

Abstract
Transcription is an essential component of basic cellular and developmental processes. However, early embryonic development occurs in the absence of transcription and instead relies upon maternal mRNAs and proteins deposited in the egg during oocyte maturation. Although the early zebrafish embryo is competent to transcribe exogenous DNA, factors present in the embryo maintain genomic DNA in a state that is incompatible with transcription. The cell cycles of the early embryo titrate out these factors, leading to zygotic transcription initiation, presumably in response to a change in genomic DNA chromatin structure to a state that supports transcription. To understand the molecular mechanisms controlling this maternal to zygotic transition, it is important to distinguish between the maternal and zygotic transcriptomes during this period. Here we use exome sequencing and RNA-seq to achieve such discrimination and in doing so have identified the first zygotic genes to be expressed in the embryo. Our work revealed different profiles of maternal mRNA post-transcriptional regulation prior to zygotic transcription initiation. Finally, we demonstrate that maternal mRNAs are required for different modes of zygotic transcription initiation, which is not simply dependent on the titration of factors that maintain genomic DNA in a transcriptionally incompetent state.
Keywords: MZT, Maternal, Paternal, Transcriptome, Zebrafish
INTRODUCTION
During the earliest steps of embryonic development there are dramatic changes in transcriptional dynamics. Initially, development proceeds in the absence of de novo transcription and depends on maternal mRNAs and proteins deposited in the egg during oocyte maturation. Following fertilisation and the completion of meiosis, the embryo undergoes rapid, synchronous and reductive cell divisions (Newport and Kirschner, 1982a; Kane and Kimmel, 1993). At a critical period thereafter, cell cycles lengthen, zygotic transcription commences and maternal mRNAs begin to degrade (Giraldez et al., 2006; Dalle Nogare et al., 2009; Tadros and Lipshitz, 2009). This period is therefore referred to as the maternal to zygotic transition (MZT). Shortly after this period, embryos acquire the ability to perform apoptosis (Stack and Newport, 1997). All animals are thought to pass through such a period of transcriptional quiescence followed by the activation of zygotic transcription, but the number of cell cycles before transcription begins varies. In the mouse, zygotic transcription commences at the 2-cell stage (Hamatani et al., 2004), whereas in the zebrafish it starts after ten cell cycles (Kane and Kimmel, 1993).
In previous work to investigate transcriptional dynamics in the early embryo, plasmid DNA was injected into fertilised Xenopus embryos (Newport and Kirschner, 1982b). The plasmid DNA was transcribed immediately after injection, then repressed, and finally reactivated at the time of normal zygotic transcription. The repression of plasmid DNA transcription coincided with a change in its chromatin structure, suggesting that the early embryo is capable of transcribing naked plasmid DNA, but that proteins in the early embryo maintain genomic DNA in a state that is incompatible with transcription. As cell division and DNA synthesis in the early embryo proceed, the factors that maintain transcriptional quiescence become titrated out: thus, after a critical number of cell cycles, genomic DNA becomes transcriptionally competent and transcription commences (Newport and Kirschner, 1982b; Newport and Kirschner, 1982a; Edgar and Schubiger, 1986; Kimelman et al., 1987; Dalle Nogare et al., 2009). Together, these experiments suggest that zygotic transcription begins because factors controlling the maintenance of transcriptional quiescence become titrated out.
Recently, new sequencing technologies have significantly improved our ability to analyse transcription (Ozsolak and Milos, 2011; Collins et al., 2012; Djebali et al., 2012). One can now assess all mRNAs present at a given developmental stage, or even in a particular cellular fraction, in a normal or disease state and in a quantifiable manner. Similarly, transcription factor binding sites can now be assessed in a genome-wide and temporal manner (Neph et al., 2012; Spitz and Furlong, 2012). Of relevance to our work, microarray analysis had previously suggested that transcription occurs prior to the MZT in the zebrafish (Mathavan et al., 2005). RNA-seq experiments now indicate that these findings were incorrect; rather, they suggest that post-transcriptional regulation rather than de novo transcription causes the observed increases in mRNA levels prior to the MZT (Aanes et al., 2011). Formal proof of this conclusion, however, and the ability to undertake more mechanistic analyses of the MZT, require the ability to distinguish maternal and zygotic transcriptomes at the genome-wide level.
Here, we report the systematic identification of maternal and zygotic transcriptomes during the zebrafish MZT, using single-nucleotide polymorphisms (SNPs) to identify maternal and paternal mRNAs. Using the appearance of paternal mRNAs as an indicator of zygotic transcription, our work demonstrates that zygotic transcription in the zebrafish first occurs after ten cell cycles, a result that is consistent with previous radioactivity incorporation experiments (Kane and Kimmel, 1993) but inconsistent with microarray experiments (Mathavan et al., 2005). We also demonstrate, for the first time (Aanes et al., 2011), that there is widespread post-transcriptional regulation of maternal mRNAs before zygotic transcription initiation, including polyadenylation, de-adenylation and cell cycle-coupled regulation. The polyadenylation of maternal mRNAs requires cytoplasmic polyadenylation elements in the 3′ UTRs of mRNAs. This post-transcriptional regulation is not required, however, for the acquisition of transcriptional competence in the early zebrafish embryo.
Finally, by performing RNA-seq analyses of embryos at time points around the MZT combined with our knowledge of maternal and paternal transcripts, we have identified the first zygotic genes to be expressed in the early zebrafish embryo. While zygotic transcription initiation depends on the establishment of a competent genomic DNA state, we now demonstrate, using inhibitors of translation, that maternal mRNAs provide the specificity that is required for the precise initiation of zygotic transcription in the early embryo. The ability to transcribe some zygotic genes is established before the 128-cell stage, whereas for other zygotic genes it is established after the 128-cell stage.
