

Abstract
To develop a functional nervous system, axons must initially navigate through a complex environment, directed by guidance ligands and receptors. These receptors must link to intracellular signaling cascades to direct axon pathfinding decisions. The Abelson tyrosine kinase (Abl) plays a crucial role in multiple Drosophila axon guidance pathways during development, though the mechanism by which Abl elicits a diverse set of guidance outputs is currently unknown. We identified Abl in a genetic screen for genes that contribute to Netrin-dependent axon guidance in midline-crossing (commissural) neurons. We find that Abl interacts both physically and genetically with the Netrin receptor Frazzled, and that disrupting this interaction prevents Abl from promoting midline axon crossing. Moreover, we find that Abl exerts its diverse activities through at least two different mechanisms: (1) a partly kinase-independent, structural function in midline attraction through its C-terminal F-actin binding domain (FABD) and (2) a kinase-dependent inhibition of repulsive guidance pathways that does not require the Abl C terminus. Abl also regulates motor axon pathfinding through a non-overlapping set of functional domains. These results highlight how a multifunctional kinase can trigger diverse axon guidance outcomes through the use of distinct structural motifs.
Keywords: Abelson, Netrin, Frazzled, DCC, Axon guidance
INTRODUCTION
During nervous system development, embryonic axons navigate through a complex environment to initiate the establishment of neural circuits. This process is accomplished by the growth cone, a motile structure at the growing end of axons. Growth cones respond to attractant and repellant cues in the environment through transmembrane guidance receptors. To alter growth cone motility, guidance receptors must signal to the underlying growth cone cytoskeleton. Thus, to understand how guidance receptors direct axon pathfinding, it is essential to identify downstream signaling molecules that allow these receptors to communicate with elements of the cytoskeleton. The bifunctional guidance cue Netrin and its attractive receptor Frazzled (Fra; also known as DCC) comprise one conserved pathway that controls axon navigation in vivo. Netrin is necessary for axon guidance in several neural cell types, but is particularly well-studied in the context of midline axon guidance of commissural neurons (Lai Wing Sun et al., 2011). Much of our understanding of Netrin signal transduction arises from in vitro studies of isolated neurons, which has led to the identification of multiple signaling pathways downstream of Netrin (Round and Stein, 2007). However, these in vitro findings have yet to be corroborated in vivo.
To identify Netrin signaling mechanisms in vivo, we sought to identify genes that contribute to midline axon crossing in the Drosophila embryonic CNS, a context in which Netrin signaling, through its Fra receptor, is essential (Kolodziej et al., 1996; Mitchell et al., 1996). In a genetic screen to identify genes that interact with the Fra pathway, we identified the cytoplasmic tyrosine kinase Abelson (Abl) as a regulator of Netrin-dependent midline axon crossing in commissural neurons. Abl plays a complex role in Drosophila axon guidance, functioning in attractive (Elkins et al., 1990; Forsthoefel et al., 2005; Gertler et al., 1989; Liebl et al., 2000), repulsive (Bashaw et al., 2000; Hsouna et al., 2003; Lee et al., 2004; Wills et al., 2002) and adhesive (Crowner et al., 2003; Wills et al., 1999a; Wills et al., 1999b) guidance pathways. In commissural neurons, Abl promotes midline axon crossing and interacts genetically with both Netrin and fra and can bind to the cytoplasmic domain of Fra (Forsthoefel et al., 2005). It is unclear from this work whether Abl functions in the Netrin pathway in commissural neurons to promote midline axon crossing or, alternatively, in a parallel pathway. In midline ipsilateral axons, Abl both promotes and inhibits Slit- and Robo-dependent midline repulsion, though the mechanism by which these apparently paradoxical functions occur is currently unknown (Bashaw et al., 2000; Hsouna et al., 2003; Lee et al., 2004; Wills et al., 2002).
We show here that Abl interacts both physically and genetically with Fra and, based on molecular genetic approaches, we suggest that it acts cell-autonomously to promote Netrin-dependent midline axon crossing through its C-terminal F-actin binding domain (FABD). We also find that the function of Abl in promoting midline axon crossing is partly kinase independent, in contrast to its role in repulsive guidance pathways. These findings shed light on how a single protein can signal diverse axon guidance responses through the use of different structural motifs.
MATERIALS AND METHODS
Abl-GFP molecular biology
All transgenic constructs were cloned into a pUAST vector containing 10xUAS and an attB site for PhiC31-mediated targeted insertion (p10UAST-attB). All Abl constructs were cloned from pUAS-AblGFP (Fox and Peifer, 2007) in frame to a C-terminal GFP epitope with the following linker sequence upstream of GFP: GGACTAGTGATTGGAGCT. All Abl constructs contain the following sequence upstream of the start codon (underlined): CACCGCGGCCGCTGGCAAATG. All Abl-GFP constructs were cloned into p10UAST-attB as NotI/XbaI fragments. AblNGFP (amino acids 1-644 of Abl) was generated by serial overlap extension PCR. AblCGFP (amino acids 645-1638) was generated as a PCR fragment by placing ATG upstream of codon 645 of Abl. Abl point mutations were generated by PCR mutagenesis: AblK417N, AblW243K, AblR297K. See supplementary material Table S2 for primer sequences. AblΔFABD (AA 1499-1638 deleted) was generated by PCR mutagenesis. Fra-Myc (Garbe and Bashaw, 2007) and Robo-Myc (Bashaw and Goodman, 1999) were described previously. Fra-9YF was generated by step-wise PCR mutagenesis of individual or multiple sites in close proximity. Mutated tyrosine residues are Y1113, Y1170, Y1189, Y1193, Y1207, Y1212, Y1247, Y1250 and Y1313 (O’Donnell and Bashaw, 2013). For p10UAST-attB-NetB-Myc, a myc-tagged NetB cDNA was amplified from genomic DNA of transgenic flies (Mitchell et al., 1996). This amplicon was cloned into p10UAST-attB using EcoR1/Xba1 sites. All constructs were fully sequenced. Transgenic flies were generated by Best Gene (Chino Hills, CA, USA).
