP is funded by the European Union.
References
- 1.Pensaert M, Ottis K, Vandeputte J, Kaplan MM, Bachmann PA. 1981. Evidence for the natural transmission of influenza A virus from wild ducks to swine and its potential importance for man. Bull. World Health Organ. 59, 75. [PMC free article] [PubMed] [Google Scholar]
- 2.Claas EC, Kawaoka Y, de Jong JC, Masurel N, Webster RG. 1994. Infection of children with avian–human reassortant influenza virus from pigs in Europe. Virology 204, 453–457 (doi:10.1006/viro.1994.1553) [DOI] [PubMed] [Google Scholar]
- 3.Guan Y, Shortridge KF, Krauss S, Li PH, Kawaoka Y, Webster RG. 1996. Emergence of avian H1N1 influenza viruses in pigs in China. J. Virol. 70, 8041–8046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ferguson NM, Fraser C, Donnelly CA, Ghani AC, Anderson RM. 2004. Public health risk from the avian H5N1 influenza epidemic. Science 304, 968–969 (doi:10.1126/science.1096898) [DOI] [PubMed] [Google Scholar]
- 5.Campitelli L, Donatelli I, Foni E, Castrucci MR, Fabiani C, Kawaoka Y, Krauss S, Webster RG. 1997. Continued evolution of H1N1 and H3N2 influenza viruses in pigs in Italy. Virology 232, 310–318 (doi:10.1006/viro.1997.8514) [DOI] [PubMed] [Google Scholar]
- 6.Zhou NN, et al. 1999. Genetic reassortment of avian, swine, and human influenza A viruses in American pigs. J. Virol. 73, 8851–8856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brown IH. 2000. The epidemiology and evolution of influenza viruses in pigs. Vet. Microbiol. 74, 29–46 (doi:10.1016/S0378-1135(00)00164-4) [DOI] [PubMed] [Google Scholar]
- 8.Vijaykrishna D, et al. 2011. Long-term evolution and transmission dynamics of swine influenza A virus. Nature 473, 519–522 (doi:10.1038/nature10004) [DOI] [PubMed] [Google Scholar]
- 9.Shortridge KF, Webster RG. 1979. Geographical distribution of swine (Hsw1N1) and Hong Kong (H3N2) influenza virus variants in pigs in southeast Asia. Intervirology 11, 9–15 (doi:10.1159/000149006) [DOI] [PubMed] [Google Scholar]
- 10.Kanegae Y, Sugita S, Shortridge KF, Yoshioka Y, Nerome K. 1994. Origin and evolutionary pathways of the H1 hemagglutinin gene of avian, swine and human influenza viruses: cocirculation of two distinct lineages of swine virus. Arch. Virol. 134, 17–28 (doi:10.1007/BF01379103) [DOI] [PubMed] [Google Scholar]
- 11.Hodder RA, Gaydos JC, Allen RG, Top FH, Nowosiwsky T, Russell PK. 1977. Swine influenza A at Fort Dix, New Jersey (January–February 1976). III. Extent of spread and duration of the outbreak. J. Infect. Dis. 136, S369–S375 (doi:10.1093/infdis/136.supplement_3.s369) [DOI] [PubMed] [Google Scholar]
- 12.Shinde V, et al. 2009. Triple-reassortant swine Influenza A (H1) in humans in the United States, 2005–2009. N. Engl. J. Med. 360, 2616–2625 (doi:10.1056/NEJMoa0903812) [DOI] [PubMed] [Google Scholar]
- 13.Ito T, et al. 1998. Molecular basis for the generation in pigs of influenza A viruses with pandemic potential. J. Virol. 72, 7367–7373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Scholtissek C. 1990. Pigs as ‘mixing vessels’ for the creation of new pandemic influenza A viruses. Med. Princ. Pract. 2, 65–71 [Google Scholar]
- 15.Ma W, Kahn RE, Richt JA. 2009. The pig as a mixing vessel for influenza viruses: human and veterinary implications. J. Mol. Genet. Med. Int. J. Biomed. Res. 3, 158–166 [PMC free article] [PubMed] [Google Scholar]
- 16.Smith GJ, et al. 2009. Origins and evolutionary genomics of the 2009 swine-origin H1N1 influenza A epidemic. Nature 459, 1122–1125 (doi:10.1038/nature08182) [DOI] [PubMed] [Google Scholar]
- 17.Gabriel G, Dauber B, Wolff T, Planz O, Klenk HD, Stech J. 2005. The viral polymerase mediates adaptation of an avian influenza virus to a mammalian host. Proc. Natl Acad. Sci. USA 102, 18 590–18 595 (doi:10.1073/pnas.0507415102) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li Z, et al. 2005. Molecular basis of replication of duck H5N1 influenza viruses in a mammalian mouse model. J. Virol. 79, 12 058–12 064 (doi:10.1128/JVI.79.18.12058-12064.2005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Matrosovich M, Tuzikov A, Bovin N, Gambaryan A, Klimov A, Castrucci MR, Donatelli I, Kawaoka Y. 2000. Early alterations of the receptor-binding properties of H1, H2, and H3 avian influenza virus hemagglutinins after their introduction into mammals. J. Virol. 