MATERIALS AND METHODS
RNA-seq
Embryos were collected 30 minutes after natural matings and then stage matched at the 2-cell, 64-cell, high (3.5 hpf), shield (6 hpf) and 90% epiboly (9 hpf) stages (Kimmel et al., 1995). One hundred embryos were collected for each stage and total RNA was extracted using TRIzol (Invitrogen) and then DNase treated. The quality of total RNA was assessed using an Agilent Bioanalyser and quantified using a Qubit (Invitrogen). Polyadenylated mRNA was then extracted from total RNA and fragmented. A 200-300 nucleotide size selection was performed and the RNA was then converted into an Illumina sequencing library according to the manufacturer’s protocol (TruSeq, Illumina). Libraries were sequenced on a Genome Analyser II as 54 bp paired-end reads. TopHat and Cufflinks were used to analyse the sequencing results (Trapnell et al., 2010).
For the cycloheximide experiment, embryos were synchronised by in vitro fertilisation. Sperm samples were collected and stored on ice in 70 μl BSMIS (75 mM NaCl, 70 mM KCl, 2 mM CaCl2, 1 mM MgSO4, 20 mM Tris pH 8.0). Eggs were then collected into a glass Petri dish by squeezing females. Sperm samples were added to the eggs with the addition of 1 ml egg water. After 2 minutes the dish was flooded with egg water. Fertilised eggs were then split into two dishes, with cycloheximide being added to one dish at a final concentration of 200 μg/ml; cell cycle progression was blocked at this concentration. Untreated and treated embryos were then collected every 15 minutes, starting just after the addition of cycloheximide. RNA-seq was performed as described above. Throughout the experiment, care was taken to incubate both dishes at 28.5°C because cell cycle progression is affected by temperature.
Exome sequencing
DNA was isolated from WIK and SAT individuals by incubating fin biopsies in 400 μl 100 μg/ml proteinase K for 10 hours at 55°C, followed by 15 minutes at 85°C to heat inactivate the proteinase K. DNA was precipitated by adding 400 μl isopropanol and centrifuging for 30 minutes at 4°C at 4000 rpm (1500 g). DNA pellets were washed twice with 400 μl 70% ethanol followed by centrifugation at 4000 rpm for 5 minutes, and resuspended in double-distilled H2O. DNA from each individual (1-2 μg) was sheared and used to construct 150-200 bp insert Illumina libraries according to the manufacturer’s protocols. To enrich for exonic regions of the genome, Agilent SureSelect baits were designed against the zebrafish genome (Zv8) (Kettleborough et al., 2013). Biotinylated RNA baits, homologous to the exon coding regions of the genome, were hybridised to the genomic DNA Illumina libraries. The hybridised fragments were purified using streptavidin-coated beads and RNA baits were digested, leaving exon-enriched genomic DNA. Each enriched sample was sequenced on an individual lane of a Genome Analyser II as 76 bp paired-end reads.
Identification of maternal and paternal mRNAs
Exome sequence variants were identified using a modified version of the 1000 Genomes Project variant calling pipeline (1000 Genomes Project Consortium, 2010; Kettleborough et al., 2013). Sequencing reads were mapped to the zebrafish genome using Burrows-Wheeler aligner (BWA) and SNPs were called by SAMtools mpileup, QCALL and the GATK Unified Genotyper. SNPs were removed if not called by all three callers and where the genotype quality was lower than 100 for GATK and lower than 50 for QCALL and SAMtools mpileup. The exome sequences of the male and female in each cross were compared and only homozygous differences were used in the identification of maternal and paternal mRNAs. Using those positions and our SNP calling pipeline, we counted the number of maternal or paternal SNPs in the RNA-seq experiments. Only positions that were informative in both crosses were used to determine whether a gene was maternally or paternally expressed. Within the RNA-seq SNP count, a count of two was used as a cut-off for determining whether an mRNA was maternal or paternal.
Polyadenylation assays
The polyadenylation tail assay was performed as described (Sallés and Strickland, 1999). Primer p1 (0.4 μg) (see supplementary material Table S13) was ligated to mRNAs by incubating with 4 μg total RNA and T4 RNA ligase (New England BioLabs) in a total volume of 10 μl. The ligase was heat inactivated by incubating at 80°C for 15 minutes. All 10 μl were used in a 50 μl cDNA synthesis reaction with primer p2 and SuperScript II reverse transcriptase (Invitrogen) according to the manufacturer’s protocol. Primer p2 and a polb gene-specific primer were used in a PCR reaction to amplify the poly(A) tail.
sox19b 3′ UTR fragments were cloned downstream of Venus in the plasmid pCS2+ using restriction enzymes XbaI and NotI with the primers listed in supplementary material Table S13. Quantitative RT-PCR was performed on cDNA generated with oligo(dT) primers and C18H16orf7 and cyclin B1 primers (supplementary material Table S13). Mutagenesis of sox19b 3′ UTR fragment 3 employed the primers listed in supplementary material Table S13. Twenty cycles of PCR were performed with Phusion DNA polymerase (New England BioLabs), followed by digestion of the template plasmid with the restriction enzyme DpnI. Following the recovery of plasmids, mutagenesis was confirmed by capillary sequencing. To test the transcriptional competence of the early embryo, fertilised eggs were injected with 50 ng plasmid DNA containing the EF1α promoter upstream of CFP. Embryos injected with plasmid were then divided into two and half of the embryos were further injected with 1.4 nl α-amanitin (Sigma-Aldrich) at 200 μg/ml (Kane et al., 1996). Fifty embryos were collected for RT-PCR, 1 hour after injection. RT-PCR of CFP and cyclin B1 was performed using the primers listed in supplementary material Table S13.
Data access
All sequencing results are accessible through the European Nucleotide Archive (http://www.ebi.ac.uk/ena/) under the accession numbers ERS017671 (WIK male), ERS017672 (SAT female), ERS017673 (SAT male), ERS017674 (WIK female) and ERP001280 (RNA-seq).