Genetics
The following alleles were used in this study: fra3, fra4, fra6, fraDf(2R)BSC3, NetABΔ, slit2, Abl1, Abl2, Abl4, egMZ360 (eg-Gal4), apGal4, exexGal4 (Hb9-Gal4). The following transgenes were used: (1) P{UAS-AblK417N}2 (Wills et al., 1999b), (2) P{UAS-Abl-GFP}86Fb, (3) P{UAS-AblKN-GFP}86Fb, (4) P{UAS-AblN-GFP}86Fb, (5) P{UAS-AblC-GFP}86Fb, (6) P{UAS-AblR297K-GFP}, (7) P{UAS-AblW243K-GFP}, (8) P{UAS-AblΔFABD-GFP}, (9) P{UAS-TauMycGFP}2, (10) P{GAL4-elav.L}3, (11) P{UAS-FraΔC-HA}8 (DNFra-weak), (12) P{UAS-FraΔC-HA}2 (DNFra-moderate). All crosses were performed using males or balanced females at 25°C. Embryos were genotyped using a combination of marked balancer chromosomes or the presence of linked transgenes. Where possible, all comparative phenotypes were analyzed in the same genetic background to limit the effects of potential modifier mutations.
Immunostaining and imaging
Dechorionated, formaldehyde-fixed, methanol-devittellinized embryos were fluorescently stained using standard methods (Kidd et al., 1998). The following antibodies were used in this study: mouse mAb BP102 [Developmental Studies Hybidoma Bank (DSHB); 1:100], mouse anti-Fasciclin-II/mAb 1D4 (DSHB; 1:100), rabbit anti-GFP (Invitrogen #A11122; 1:500), rabbit anti-c-Myc (Sigma C3956; 1:500), Alexa-647 conjugated goat-anti-HRP (Jackson ImmunoResearch #123-605-021; 1:250), Cyanine 3-conjugated goat anti-mouse (Jackson ImmunoResearch #115-165-003; 1:1000), Alexa-488-conjugated goat anti-rabbit (Molecular Probes #A11008; 1:500). Embryos were mounted in 70% glycerol in PBS. Fluorescent mRNA in situ hybridization was performed as described (Garbe and Bashaw, 2007). Phenotypes were analyzed and images were acquired using a spinning disk confocal system (Perkin Elmer) built on a Nikon Ti-U inverted microscope using a Nikon OFN25 60× objective with a Hamamatsu C10600-10B CCD camera and Yokogawa CSU-10 scanner head with Volocity imaging software. Images were processed using ImageJ. Immunofluorescence quantification was performed using ImageJ.
Cell culture and biochemistry
For co-immunoprecipitations, 106 Drosophila S2R+ cells were transfected at 25°C in Schneider’s medium containing 10% fetal calf serum with indicated constructs (0.5 μg Fra-Myc/Robo-Myc, 1 μg Abl-FL-GFP, 0.25 μg Abl-N-GFP) along with 0.5 μg pRmHA3-Gal4 using the Effectene Transfection Reagent (Qiagen). After 24 hours, cells were induced with 1 mM CuSO4. Forty-eight hours post-transfection, cells were washed once with PBS, then lysed in 1 ml lysis buffer [50 mM Tris, pH 8.0, 100 mM NaCl, 1 mM MgCl2, 0.5% NP-40, 10 mM NaF, 2 mM Na3VO4, 1 mM phenylmethanesulphonylfluoride (PMSF), 1× Complete protease inhibitor cocktail (Roche)]. Lysates were pre-cleared with protein-G agarose (Invitrogen), followed by addition of 2.5 μl of rabbit anti-Myc antibody (Millipore, 06-549) for 2 hours at 4°C. Protein G agarose was added (30 μl per immunoprecipitation), followed by three washes in lysis buffer. For western blots, we used mouse anti-Myc (9E10, DSHB; 1:1000) and mouse anti-GFP (Roche; 1:1000). Quantitative analysis of biochemical results was performed using ImageJ. Statistical comparisons were performed using Student’s t-test.
Phenotypic quantification
For EW neuron crossing phenotypes, whole-mount embryos were analyzed at stage 15 and 16. Eight abdominal segments were analyzed per embryo, and for each embryo the percentage of non-crossing segments was calculated. A segment was considered ‘non-crossing’ when both clusters of EW axons (six axons per segment) failed to reach the midline. Standard error (s.e.m.) was based on the number of embryos per genotype. For apterous ectopic axon crossing phenotypes, whole-mount embryos were analyzed at stage 17. Eight abdominal segments were scored per embryo. When a segment contained a continuous crossing projection of at least the thickness of incoming axons from ap cell bodies, it was considered an ectopic cross. For ISNb stalling, stage 17 embryos were filleted. A segment was considered to have stalled when ISNb axons could clearly be found within the ventral muscle field but had not reached the muscle 12/13 cleft. Segments with a ‘bypass’ phenotype were not scored. For ISNb bypass phenotypes, a segment was considered ‘full bypass’ when no ISNb axons entered the ventral muscle field and the dorsal intersegmental motor nerve (ISN) was substantially thickened. A ‘partial bypass’ was scored when one or more axons were seen entering the ventral muscle field and ISN thickening could be observed. Ten abdominal hemisegments were analyzed per embryo. Muscles were identified using differential interference contrast optics. For statistical analysis, comparisons were made using generalized estimate equations (GEE) for clustered binary data, using ‘geepack’ within R software (Halekoh et al., 2006; Yan and Fine, 2004). Correlation structure was chosen based on calculation of quasi-log-likelihood under the independence model information criterion (QIC) and correlation information criterion (CIC) as described (Pan:2001vh; Pan, 2001; Hin and Wang, 2009). For multiple comparisons, a post-hoc Bonferroni correction was applied. P-values are based on corresponding Wald statistics.