74, 8502–8512 (doi:10.1128/JVI.74.18.8502-8512.2000) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Imai M, et al. 2012. Experimental adaptation of an influenza H5 HA confers respiratory droplet transmission to a reassortant H5 HA/H1N1 virus in ferrets. Nature 486, 420–428 (doi:10.1038/nature10831) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Herfst S, et al. 2012. Airborne transmission of influenza A/H5N1 virus between ferrets. Science 336, 1534–1541 (doi:10.1126/science.1213362) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sharp PM, Bailes E, Chaudhuri RR, Rodenburg CM, Santiago MO, Hahn BH. 2001. The origins of acquired immune deficiency syndrome viruses: where and when? Phil. Trans. R. Soc. Lond. B 356, 867–876 (doi:10.1098/rstb.2001.0863) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Antia R, Regoes RR, Koella JC, Bergstrom CT. 2003. The role of evolution in the emergence of infectious diseases. Nature 426, 658–661 (doi:10.1038/nature02104) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dunham EJ, Dugan VG, Kaser EK, Perkins SE, Brown IH, Holmes EC, Taubenberger JK. 2009. Different evolutionary trajectories of European avian-like and classical swine H1N1 influenza A viruses. J. Virol. 83, 5485–5494 (doi:10.1128/JVI.02565-08) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bhatt S, Holmes EC, Pybus OG. 2011. The genomic rate of molecular adaptation of the human influenza A virus. Mol. Biol. Evol. 28, 2443–2451 (doi:10.1093/molbev/msr044) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bowden TA, Crispin M, Graham SC, Harvey DJ, Grimes JM, Jones EY, Stuart DI. 2009. Unusual molecular architecture of the machupo virus attachment glycoprotein. J. Virol. 83, 8259–8265 (doi:10.1128/JVI.00761-09) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Corpet F. 1988. Multiple sequence alignment with hierarchical clustering. Nucleic Acids Res. 16, 10 881–10 890 (doi:10.1093/nar/16.22.10881) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gouet P, Courcelle E, Stuart DI, Metoz F. 1999. ESPript: analysis of multiple sequence alignments in PostScript. Bioinformatics 15, 305–308 (doi:10.1093/bioinformatics/15.4.305) [DOI] [PubMed] [Google Scholar]
- 29.Williamson S. 2003. Adaptation in the env gene of HIV-1 and evolutionary theories of disease progression. Mol. Biol. Evol. 20, 1318–1325 (doi:10.1093/molbev/msg144) [DOI] [PubMed] [Google Scholar]
- 30.Bhatt S, Katzourakis A, Pybus OG. 2010. Detecting natural selection in RNA virus populations using sequence summary statistics. Infect. Genet. Evol. 10, 421–430 (doi:10.1016/j.meegid.2009.06.001) [DOI] [PubMed] [Google Scholar]
- 31.Arlot S, Celisse A. 2010. A survey of cross-validation procedures for model selection. Stat. Surv. 4, 40–79 (doi:10.1214/09-SS054) [Google Scholar]
- 32.Finkelstein DB, Mukatira S, Mehta PK, Obenauer JC, Su X, Webster RG, Naeve CW. 2007. Persistent host markers in pandemic and H5N1 influenza viruses. J. Virol. 81, 10 292–10 299 (doi:10.1128/JVI.00921-07) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.De Jong JC, et al. 2001. Antigenic and molecular heterogeneity in recent swine influenza A (H1N1) virus isolates with possible implications for vaccination policy. Vaccine 19, 4452–4464 (doi:10.1016/S0264-410X(01)00190-6) [DOI] [PubMed] [Google Scholar]
- 34.Vincent AL, Lager KM, Ma W, Lekcharoensuk P, Gramer MR, Loiacono C, Richt JA. 2006. Evaluation of hemagglutinin subtype 1 swine influenza viruses from the United States. Vet. Microbiol. 118, 212–222 (doi:10.1016/j.vetmic.2006.07.017) [DOI] [PubMed] [Google Scholar]
- 35.Brown IH, et al. 1997. Antigenic and genetic analyses of H1N1 influenza A viruses from European pigs. J. Gen. Virol. 78, 553. [DOI] [PubMed] [Google Scholar]
- 36.Skehel JJ, Stevens DJ, Daniels RS, Douglas AR, Knossow M, Wilson IA, Wiley DC. 1984. A carbohydrate side chain on hemagglutinins of Hong Kong influenza viruses inhibits recognition by a monoclonal antibody. Proc. Natl Acad. Sci. USA 81, 1779–1783 (doi:10.1073/pnas.81.6.1779) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hensley SE, Das SR, Gibbs JS, Bailey AL, Schmidt LM, Bennink JR, Yewdell JW. 2011. Influenza A virus hemagglutinin antibody escape promotes neuraminidase antigenic variation and drug resistance. PLoS ONE 6, e15190 (doi:10.1371/journal.pone.0015190) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hensley SE, et al. 2009. Hemagglutinin receptor binding avidity drives influenza A virus antigenic drift. Science 326, 734–736 (doi:10.1126/science.1178258) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chothia C, Lesk AM. 1986. The relation between the divergence of sequence and structure in proteins. EMBO J. 5, 823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ludwig S, Stitz L, Planz O, Van H, Fitch WM, Scholtissek C. 1995. European swine virus as a possible source for the next influenza pandemic? Virology 212, 555–561 (doi:10.1006/viro.1995.1513) [DOI] [PubMed] [Google Scholar]
- 41.Stech J, Xiong X, Scholtissek C, Webster RG. 1999. Independence of evolutionary and mutational rates after transmission of avian influenza viruses to swine. J. Virol. 73, 1878–1884 [DOI] [PMC free article] [PubMed] [Google Scholar]