RESULTS
Distinguishing the maternal and zygotic transcriptomes
To distinguish maternal and zygotic mRNAs during the MZT we performed RNA-seq on embryos obtained from crosses of the SAT and WIK zebrafish strains (Fig. 1A). Embryos were collected after 30 minutes of natural mating of a cross of a female SAT and male WIK (SATf/WIKm) and also a reciprocal cross of a female WIK and male SAT (WIKf/SATm). Only four adult zebrafish were used in the described experiment. For each cross, 100 embryos were collected at five different developmental stages that span the MZT: 2-cell [0.75 hours post-fertilisation (hpf)], 64-cell (2 hpf), high stage (3.5 hpf), shield (6 hpf) and 90% epiboly (9 hpf) (Kimmel et al., 1995). The later developmental stages also mark a period when the embryo has been patterned into the three germ layers and is near to completing gastrulation. For each sample, polyadenylated RNA was extracted from total RNA and then subjected to Illumina sequencing. Each sample was sequenced on an individual lane of a Genome Analyser II, generating a total of 697,595,232×54 bp reads (supplementary material Table S1). Reads were mapped to the zebrafish genome (Zv9) using TopHat (Trapnell et al., 2010; Collins et al., 2012; Howe et al., 2013) and mRNA levels were quantified as fragments per kb per million reads (FPKM) using Cufflinks (supplementary material Table S2) (Trapnell et al., 2010).
Fig. 1.
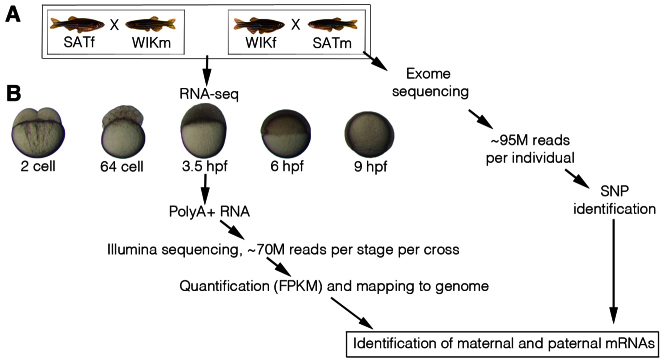
Identification of maternal and paternal mRNAs. (A) To identify maternal and paternal mRNAs, crosses of two different strains of zebrafish were used. A female SAT was crossed with a male WIK and the reciprocal cross of female WIK and male SAT was also performed. The four adults used were fin clipped and then subjected to exon enrichment and Illumina sequencing. On average, ∼95 million (M) reads were obtained for each sample/individual. Each sample was then run through our single-nucleotide polymorphism (SNP) calling pipeline. The exon-enriched sequences for the male and female in each cross were compared to identify homozygous SNPs that distinguished male and female alleles. (B) One hundred zebrafish embryos were collected at five different developmental stages from each cross: 2-cell, 64-cell, 3.5 hpf, 6 hpf and 9 hpf. Polyadenylated RNA was then extracted from total RNA and subjected to Illumina sequencing to produce ∼70 million reads per stage per cross. Reads were mapped to the zebrafish genome (Zv9) using TopHat and quantified using Cufflinks. SNPs identified were then used to identify maternal and paternal mRNAs. FPKM, fragments per kb per million reads; m, male; f, female.
To distinguish the maternal and zygotic transcriptomes during the MZT, we chose to use SNPs within the genomic sequences of the four WIK and SAT adults used (Fig. 1). The identification of homozygous genomic differences between the male and female in each cross could be used to compare variation within the RNA-seq data and thus distinguish between maternal mRNAs and paternal mRNAs, which provide an indicator of zygotic transcription. To identify SNPs, we used Agilent SureSelect baits to enrich the exonic regions of the genomes (exomes) of the four adults used (Kettleborough et al., 2013). Each sample was sequenced on an individual lane of a Genome Analyser II, generating a total of 380,286,918×76 bp reads (supplementary material Table S1). On average, 82.22% of target exon sequence was covered at 20×, a depth required for robust SNP calling (Nielsen et al., 2011). Using a modified version of the 1000 Genomes Project variant calling pipeline, we first identified homozygous genomic differences between the male and female in each cross (1000 Genomes Project Consortium, 2010; Kettleborough et al., 2013). We identified 43,039 homozygous genomic differences, representing 10,893 genes, that distinguished the SAT male and WIK female; 35,055 positions, corresponding to 9764 genes, distinguished the WIK male and SAT female. In total, there were 19,280 SNPs that distinguished male and female genomic sequences in both crosses, corresponding to 6721 protein-coding genes (supplementary material Table S3).
During the MZT, paternal mRNAs should only be present after zygotic transcription has begun. To distinguish between maternal and paternal mRNAs, we only used positions that were informative in both crosses (SATf/WIKm and WIKf/SATm). The first developmental stage at which paternal expression could be identified, in both crosses, was at 3.5 hpf (Fig. 2A). No paternal expression was observed at the 2-cell or 64-cell stage. This result is consistent with previous radioactive UTP incorporation experiments, which concluded that zygotic expression first starts at 3 hpf (Kane and Kimmel, 1993). Within our RNA-seq dataset we could distinguish maternal or zygotic expression for 5045 protein-coding genes. Of these, 1703 genes (34%) were only maternally expressed (Fig. 2B,C; supplementary material Table S4), 3094 genes (61%) had maternal and zygotic expression (Fig. 2D,E; supplementary material Table S5), and 248 genes (5%) were only zygotically expressed (Fig. 2F,G; supplementary material Table S6). Therefore, the majority of genes expressed in the early embryo are both maternally and zygotically expressed, with surprisingly few genes only zygotically expressed. Of the 24,040 protein-coding genes, 19,020 had an FPKM of greater than 0 at one or more of the measured stages (supplementary material Table S2). Extrapolating from this we estimate that, in total, 6467 genes are only maternally expressed, 11,602 are maternally and zygotically expressed, and 951 genes are only zygotically expressed.