RESULTS
Abl mutations enhance frazzled loss of function in commissural axon pathfinding
To identify genes involved in commissural axon guidance, we performed a genetic screen in Drosophila embryos in which midline Netrin signaling is reduced. We reduced Netrin signaling through expression of a dominant-negative Fra receptor (DN-Fra) (Garbe et al., 2007) in a population of commissural neurons that are eagle-Gal4-positive. We have seen that in this genetic background, heterozygous mutations in genes that normally promote midline axon crossing exacerbate crossing defects in a subset these eagle-Gal4-positive neurons, the EW neurons (Yang et al., 2009). We employed this sensitized background to screen heterozygous deletions on the Drosophila third chromosome. From this screen, we identified Abl as a dominant enhancer of midline crossing defects (supplementary material Fig. S1, Table S1). We found that Abl mutations also dominantly enhance midline-crossing defects in EW neurons when endogenous fra function is reduced using a hypomorphic allelic combination (frahypo; Fig. 1A,B). We observed this enhancement using multiple Abl loss-of-function alleles, suggesting that Abl is the gene contributing to midline crossing in this background (Fig. 1B,C; supplementary material Fig. S1G). Additionally, we found that homozygous loss of Abl results in significant midline-crossing defects in embryos expressing a weak DN-Fra construct (Fig. 2B; supplementary material Fig. S1H). The contribution of Abl to midline crossing has been previously reported; however, the phenotype of fra;Abl double mutants, which is stronger than either fra or Abl alone, suggests that Abl functions at least in part through a parallel pathway to promote midline crossing (Forsthoefel et al., 2005). This is not surprising given the proposed dual role of Abl in Slit- and Robo-dependent midline repulsion (Bashaw et al., 2000; Hsouna and VanBerkum, 2008; Hsouna et al., 2003; Lee et al., 2004; Wills et al., 2002). However, this interpretation is complicated owing to a substantial maternal contribution of Abl mRNA, which may compensate for zygotic loss of Abl in the embryonic CNS (Bennett and Hoffmann, 1992; Grevengoed et al., 2001). Thus, a role for Abl in the Netrin/Fra pathway has not been clearly demonstrated in commissural neurons. To determine whether the dominant interactions we observe in fra hypomorphs reflect a role for Abl in Netrin signaling, we performed the same manipulations in Netrin null mutants (NetABΔ) (Brankatschk and Dickson, 2006). We reasoned that if Abl functions in the Netrin/Fra pathway, Abl mutations would not enhance NetAB null mutants (in contrast to frahypo mutants) because Netrin signaling is eliminated in this background. Indeed, we observe that Abl mutations do not dominantly enhance EW crossing defects in NetAB mutants (Fig. 1C). Similarly, when Abl mutations are introduced into fra null mutants, we do not observe enhancement of EW crossing defects, suggesting that the dominant interactions we observe in fra hypomorphs are likely to be due to a role in the Netrin/Frazzled pathway in these neurons (Fig. 1C).
Fig. 1.
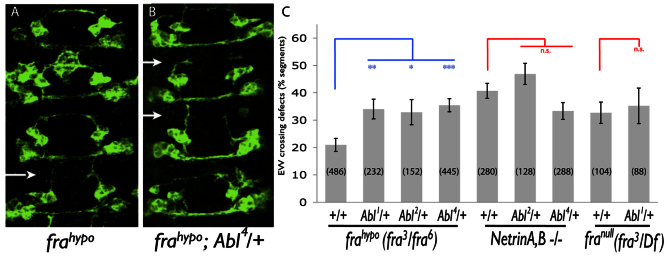
Abl functions in the Netrin pathway to promote EW midline axon crossing. (A,B) Stage 15 embryos expressing TauMycGFP in eagle-positive EW neurons. Axons are visualized with anti-GFP. Four segments are shown. Anterior is up. (A) A fra3/fra6 hypomorphic mutant embryo. Occasional defects in EW midline axon crossing occur (arrow). (B) A fra3/fra6; Abl4/+ embryo. EW crossing defects are enhanced (arrows). (C) Quantification of EW midline-crossing defects in fra and Netrin mutants. *P<0.05, **P<0.01, ***P<0.001. n.s., not significant. Error bars indicate s.e.m. Number of segments scored is shown in parentheses.
Fig. 2.
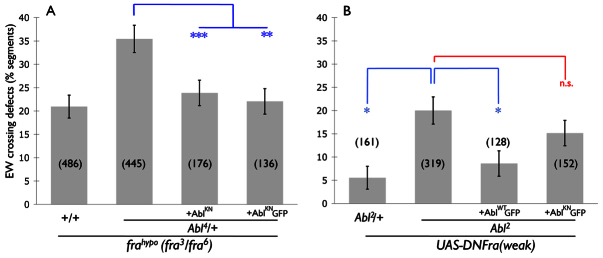
The pro-crossing function of Abl relies partly on its kinase activity. (A,B) Quantification of EW axon crossing defects in Abl mutants. (A) frahypo embryos. Expression of kinase-inactive Abl (AblKN) in EW neurons rescues the dominant interaction between fra and Abl. Data from two independent transgenic UAS-AblKN lines are shown. (B) Embryos expressing a weak DN-Fra transgene, which causes no defects in WT embryos (data not shown). AblWTGFP rescues midline-crossing defects in Abl2 homozygous mutants, though AblKNGFP does not. *P<0.05, **P<0.01, ***P<0.001. n.s., not significant. Error bars indicate s.e.m. Number of segments scored is shown in parentheses. Genotypes are as follows: (A) frahypo denotes fra3,[UAS-TauMycGFP]/fra6; [eg-Gal4]/+; Abl4/+ denotes fra3,[UAS-TauMycGFP]/fra6; Abl4,[eg-Gal4]/+; these two are a repetition of data shown in Fig. 1. +AblKN denotes fra3,[UAS-TauMycGFP]/fra6, [UAS-AblKN]2; Abl4,[eg-Gal4]/+; and +AblKNGFP denotes fra3,[UAS-TauMycGFP]/fra6; Abl4,[eg-Gal4]/[UAS-AblKNGFP]. (B) abl2/+ denotes [UAS-DN-Fra]#8/[UAS-TauMycGFP]; Abl2, [eg-Gal4] / +. On the right, all are [UAS-DN-Fra]#8/[UAS-TauMycGFP]; Abl2, [eg-Gal4]/Abl2, [UAS-AblXGFP].