Fig. 2.

Maternal and zygotic expression. (A) The developmental stage at which paternal SNPs (green) and the corresponding transcripts (purple) are first detected. Paternal mRNAs, which are an indicator of zygotic transcription, are first detected at 3.5 hpf. The SNPs counted displayed the same expression pattern in both the SATm/WIKf and WIKm/SATf crosses. (B-G) mRNA maternal (red) and paternal (blue) SNP count for three genes displaying only maternal expression (B,C; ehd1b ENSDARG00000014793), maternal and zygotic expression (D,E; tsr2 ENSDARG00000005772) and only zygotic expression (F,G; wnt11 ENSDARG00000004256). (B,D,F) mRNA SNP counts from the SATf and WIKm cross. (C,E,G) mRNA SNP counts from the WIKf and SATm cross. M, maternal; Z, zygotic.
Although the distinction of maternal and zygotic expression focused on protein-coding genes, within the Ensembl 60 annotation there were 4471 non-coding genes (supplementary material Table S2). As zygotic transcription is not detectable at the 2-cell and 64-cell stages, we used these stages to estimate maternal or zygotic non-coding expression. Two hundred and seventy-eight genes had an FPKM of greater than 0 at one or more of the stages. One hundred and fifty-eight genes were expressed at either the 2-cell or 64-cell stage and therefore maternally expressed - although this does not distinguish maternal-only from maternal and zygotic expression. One hundred and twenty genes were not expressed at the 2-cell or 64-cell stage, but were expressed at 3.5, 6 or 9 hpf and were therefore only zygotically expressed. This suggests that the non-coding genes displayed nearly equal contributions of maternal and zygotic-only expression, in contrast to the protein-coding genes.
Post-transcriptional regulation of maternal mRNAs
Although zygotic transcription does not commence until 3 hpf, we observed a number of mRNAs with different maternal mRNA SNP counts and FPKM between the 2-cell and 64-cell stages (Fig. 3A-C; supplementary material Table S2). Using Cuffdiff (Trapnell et al., 2010) and a P-value cut-off of 1×10-6 there were 470 genes with an increased FPKM between the 2-cell and 64-cell stages (supplementary material Table S7) and 570 with a decreased FPKM (supplementary material Table S8).
Fig. 3.
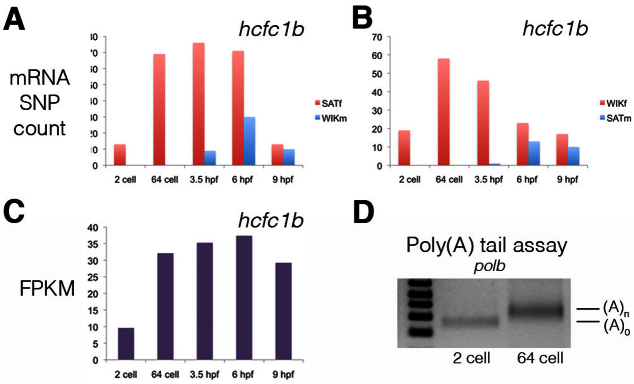
Polyadenylation of maternal mRNAs. (A,B) Significant increases in the maternal mRNA SNP count and FPKM levels were observed from the 2-cell to 64-cell stage. Because these changes occur in the absence of de novo transcription they represent the post-transcriptional regulation of maternal mRNAs. The SNP count of maternal and paternal mRNAs for the gene hcfc1b (ENSDARG00000012519) in the (A) SATf/WIKm cross and (B) WIKf/SATm cross is shown. (C) FPKM for hcfc1b. (D) Polyadenylation tail assay for the gene polb (ENSDARG00000003749). A primer (p1) was first ligated onto the 3′ end of mRNAs and cDNA was generated using a complementary primer (p2). A polb gene-specific primer and p2 were then used to amplify the poly(A) tail. As shown, there was an increase in the poly(A) tail for polb from the 2-cell to the 64-cell stage.
During the generation of the Illumina sequencing libraries, polyadenylated RNA was purified from total RNA. Thus, our results suggest that in the absence of transcription there is widespread post-transcriptional regulation of maternal mRNAs. To test this we performed a polyadenylation tail assay (Sallés and Strickland, 1999). First, a primer (p1) was ligated onto the 3′ end of mRNAs and cDNA was generated using a complementary primer (p2). A gene-specific primer and p2 were then used to amplify the poly(A) tail. This assay demonstrated an increase in the poly(A) tails of maternal mRNAs between the 2-cell and 64-cell stages (Fig. 3D). Consistent with this result, quantitative RT-PCR demonstrated an increase in the amount of poly(A)+ RNA between the 2-cell and 64-cell stages (not shown). Using microarrays, Mathavan et al. (Mathavan et al., 2005) identified increased mRNA levels between the 1- to 4-cell and 64-cell stages. Of the 118 genes identified by Mathavan et al., 107 had an increase in FPKM from the 2-cell to 64-cell stage in the results presented here, and 13 were also identified in our analysis but under the described statistical cut-off (supplementary material Table S7).
Although changes in mRNA levels in the early zebrafish embryo have been identified previously (Mathavan et al., 2005; Aanes et al., 2011), our results are the first to demonstrate that such changes occur in the absence of de novo transcription, and therefore provide the first comprehensive, genome-wide analysis of maternal mRNA post-transcriptional regulation.
Cytoplasmic polyadenylation controls the post-transcriptional regulation of maternal mRNAs
To map the elements controlling the polyadenylation of maternal mRNAs we studied the 3′ UTR of one such gene, sox19b. In an unbiased approach, we cloned different fragments of the sox19b 3′ UTR downstream of the cDNA for the fluorescent protein Venus (Fig. 4A). Embryos were injected with RNA for the Venus-sox19b 3′ UTR fragments and embryos were harvested at the 2-cell and 64-cell stages. We studied the changing polyadenylation levels of these 3′ UTR fragments by quantitative RT-PCR of cDNA generated with oligo(dT) primers. Using this approach we identified only one 3′ UTR fragment (fragment 3) that displayed increased polyadenylation levels from the 2-cell to 64-cell stage (Fig. 4B). An in silico analysis of this UTR fragment revealed the presence of a cytoplasmic polyadenylation element (CPE). Previous work has demonstrated that during oocyte maturation CPEs maintain maternal mRNAs in a dormant state and then release these mRNAs when required (Richter, 2007; Zhang and Sheets, 2009). Deletion of the CPE from the sox19b 3′ UTR fragment abolished the polyadenylation from the 2-cell to 64-cell stages (Fig. 4C).
Fig. 4.

Cytoplasmic polyadenylation elements drive the post-transcriptional regulation of maternal mRNAs. (A) To map the elements controlling polyadenylation of the maternal mRNA sox19b, four different fragments of the 3′ UTR were cloned downstream of the coding sequence of the fluorescent protein Venus. The positions of a cytoplasmic polyadenylation element (CPE) and hexamer, which were contained within fragment 3, are shown. (B) Injection of fertilised zebrafish embryos with Venus-sox19b 3′ UTR RNA demonstrated that only fragment 3 is polyadenylated. The ratio of quantitative PCR levels at the 64-cell stage to 2-cell stage are shown. cDNA was generated with oligo(dT) primers. As a negative control (neg. con.), embryos were injected with Venus RNA without a 3′ UTR. Error bars indicate s.d. (C) A CPE is present within fragment 3 of the sox19b 3′ UTR, which when deleted abolishes polyadenylation. (D) To study the cell cycle-dependent post-transcriptional regulation of cyclin B1, quantitative PCR was performed on cDNA, generated with oligo(dT) primers, derived from embryos synchronised by IVF. Embryos were collected every 3 minutes, starting just after the 2-cell stage. cyclin B1 polyadenylation levels are regulated in a cell cycle-dependent manner, with levels peaking during mitosis.
Intriguingly, during Xenopus early embryonic development CPEs control the cell cycle-dependent post-transcriptional regulation of cyclin B1 and such regulation is required for early embryonic cell cycles (Groisman et al., 2002). The RNA-seq experiments described above were performed on clutches of stage-matched embryos collected after 30 minutes of natural mating. As the cell cycles of the early embryo are ∼15 minutes long (Kane and Kimmel, 1993), these batches of embryos were therefore not perfectly synchronised. The post-transcriptional regulation that we identified (Fig. 3) therefore represents a gradual increase in polyadenylation levels as development progresses. To determine whether cell cycle-dependent post-transcriptional regulation of maternal mRNAs also exists in the early zebrafish embryo, we studied the polyadenylation levels of cyclin B1 by quantitative RT-PCR of cDNA generated with oligo(dT) primers. Embryos were synchronised using in vitro fertilisation (IVF) and then harvested every 3 minutes, starting just after the 2-cell stage (Fig. 4D). This demonstrated that, consistent with previous findings (Groisman et al., 2002), cyclin B1 polyadenylation levels are regulated in a cell cycle-dependent manner, with highest levels during mitosis.
Therefore, in the early embryo there are several different modes of maternal mRNA post-transcriptional regulation, which are regulated by CPEs in the 3′ UTRs of such mRNAs. As development proceeds, CPEs lead to a gradual increase in the polyadenylation levels of mRNAs such as sox19b, whereas CPEs in the 3′ UTR of cyclin B1 are required for cell cycle-dependent regulation (Groisman et al., 2002).
Zygotic transcription initiation
To determine the transcriptional competence of the early zebrafish embryo, we injected fertilised embryos with plasmid DNA containing the EF1α promoter upstream of the coding sequence for the fluorescent protein CFP (Fig. 5A). Transcription of the plasmid DNA was detected 1 hour after injection, at approximately the 16-cell stage, and co-injection with the RNA polymerase II inhibitor α-amanitin abolished transcription. These results demonstrated that, consistent with findings for fertilised Xenopus embryos (Newport and Kirschner, 1982b), the early zebrafish embryo is capable of transcribing naked plasmid DNA. However, the chromatin state of genomic DNA is incompatible with transcription and therefore maintains transcriptional quiescence (Newport and Kirschner, 1982b). Treatment of embryos with the translation inhibitor cycloheximide blocks cell cycle progression and has previously been used to demonstrate that the cell cycles of the early embryo titrate out factors that maintain genomic DNA in a chromatin state that is incompatible with transcription (Newport and Kirschner, 1982b; Kimelman et al., 1987). This implies a model in which cell cycle progression leads to a switch in genomic DNA to a transcriptionally competent state and, subsequently, to the activation of zygotic transcription.
Fig. 5.

Zygotic transcription initiation in the zebrafish embryo. (A) To test the transcriptional competence of the early zebrafish embryo, fertilised embryos were injected with plasmid DNA containing the EF1α promoter upstream of the coding sequence for the fluorescent protein CFP. Lane 1, size ladder; lane 2, uninjected embryos; lane 3, embryos injected with plasmid; lane 4, embryos co-injected with plasmid DNA and the RNA polymerase II inhibitor α-amanitin. RT-PCR for CFP is shown on the left, with control cyclin B1 RT-PCR on the right. (B) Experimental design to identify the first zygotic genes to be expressed. Embryos were synchronised by IVF and half of the clutch was exposed to the translation blocker cycloheximide at the 128-cell stage. Untreated and treated embryos were then collected every 15 minutes, starting just after the 128-cell stage. RNA-seq was then performed on the samples. (C) FPKM for a sample of genes that are expressed only zygotically (supplementary material Table S6) demonstrates that the sampled stages (labelled A-F) span the period of normal zygotic transcription initiation. (D,E) In embryos treated with cycloheximide (128c), some of the zygotically expressed genes, such as vox (D), are unaffected by treatment. However, some zygotic genes, such as claudin e (E), fail to be expressed in the treated embryos. WT, wild type, untreated with cycloheximide.