Abl promotes EW commissural axon crossing cell-autonomously
To date, all of the identified axon guidance functions of Abl in Drosophila have been attributed to its kinase activity. Though a kinase-independent role for Abl has been identified in Drosophila, the nature of this function is unknown (Henkemeyer et al., 1990). Additionally, as rescue experiments have not been performed in commissural neurons, an autonomous role in neurons for the pro-midline crossing function of Abl has not been demonstrated. To address these issues, we constructed transgenic flies that express either wild-type (AblWT) or kinase-inactive (AblKN) Abl (Henkemeyer et al., 1990) fused to GFP under the control of Gal4. We sought to rescue EW axon midline crossing defects cell-autonomously in embryos in which Abl function is reduced. For technical reasons, we have been unable to analyze EW pathfinding in fra mutants expressing AblWT (supplementary material Fig. S2C). Therefore, we expressed AblKN in EW neurons using eg-Gal4. Interestingly, we find that expression of AblKN in frahypo; Abl/+ embryos rescues the portion of EW midline-crossing defects that is presumably due to loss of Abl function (Fig. 2A). These data suggest that Abl might perform a kinase-independent function in commissural neurons to promote midline axon crossing.
Our observations in Abl heterozygous mutants do not preclude a role for Abl kinase activity in midline axon attraction. It is possible that in these experiments, kinase-dead Abl could cooperate with endogenous Abl in some way to promote midline axon crossing. To address whether Abl kinase function is required for midline axon attraction, we performed similar rescue experiments in Abl homozygous mutants expressing DN-Fra (Fig. 2B; supplementary material Fig. S3). In this genetic background, expression of AblWT rescues EW midline-crossing defects. However, in contrast to what we see in Abl heterozygous embryos, AblKN does not rescue midline-crossing defects in these embryos, suggesting that endogenous Abl is required for kinase-dead Abl to signal. Together, these observations indicate a basal requirement for Abl kinase activity to promote commissural axon crossing; however, Abl might have additional functions that are revealed when Abl activity is only partly limited.
Abl physically interacts with Frazzled through its N-terminal SH2 motif
It has been shown previously that Abl can physically interact with Fra, but the functional consequences of this interaction are not known (Forsthoefel et al., 2005). To determine how Abl functions in commissural guidance, we sought to determine the structural motifs in Abl that regulate this interaction. When expressed in Drosophila S2R+ cells, co-immunoprecipitation shows that Abl-GFP interacts with full length Fra-Myc. This interaction maps to the N terminus of Abl, which contains the SH3, SH2 and kinase domains (Fig. 3A,D,E).
Fig. 3.

Abl binds to Fra through its N-terminal SH2 motif. (A-C) Co-immunoprecipitation of GFP-Abl fusions expressed in S2R+ cells with anti-Myc antibodies. Western blots representative of at least three independent experiments are shown. Mouse-anti GFP and Mouse anti-Myc (indicated on the right) were used. Constructs are indicated at the top. (A) Abl-FL and Abl-N, but not Abl-C, bind to Fra (compare lanes 2 and 3 to lane 4). Positive control (lane 5) shows that Abl binds Robo-myc. (B) AblR297KGFP does not bind to Fra (lane 5). (C) Tyrosine phosphorylation of Fra is not required for the Abl-Fra interaction, as Fra-9YF pulls down Abl-GFP (lane 3). (D) Quantification of co-immunoprecipitation experiments. Shown are the ratio of immunoprecipitated Abl-GFP to input, normalized to the control (Abl-GFP alone). Number of experiments used for quantification are shown in parentheses. **P<0.01. Error bars indicate s.e.m. (E) Schematic depicting Abl indicating relevant features. Mutations and truncations used in this figure are labeled.
To identify the domains that are required for this interaction, we mutated conserved residues in these motifs that are known to facilitate substrate binding (Fig. 3E). For the SH3 domain, we mutated the conserved Trp243 to lysine (AblW243KGFP), which is predicted to eliminate binding to proline-rich ligands (Musacchio et al., 1994; Yu et al., 1994). For the Abl SH2 motif, we changed the conserved Arg297 to lysine (AblR297KGFP) to eliminate phosphotyrosine binding (Mayer et al., 1992; Waksman et al., 1992; Waksman et al., 1993; Zhu et al., 1993). We also tested whether Abl kinase activity is necessary for Fra interaction using the kinase-inactive AblK417NGFP mutant. Through co-immunoprecipitation, we find that only the SH2 domain mutant, AblR297KGFP, is deficient in Fra binding, suggesting that the physical interaction could be mediated by a phosphorylated tyrosine (Fig. 3B,D). To determine whether this interaction occurs through tyrosine residue in the cytoplasmic domain of Fra, we tested for binding with a Fra receptor in which all cytoplasmic tyrosines are mutated to phenylalanine (Fra9YF) (O’Donnell and Bashaw, 2013). We find that Abl can still bind to this mutant Fra receptor (Fig. 3C,D). This result suggests that if binding occurs through phosphorylated tyrosines, this interaction could be indirect. However, owing to the variability in these assays, we cannot exclude the possibility that Fra9YF binds to Abl with lower affinity than wild-type Fra. Nonetheless, Abl binds to Frazzled through its SH2 motif, probably involving an indirect, phosphotyrosine-dependent interaction.
The Abl C-terminal FABD is necessary for Netrin-dependent commissural axon guidance
In vertebrates, the Abl C terminus contains multiple cytoskeletal interaction domains, which mediate actin binding, bundling and actin-microtubule crosslinking (Bradley and Koleske, 2009; Van Etten et al., 1994; Wang et al., 2001). As we have seen evidence for a partially kinase-independent function of Drosophila Abl, we hypothesize that this activity might be conferred by its C terminus, which has been shown to contain cytoskeletal interaction motifs in vertebrates (Lapetina et al., 2009; Miller et al., 2004). Our observation that Abl binds to Fra through its N terminus allows us to test whether C-terminal sequences are also required for Netrin-dependent responses. If the Abl-Fra physical interaction is involved in Netrin signaling, we predict that expression of the Abl-N terminus (AblN, Fig. 4G) would compete with endogenous Abl for Fra binding. If C-terminal sequences are necessary for the function of Abl in midline axon crossing, then we would expect this truncation to interfere with Netrin-dependent responses.