To investigate the regulation of zygotic transcription initiation we sought to identify the first zygotic genes to be expressed. A large clutch of zebrafish embryos were synchronised by IVF and half were then treated with the translation inhibitor cycloheximide at the 128-cell stage, which efficiently blocked cell cycle progression. Untreated (supplementary material Table S9) and cycloheximide-treated embryos (supplementary material Table S10) were then collected every 15 minutes at six time points, starting just after the 128-cell stage, and subjected to RNA-seq (Fig. 5C). Focussing on those genes that are only zygotically expressed (supplementary material Table S6), we observed that zygotic transcription commenced normally in the untreated embryos (Fig. 5C; supplementary material Table S11). In the cycloheximide-treated embryos (supplementary material Table S12), we observed that some zygotic genes, such as vox, appeared to be activated normally (Fig. 5D), whereas others, such as claudin e, failed to be expressed (Fig. 5E). These results demonstrate that, although the activation of zygotic transcription depends on the establishment of a particular chromatin state, the ability to activate zygotic transcription is established before the 128-cell stage for some genes and after for others, in a manner that requires protein synthesis.
DISCUSSION
The development of new sequencing technologies permits the study of mRNA expression to an unprecedented level. Recent RNA-seq experiments have analysed the changing transcriptome in the early zebrafish embryo (Aanes et al., 2011; Vesterlund et al., 2011; Pauli et al., 2012), but the work described here provides the first systematic identification of maternal and paternal mRNAs in this species. Previous microarray analyses had suggested that transcription occurs before the MZT in the zebrafish (Mathavan et al., 2005) and RNA-seq experiments have also described changes in mRNA levels prior to the MZT, suggesting that such changes were due to the post-transcriptional regulation of mRNAs (Aanes et al., 2011). By identifying zygotically expressed genes (Fig. 2) and then applying tight temporal resolution (Fig. 5B,C), our work confirms that, consistent with previous radioactivity incorporation experiments (Kane and Kimmel, 1993), zygotic transcription begins after ten cell cycles. Through the identification of maternal and paternal mRNAs our work is the first to demonstrate that, in the early embryo prior to zygotic transcription initiation, changes in mRNA levels are due to the post-transcriptional regulation of maternal mRNAs and not to transcription.
Our work indicates that there are several different types of post-transcriptional regulation in the early embryo (Fig. 4). As development proceeds there is a gradual increase in the polyadenylation levels of certain maternal mRNAs (supplementary material Table S7), whereas others gradually become de-adenylated (supplementary material Table S8). On top of such regulation, mRNAs such as cyclin B1 are polyadenylated and de-adenylated in a cell cycle-coupled manner (Fig. 4D). CPEs in the 3′ UTRs of mRNAs are required for the gradual polyadenylation of maternal mRNAs following fertilisation (Fig. 4C). Intriguingly, in Xenopus such elements are also required for the cell cycle-coupled polyadenylation of cyclin B1 (Groisman et al., 2002). Thus, CPEs and the proteins that bind to them play a central role in controlling different modes of post-transcriptional regulation in the early embryo.
Although the early embryo is transcriptionally competent (Fig. 5A), the state of genomic DNA is incompatible with transcription (Newport and Kirschner, 1982b). Early embryonic cell cycles titrate out the factors that maintain transcriptional quiescence, leading ultimately to zygotic transcription initiation. The nucleocytoplasmic ratio is a critical determinant of cell cycle progression in the early embryo and therefore of the titration of the repressive factors (Newport and Kirschner, 1982a; Newport and Kirschner, 1982b; Kane and Kimmel, 1993). Similarly, maternal mRNAs are crucial for this, as inhibition of mRNA translation blocks cell cycle progression. However, if zygotic transcription initiation solely required the establishment of a transcriptionally competent state then, depending on the stage at which embryos are exposed to cycloheximide, zygotic transcription would either all initiate or all fail to commence. Instead, we observed that when embryos were exposed to cycloheximide at the 128-cell stage the expression of some zygotic genes is unaffected whereas others fail to be expressed (Fig. 5D,E). Therefore, although the translation of maternal mRNAs is required for cell cycle progression and for the subsequent establishment of transcriptional competence, maternal mRNAs are also required for different modes of zygotic transcription initiation. For some genes, the ability to initiate zygotic transcription is established before the 128-cell stage, whereas for others it is only established afterwards. We do not yet know how this differential regulation is achieved.
A second model for the control of the MZT suggests that a cell cycle-independent clock, which is triggered following fertilisation, is crucial. In support of this model, the degradation of Cyclin A and Cyclin E1 protein during Xenopus development is independent of the nucleocytoplasmic ratio and occurs at a given time or developmental stage post-fertilisation (Howe et al., 1995; Howe and Newport, 1996). Similarly, by manipulating DNA content in the Drosophila embryo, it has been demonstrated that although the initiation of zygotic transcription is dependent on the nucleocytoplasmic ratio for some genes, the majority of zygotic expression is dependent on time post-fertilisation or developmental stage (Lu et al., 2009). Amongst the zebrafish mRNAs that display gradual polyadenylation after fertilisation (supplementary material Table S7), there are several genes, including sox19b, with GO terms that are associated with transcription. The gradual accumulation of these factors following fertilisation could be critical to the different modes of zygotic transcription initiation. However, although it is possible that a threshold accumulation of such factors is required for zygotic transcription initiation, the experiments described here do not distinguish this model from one in which certain loci are more affected than others by the cell cycle-dependent depletion of factors that maintain transcriptional quiescence.