Fig. 4.
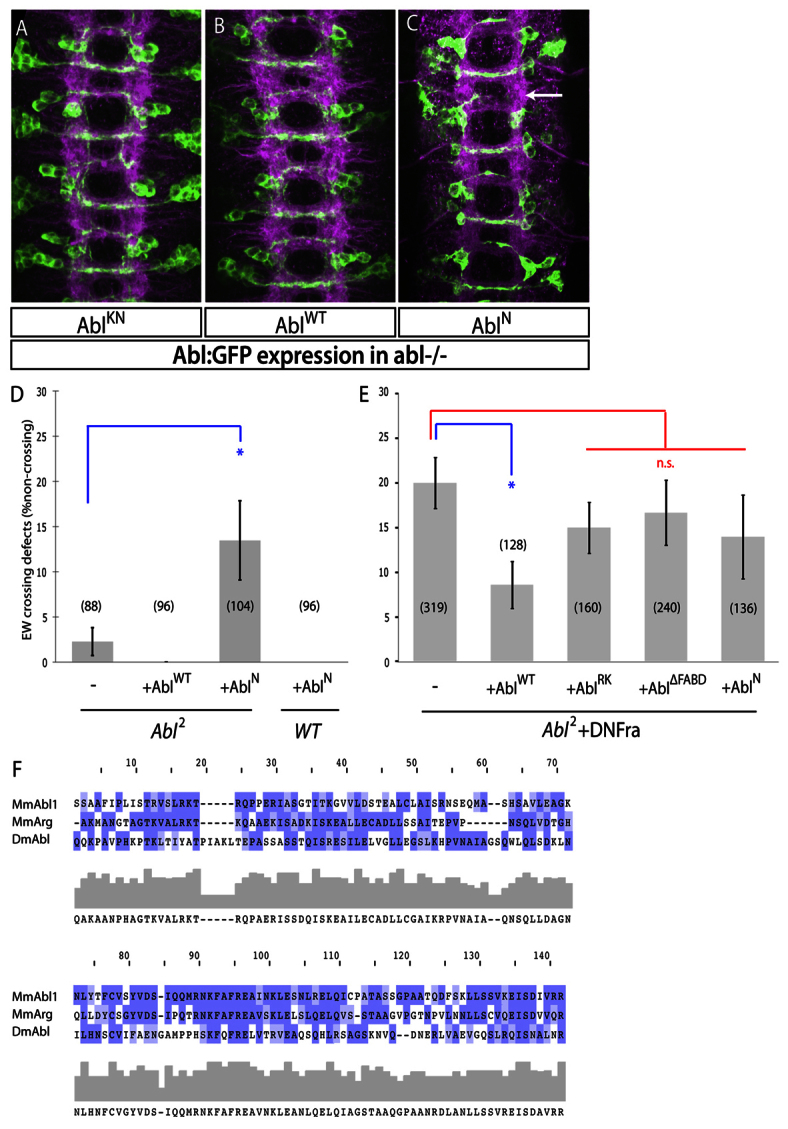
The Abl FABD and SH2 motifs are necessary for commissural axon guidance. (A-C) Stage 16 Abl2 mutant embryos expressing the indicated Abl-GFP fusions in EW neurons using eg-Gal4. Four segments are shown. Anterior is up. (A) AblKNGFP has no effect in these embryos. (B) AblWTGFP expression has no effect. (C) AblNGFP interferes with midline crossing of EW neurons (arrow). (D) Quantification of EW crossing defects in Abl2 mutants expressing the indicated Abl constructs. AblN, unlike AblWT, interferes with EW midline axon crossing. (E) Quantification of EW axon crossing defects in Abl2 mutant embryos expressing DNFra along with the indicated constructs. AblWT rescues midline crossing defects though AblRK and AblΔFABD do not. Data from control and AblWT rescue are also presented in Fig. 2B. For D and E, error bars indicate s.e.m.; number of segments scored is shown in parentheses; *P<0.05. n.s., not significant. (F) Multiple sequence alignment of the predicted FABD of Mus musculus (Mm) Abl1 and Arg (Abl2) along with that of Drosophila melanogaster (Dm) Abl using ClustalW. Identical residues are shaded in dark blue, similar residues are shaded in light blue. Similarity plot and consensus sequence are shown below. Amino acids 1499-1638 are shown for D. melanogaster Abl.
To address whether AblN can interfere with commissural axon guidance, we expressed this construct in the EW neurons of Abl2 mutant embryos, which have few commissural defects (Fig. 4D). As Abl mRNA is supplied maternally, the relatively mild defects in zygotic Abl mutants are likely to result from maternal compensation (Bennett and Hoffmann, 1992; Grevengoed et al., 2001). When AblN is expressed in these embryos, EW defects are dramatically increased (Fig. 4C,D), suggesting that AblN might interfere with residual maternal Abl in EW neurons. Importantly, AblWT has no effect in this context (Fig. 4B,D). These results suggest that removal of Abl C-terminal sequence interferes with Netrin- and Fra-dependent commissural axon guidance.
We have seen that AblWT can rescue midline-crossing defects in Abl2 mutants when Fra function is partially inhibited (Fig. 2B). AblN, however, does not rescue EW midline-crossing defects in these embryos (Fig. 4E), consistent with our results in gain-of-function experiments. To determine whether interaction with Fra is necessary for the pro-crossing function of Abl in EW neurons, we generated transgenic flies expressing the Abl SH2 mutant AblRK, which is deficient in Fra binding. We find that unlike AblWT, AblRK fails to rescue EW midline-crossing defects in Abl2 mutants, which suggests that the Abl-Fra physical interaction might be important for midline axon attraction (Fig. 4E). Though these constructs are expressed at similar levels in EW neurons (supplementary material Fig. S4), we cannot rule out the possibility that mutation of the Abl SH2 motif interferes with other Abl functions in this context.
The Drosophila Abl C terminus contains a highly conserved FABD (Fig. 4F), which could potentially serve as a physical link between Fra and the cytoskeleton. To address the role of the Abl FABD in axon guidance, we generated transgenic flies expressing Abl:GFP bearing a deletion of the FABD, AblΔFABD, under Gal4-dependent control. When driven in EW neurons, AblΔFABD is expressed at levels indistinguishable from those of AblWT and appears to be properly localized to axons (supplementary material Fig. S4). To determine whether the Abl FABD is important for commissural axon attraction, we expressed AblΔFABD in EW neurons of Abl2 mutant embryos. AblΔFABD fails to rescue EW midline-crossing defects in these embryos (Fig. 4E), suggesting that the Abl FABD is essential for midline guidance of EW neurons. Taken together, these results suggest that Abl associates with the Fra receptor through its SH2 domain and then, through its C-terminal FABD, promotes Netrin-dependent commissural axon guidance.
Abl promotes motor axon pathfinding through its C terminus
A well-characterized function of Abl in Drosophila is to promote growth of embryonic motor axons (Wills et al., 1999b). This function is evident in RP motoneurons of the intersegmental nerve (ISNb), which normally innervate ventral body wall muscles 6, 7, 12 and 13 (Fig. 5A,F) (Sink and Whitington, 1991). In Abl mutants, these axons frequently stall prior to reaching their muscle targets (Fig. 5B,F). These defects are presumably due to a reduction in axon growth but could also reflect aberrant pathfinding in response to an unidentified guidance pathway. We therefore refer to these defects as axon stalling. To determine how Abl promotes motor axon pathfinding, we rescued ISNb motor axon defects in Abl mutants by restoring Abl expression in RP motoneurons using Hb9Gal4. When AblWTGFP is expressed in Abl2 mutants, this stalling phenotype is rescued (Fig. 5C,G). However, when AblNGFP is expressed, ISNb stalling phenotypes are exacerbated rather than rescued (Fig. 5D,G). This suggests that, like in commissural neurons, the C-terminal domain of Abl is necessary for axon growth in motoneurons. We hypothesized that Abl might regulate motor axon pathfinding in the same way that it does in commissural neurons, namely through its FABD and SH2 motif. However, AblΔFABDGFP fully rescues motor axon stalling, in contrast to AblNGFP (Fig. 5E,G), suggesting that the Abl FABD is dispensable for RP motor axon pathfinding and that additional, unidentified C-terminal motifs are likely to be required. Furthermore, AblRKGFP rescues motor axon stalling in Abl2 mutants, which also distinguishes its role in motor axon pathfinding from that of commissural neurons (Fig. 5G).
Fig. 5.

Abl C terminus is required for ISNb axon pathfinding. (A-D) Stage 17 embryos of indicated genotypes. Motor axons are visualized with anti-FasII (magenta). Abl-GFP fusions are expressed in RP motoneurons with Hb9Gal4 and visualized with anti-GFP (green). Anterior is left. (A) Control embryo. All three ventral muscle clefts are innervated (arrows). (B) Abl2 mutant embryo. Occasional stalling occurs prior to reaching muscle 12 (asterisk). Arrows indicate properly innervated muscle clefts. (C) An Abl2 embryo rescued with AblWTGFP. All three ventral muscle clefts are innervated (arrows). (D) An Abl2 embryo expressing AblNGFP. No rescue of ventral motor stalling. Stalling phenotype is enhanced in these embryos (asterisks). (E) An Abl2 embryo rescued with AblΔFABDGFP. Stalling is fully rescued. All three ventral muscle clefts are innervated (arrows). (F) Schematic showing typical innervation pattern of ventral muscles by RP axons in a wild type (WT; left) and Abl mutant (right). RP axons are depicted in red; other motor axons are gray. Note the absence of innervation of the muscle 12/13 cleft in Abl mutants, termed stalling. (G) Quantitative analysis of ISNb rescue experiments. Error bars indicate s.e.m. Number of segments scored is shown in parentheses. *P<0.05, ***P<0.001. n.s., not significant.
The Abl C terminus is dispensable for inhibition of Slit/Robo signaling
Based on our results in commissural and motoneurons, we wondered if there are contexts in which Abl functions through entirely different mechanisms. In addition to promoting axon growth and commissural axon crossing, Abl plays a complex role in other midline and motor axon guidance pathways. Depending on the context, Abl can either promote or inhibit Slit- and Robo-dependent midline repulsive signaling (Bashaw et al., 2000; Hsouna et al., 2003; Lee et al., 2004; Wills et al., 2002). Abl also antagonizes the receptor-protein tyrosine phosphatase Lar, which is required for RP axon defasciculation towards ventral muscles (Wills et al., 1999a). A common mechanism by which Abl inhibits both Robo and Lar is through antagonism of enabled (ena). To address whether, as in commissural neurons, the C-terminal elements of Abl are necessary for these functions, we expressed Abl in neurons that normally require Slit/Robo signaling or Lar signaling.
In slit/+ embryos, the normally ipsilateral-projecting apterous neurons show occasional defects (Fig. 6A,D). These ectopic midline-crossing defects occur as a result of a reduction in Slit- and Robo-dependent repulsion. When AblWTGFP is expressed in these neurons using apGal4, these defects are enhanced, suggesting that Abl can antagonize Slit/Robo signaling in these neurons (Fig. 6B,D). We then investigated whether the Abl C terminus is required for this activity. Strikingly, AblNGFP causes a similarly penetrant ectopic crossing phenotype in these embryos (Fig. 6C,D). Thus, the Abl C terminus is dispensable for the ability of Abl to inhibit Slit/Robo signaling and induce ectopic-ap axon midline crossing. Interestingly, however, neither AblRKGFP nor AblΔFABDGFP can increase ectopic crossing in this background, suggesting that these constructs are either not active in these neurons or are preferentially targeted to a different pathway (Fig. 6D).
Fig. 6.