The Wnt signalling pathway is an early regulator of zygotic transcription in the embryo. Wnt signalling, through β-catenin, is required for the expression of genes on the dorsal side of the early embryo (Kelly et al., 2000; Dougan et al., 2003; Tao et al., 2005; Bellipanni et al., 2006), and a combination of Wnt and BMP signalling acts as an organiser on the ventral side of the embryo (Agathon et al., 2003; Harvey et al., 2010). Wnt signalling could therefore influence the induction of zygotic transcription. Under this model one would predict that the first zygotically expressed genes would be spatially restricted to regions of the embryo where Wnt signalling is present. Indeed, bozozok (also known as dharma), a dorsally expressed gene that is a known target of the Wnt signalling pathway (Bellipanni et al., 2006), is one of the first genes to be expressed in the early embryo. However, foxi1 (ENSDARG00000019659), which is also one of the first zygotically expressed genes, is spatially restricted to the presumptive epidermis, repressed by Wnt signalling and induced by the BMP signalling pathway (Rhinn et al., 2005; Hans et al., 2007). These observations suggest that the differential regulation of zygotic transcription initiation is not simply achieved by one particular signalling pathway.
Acknowledgments
We thank Pete Ellis, Nathalie Bason and members of the Wellcome Trust Sanger Institute Research Support Facility for technical support, and members of the D.S. laboratory for advice, especially John Collins.
Footnotes
Funding
This work was funded by a Wellcome Trust grant and Medical Research Council core support [programme U117597140] to J.C.S., and by the Wellcome Trust Sanger Institute. Deposited in PMC for immediate release.
Competing interests statement
The authors declare no competing financial interests.
Author contributions
This project was conceived and executed by S.H. Experiments were designed by S.A.H., D.S. and J.C.S. Bioinformatics were performed by I.S. and R.W. R.K. and F.F. provided assistance with exome sequencing and IVF, respectively.
Supplementary material
Supplementary material available online at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.095091/-/DC1
References
- Aanes H., Winata C. L., Lin C. H., Chen J. P., Srinivasan K. G., Lee S. G., Lim A. Y., Hajan H. S., Collas P., Bourque G., et al. (2011). Zebrafish mRNA sequencing deciphers novelties in transcriptome dynamics during maternal to zygotic transition. Genome Res. 21, 1328–1338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agathon A., Thisse C., Thisse B. (2003). The molecular nature of the zebrafish tail organizer. Nature 424, 448–452 [DOI] [PubMed] [Google Scholar]
- Bellipanni G., Varga M., Maegawa S., Imai Y., Kelly C., Myers A. P., Chu F., Talbot W. S., Weinberg E. S. (2006). Essential and opposing roles of zebrafish beta-catenins in the formation of dorsal axial structures and neurectoderm. Development 133, 1299–1309 [DOI] [PubMed] [Google Scholar]
- Collins J. E., White S., Searle S. M., Stemple D. L. (2012). Incorporating RNA-seq data into the zebrafish Ensembl genebuild. Genome Res. 22, 2067–2078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalle Nogare D. E., Pauerstein P. T., Lane M. E. (2009). G2 acquisition by transcription-independent mechanism at the zebrafish midblastula transition. Dev. Biol. 326, 131–142 [DOI] [PubMed] [Google Scholar]
- Djebali S., Davis C. A., Merkel A., Dobin A., Lassmann T., Mortazavi A., Tanzer A., Lagarde J., Lin W., Schlesinger F., et al. (2012). Landscape of transcription in human cells. Nature 489, 101–108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougan S. T., Warga R. M., Kane D. A., Schier A. F., Talbot W. S. (2003). The role of the zebrafish nodal-related genes squint and cyclops in patterning of mesendoderm. Development 130, 1837–1851 [DOI] [PubMed] [Google Scholar]
- Edgar B. A., Schubiger G. (1986). Parameters controlling transcriptional activation during early Drosophila development. Cell 44, 871–877 [DOI] [PubMed] [Google Scholar]
- 1000 Genomes Project Consortium (2010). A map of human genome variation from population-scale sequencing. Nature 467, 1061–1073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giraldez A. J., Mishima Y., Rihel J., Grocock R. J., Van Dongen S., Inoue K., Enright A. J., Schier A. F. (2006). Zebrafish MiR-430 promotes deadenylation and clearance of maternal mRNAs. Science 312, 75–79 [DOI] [PubMed] [Google Scholar]
- Groisman I., Jung M. Y., Sarkissian M., Cao Q., Richter J. D. (2002). Translational control of the embryonic cell cycle. Cell 109, 473–483 [DOI] [PubMed] [Google Scholar]
- Hamatani T., Carter M. G., Sharov A. A., Ko M. S. (2004). Dynamics of global gene expression changes during mouse preimplantation development. Dev. Cell 6, 117–131 [DOI] [PubMed] [Google Scholar]
- Hans S., Christison J., Liu D., Westerfield M. (2007). Fgf-dependent otic induction requires competence provided by Foxi1 and Dlx3b. BMC Dev. Biol. 7, 5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey S. A., Tümpel S., Dubrulle J., Schier A. F., Smith J. C. (2010). No tail integrates two modes of mesoderm induction. Development 137, 1127–1135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe K., Clark M. D., Torroja C. F., Torrance J., Berthelot C., Muffato M., Collins J. E., Humphray S., McLaren K., Matthews L., et al. (2013). The zebrafish reference genome sequence and its relationship to the human genome. Nature 496, 498–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe J. A., Newport J. W. (1996). A developmental timer regulates degradation of cyclin E1 at the midblastula transition during Xenopus embryogenesis. Proc. Natl. Acad. Sci. USA 93, 2060–2064 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe J. A., Howell M., Hunt T., Newport J. W. (1995). Identification of a developmental timer regulating the stability of embryonic cyclin A and a new somatic A-type cyclin at gastrulation. Genes Dev. 9, 1164–1176 [DOI] [PubMed] [Google Scholar]
- Kane D. A., Kimmel C. B. (1993). The zebrafish midblastula transition. Development 119, 447–456 [DOI] [PubMed] [Google Scholar]
- Kane D. A., Hammerschmidt M., Mullins M. C., Maischein H. M., Brand M., van Eeden F. J., Furutani-Seiki M., Granato M., Haffter P., Heisenberg C. P., et al. (1996). The zebrafish epiboly mutants. Development 123, 47–55 [DOI] [PubMed] [Google Scholar]
- Kelly C., Chin A. J., Leatherman J. L., Kozlowski D. J., Weinberg E. S. (2000). Maternally controlled (beta)-catenin-mediated signaling is required for organizer formation in the zebrafish. Development 127, 3899–3911 [DOI] [PubMed] [Google Scholar]
- Kettleborough R., Busch-Nentwich E. M., Harvey S. A., de Bruijn E., van Eeden F., Sealy I., White R., Dooley C., Herd C., Nijman I. J., et al. (2013). A systematic genome-wide analysis of zebrafish protein-coding gene function. Nature 496, 494–497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimelman D., Kirschner M., Scherson T. (1987). The events of the midblastula transition in Xenopus are regulated by changes in the cell cycle. Cell 48, 399–407 [DOI] [PubMed] [Google Scholar]
- Kimmel C. B., Ballard W. W., Kimmel S. R., Ullmann B., Schilling T. F. (1995) Stages of embryonic development of the zebrafish. Dev. Dyn. 203, 253–310 [DOI] [PubMed] [Google Scholar]
- Lu X., Li J. M., Elemento O., Tavazoie S., Wieschaus E. F. (2009). Coupling of zygotic transcription to mitotic control at the Drosophila mid-blastula transition. Development 136, 2101–2110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathavan S., Lee S. G., Mak A., Miller L. D., Murthy K. R., Govindarajan K. R., Tong Y., Wu Y. L., Lam S. H., Yang H., et al. (2005). Transcriptome analysis of zebrafish embryogenesis using microarrays. PLoS Genet. 1, 260–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neph S., Vierstra J., Stergachis A. B., Reynolds A. P., Haugen E., Vernot B., Thurman R. E., John S., Sandstrom R., Johnson A. K., et al. (2012). An expansive human regulatory lexicon encoded in transcription factor footprints. Nature 489, 83–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newport J., Kirschner M. (1982a). A major developmental transition in early Xenopus embryos: I. characterization and timing of cellular changes at the midblastula stage. Cell 30, 675–686 [DOI] [PubMed] [Google Scholar]
- Newport J., Kirschner M. (1982b). A major developmental transition in early Xenopus embryos: II. Control of the onset of transcription. Cell 30, 687–696 [DOI] [PubMed] [Google Scholar]
- Nielsen R., Paul J. S., Albrechtsen A., Song Y. S. (2011). Genotype and SNP calling from next-generation sequencing data. Nat. Rev. Genet. 12, 443–451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ozsolak F., Milos P. M. (2011). RNA sequencing: advances, challenges and opportunities. Nat. Rev. Genet. 12, 87–98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pauli A., Valen E., Lin M. F., Garber M., Vastenhouw N. L., Levin J. Z., Fan L., Sandelin A., Rinn J. L., Regev A., et al. (2012). Systematic identification of long noncoding RNAs expressed during zebrafish embryogenesis. Genome Res. 22, 577–591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhinn M., Lun K., Luz M., Werner M., Brand M. (2005). Positioning of the midbrain-hindbrain boundary organizer through global posteriorization of the neuroectoderm mediated by Wnt8 signaling. Development 132, 1261–1272 [DOI] [PubMed] [Google Scholar]
- Richter J. D. (2007). CPEB: a life in translation. Trends Biochem. Sci. 32, 279–285 [DOI] [PubMed] [Google Scholar]
- Sallés F. J., Strickland S. (1999). Analysis of poly(A) tail lengths by PCR: the PAT assay. Methods Mol. Biol. 118, 441–448 [DOI] [PubMed] [Google Scholar]
- Spitz F., Furlong E. E. (2012). Transcription factors: from enhancer binding to developmental control. Nat. Rev. Genet. 13, 613–626 [DOI] [PubMed] [Google Scholar]
- Stack J. H., Newport J. W. (1997). Developmentally regulated activation of apoptosis early in Xenopus gastrulation results in cyclin A degradation during interphase of the cell cycle. Development 124, 3185–3195 [DOI] [PubMed] [Google Scholar]
- Tadros W., Lipshitz H. D. (2009). The maternal-to-zygotic transition: a play in two acts. Development 136, 3033–3042 [DOI] [PubMed] [Google Scholar]
- Tao Q., Yokota C., Puck H., Kofron M., Birsoy B., Yan D., Asashima M., Wylie C. C., Lin X., Heasman J. (2005). Maternal wnt11 activates the canonical wnt signaling pathway required for axis formation in Xenopus embryos. Cell 120, 857–871 [DOI] [PubMed] [Google Scholar]
- Trapnell C., Williams B. A., Pertea G., Mortazavi A., Kwan G., van Baren M. J., Salzberg S. L., Wold B. J., Pachter L. (2010). Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28, 511–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vesterlund L., Jiao H., Unneberg P., Hovatta O., Kere J. (2011). The zebrafish transcriptome during early development. BMC Dev. Biol. 11, 30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y., Sheets M. D. (2009). Analyses of zebrafish and Xenopus oocyte maturation reveal conserved and diverged features of translational regulation of maternal cyclin B1 mRNA. BMC Dev. Biol. 9, 7 [DOI] [PMC free article] [PubMed] [Google Scholar]