The Abl N terminus is sufficient to inhibit multiple guidance pathways. (A-C) Stage 17 slit/+ embryos expressing TauMycGFP and the indicated transgenes in ipsilateral apterous neurons under control of apGal4. Five abdominal segments are shown. apterous axons are visualized with anti-GFP (green). FasII-positive ipsilateral axons are in magenta. Anterior is up. (A) slit/+ embryo. Occasional ectopic crossing occurs (arrows). (B) slit/+ embryo expressing AblWTGFP. Ectopic crossing (arrows) is significantly enhanced. (C) slit/+ embryo expressing AblNGFP. Ectopic crossing (arrows) is enhanced. (D) Quantification of ap ectopic crossing defects. Expression of AblWTGFP and AblNGFP results in a similarly penetrant phenotype. Error bars indicate s.e.m. Number of segments scored is shown in parentheses. *P<0.05, ***P<0.001. (E-G) Stage 17 embryos expressing the indicated Abl-GFP fusions under control of pan-neural Elav-Gal4. Motor axons are visualized with anti-FasII. Three segments are shown. Anterior is up. Approximate location of muscle clefts and muscle numbers are indicated. (E) AblKNGFP has no effect when overexpressed. All three muscle clefts are properly innervated. (F) AblWTGFP expression results in full ISNb bypass phenotype. (G) AblNGFP expression generates ISNb bypass, although to a lesser degree than AblWTGFP.
To address whether Abl acts through a similar mechanism in motor axon defasciculation, we expressed Abl-GFP fusions pan-neurally using elav-Gal4. AblWTGFP expression results in an almost fully penetrant bypass phenotype (Fig. 6F). This bypass phenotype occurs when ISNb axons fail to defasciculate, and instead continue to grow dorsally past their muscle targets. When we express AblNGFP or AblΔFABDGFP, this results in a partially penetrant bypass phenotype, suggesting that the Abl N terminus is sufficient for this activity, although to a lesser degree than full-length Abl (Fig. 6G; data not shown). We also observe occasional bypass events when AblNGFP is expressed in Abl2 mutants using Hb9-Gal4, a phenotype never observed in Abl mutants, suggesting that this effect is not likely to require normal levels of endogenous Abl. It has been shown previously that Abl kinase activity is required for ISNb bypass and, consistently, AblKNGFP has no effect in this assay (Wills et al., 1999a). Thus, Abl, through its N-terminal kinase activity, can inhibit Slit/Robo signaling in midline neurons and inhibit defasciculation in motor axons. These activities, in contrast to its role in commissural guidance and RP motor axon growth, do not strictly require its C-terminal cytoskeletal interaction motifs.
DISCUSSION
In this work, we have shown that Abl contributes to midline axon crossing in Drosophila commissural neurons, through a mechanism that relies on the Abl C-terminal FABD and that is partially kinase independent. The genetic and physical interactions between Abl and Fra we have observed, as well as the rescue and gain-of-function experiments, suggest that Abl functions in the Netrin pathway to promote EW neuron midline crossing through interaction with the cytoplasmic domain of Fra. Both commissural neurons and motoneurons require the Abl C terminus for axon growth and pathfinding, though probably through different mechanisms. We also show that some of the functions of Abl, namely inhibition of Slit- and Robo-dependent repulsion and inhibition of motor axon defasciculation occur through a distinct, strictly kinase-dependent mechanism that does not require the Abl C terminus. Here, we discuss these findings in light of our current knowledge of the cell biological functions of Abl and we speculate about the mechanism by which Abl, through its C terminus, promotes axon growth and pathfinding.
We have shown that Abl interacts genetically and physically with the Netrin receptor Frazzled. The genetic interactions we have observed are consistent with earlier work suggesting that Abl promotes commissural axon guidance and physically interacts with Fra (Forsthoefel et al., 2005). Based on earlier work, it has been unclear whether the pro-crossing function of Abl reflects a role in Netrin signal transduction in commissural neurons. Forsthoefel and colleagues observed enhancement of both NetAB and frazzled mutants in Abl heterozygotes by analyzing all commissural neurons using mAb BP102, suggesting that Abl might function in parallel to Netrin to promote midline crossing. However, not all Drosophila commissural neurons require Netrin function for midline axon crossing (Mitchell et al., 1996); thus, a more severe commissural phenotype in NetAB, Abl/+ mutants could reflect Abl function in Netrin-independent commissural neurons. Here, we have analyzed the contribution of Abl to commissural guidance in EW neurons, which require Netrin signaling and in which Frazzled functions autonomously to direct midline axon crossing (Brankatschk and Dickson, 2006; Garbe et al., 2007). We find that in EW neurons, in contrast to earlier observations, reduction in Abl gene dose leads to increased EW midline-crossing defects only in hypomorphic fra allelic combinations, not in NetAB or fra null mutants. These results are consistent with Abl functioning in the Netrin pathway in commissural neurons.
How does Abl promote Netrin-dependent commissural axon guidance? Though we have replicated the findings of Forsthoefel and colleagues in demonstrating that Abl binds to Fra, our results suggest that in the context of Drosophila cells, this interaction might be indirect. Because we have seen that Abl requires a functional SH2 motif to interact with Fra when expressed in S2R+ cells, and that the cytoplasmic tyrosines of Fra are largely dispensable for binding, this physical interaction is likely to involve an intermediate, phosphotyrosine-containing protein. This hypothetical protein could theoretically be a direct target for Abl kinase activity, or might be regulated by a different kinase. Currently, we cannot distinguish between these two possibilities. AblKN appears to bind Fra in our assays, though in this case endogenous Abl kinase activity might be sufficient to promote this interaction. We hypothesize that this physical interaction is necessary for Netrin-dependent midline axon attraction and, consistent with this idea, we have seen that the Abl SH2 mutant AblRK cannot replace Abl function in EW commissural neurons.
Our results suggest that once Abl binds to Fra, the C-terminal FABD of Abl is necessary to promote midline axon crossing. Expression of the Abl N terminus, which can bind to Fra, does not rescue midline-crossing defects in Abl mutants and, instead, appears to act as a dominant-negative when expressed in EW neurons. We speculate that this occurs by occluding the interaction between Fra and endogenous Abl, though we cannot rule out the possibility that AblN has neomorphic activity in this context. Multiple functions have been attributed to the C terminus of Abl family members in vertebrates, including F- and G-actin binding, F-actin bundling, microtubule binding, actin-microtubule cross-linking, DNA binding, and scaffolding functions (Bradley and Koleske, 2009). It is unclear which, if any, of these activities are retained in the Drosophila kinase, though primary sequence homology suggests that the F-actin-binding activity is likely to be conserved. Our results suggest that this FABD is necessary for the ability of Abl to promote midline axon crossing in EW neurons. It is tempting to speculate that Abl, through its C-terminal FABD, could link Fra to actin filaments and promote the assembly of a complex to regulate cytoskeletal motility. Alternatively, the Abl FABD could be used to regulate kinase activity in certain contexts. Evidence for this possibility comes from work in mammalian Abl kinase. F-actin can inhibit Abl kinase activity directly, and the Abl FABD is necessary for this modulation (Woodring et al., 2001). Perhaps when bound to Fra and in the proximity of actin filaments, attenuation of Abl kinase activity through interaction with F-actin might be necessary for appropriate signal transduction. Further work is necessary to distinguish these possibilities.
Although the Abl FABD is crucial for commissural axon pathfinding, additional C-terminal motifs are clearly required to promote motor axon growth and targeting. When the Abl FABD is deleted, growth cone localization is altered. Qualitatively, this construct appears to be expressed at lower levels and cannot often be seen in filopodia of motor axons (supplementary material Fig. S4J). Despite this altered localization, AblΔFABDGFP fully rescues motor axon defects in RP neurons. This result is dramatically different to what we observe after deletion of the entire C terminus, suggesting that different elements in the C terminus are necessary for axon pathfinding in RP motoneurons. Additionally, AblΔFABDGFP is not as potent an inhibitor of EW axon crossing as is AblNGFP (data not shown). Thus additional C-terminal motifs might play a role in both commissural and motor axon guidance. These elements could be additional cytoskeletal interaction motifs, or alternatively scaffolding domains, a possibility we discuss here.
There are several proline-rich regions in the Drosophila Abl C terminus, which in vertebrate Abl family members allow for interaction with proteins involved in Abl-dependent cell protrusions. For M. musculis Arg, these include the Arp2/3 regulator cortactin (Lapetina et al., 2009), and for Abl1, these include the scaffolding protein Nck and the Rac activator CrkII (Antoku et al., 2008; Ren et al., 1994). It is interesting to note that Nck has been shown to interact with DCC in a complex that includes Wasl (N-Wasp) and is necessary for Netrin-dependent Rac activation (Li et al., 2002; Shekarabi et al., 2005). It is presently unclear whether the Drosophila ortholog of Nck, Dreadlocks (Dock), performs a similar function. The Arg C terminus interacts with the cortactin SH3 domain through a PXXP motif (Lapetina et al., 2009). Cortactin would also be a good candidate to mediate some of the additional C-terminal functions of Abl, and this would be straightforward to test in Drosophila commissural and motoneurons. Abl might simply link Fra (or other receptors) to the cytoskeleton, or it could act as a scaffold for the assembly of cytoskeletal regulators, such as cortactin, during guidance receptor signaling or axon growth. Molecular genetic approaches in commissural neurons, as well as the identification of cytoskeletal interaction motifs in the Drosophila kinase should help to elucidate the role of the Abl C terminus in commissural and motor axon guidance.
Our results in commissural neurons suggest that Abl might serve a partly kinase-independent function to promote midline axon crossing, as we have seen in Abl heterozygous mutant embryos. However, a kinase-inactive Abl cannot functionally compensate for wild-type Abl in embryos lacking all zygotic Abl function, suggesting that Abl kinase activity is not absolutely dispensable for the pro-crossing function of Abl. Similarly, it is important to consider that endogenous Abl gene dose might also affect the activity of other mutant Abl transgenes we have tested here. Kinase-independent functions of Abl have been reported previously (Henkemeyer et al., 1990; Lapetina et al., 2009). Although these results could potentially reflect multimerization activity of Abl kinase, to our knowledge there is no direct evidence for an intrinsic dimerization activity of Abl kinase. Rather, our results are reminiscent of what has been seen in mammalian cells lacking the Abl family member Arg (Abl2). In fibroblasts, for example, the Arg C terminus, through interaction with cortactin and Nck1, promotes adhesion-dependent protrusions. In arg null cells, expression of the Arg C terminus (lacking the kinase domain) or a kinase-inactive Arg can rescue adhesion-dependent protrusive activity, but only in conditions in which some Abl kinase activity remains, provided by Abl1. A kinase-inactive Arg or a C-terminal fragment can rescue protrusion defects in Arg-/- cells, but not in Arg-/-;Abl-/- double knockout cells. In Arg-/- cells, Abl, acting in trans, can substitute for Arg kinase activity. Thus, when Abl function is reduced below a threshold level, these kinase-independent functions can no longer be observed. Our results are consistent with these reports and the model for stepwise scaffolding and kinase functions of Abl in cell motility (Lapetina et al., 2009) and suggest that Abl might serve a similar function in Drosophila commissural neurons to promote Netrin-dependent axon attraction.
We have previously shown that Fra also promotes commissural axon guidance through Netrin-independent inhibition of the Slit/Robo pathway by increasing the expression of the Robo inhibitor commissureless (Yang et al., 2009). It is conceivable that AblKN promotes midline crossing through inhibition of the Robo pathway, as has been suggested before (Hsouna et al., 2003), which could in principle explain our observations in EW neurons. Two of our observations suggest that this is not likely to be the case. First, when AblWT is expressed in ap neurons, this results in ectopic midline axon crossing, presumably through inhibition of Robo signaling. AblKN does not induce this phenotype, suggesting that Abl kinase activity is necessary for inhibition of Slit/Robo signaling, consistent with earlier work (Bashaw et al., 2000). Second, heterozygosity of Abl results in enhancement of EW midline-crossing defects in fra hypomorphic embryos, but not in NetAB or fra null mutants, suggesting that this genetic interaction is not likely to occur through an effect on the Slit/Robo pathway. We suggest, based on these observations, that Abl probably promotes Netrin-dependent commissural axon guidance through a mechanism that is partly kinase independent.
Finally, we show here that some of the functions of Abl do not depend on the C-terminal domain, arguing that Abl must affect guidance pathways through different mechanisms. We have seen that, similar to full-length Abl, AblN can inhibit Slit- and Robo-dependent repulsion in ipsilaterally projecting apterous neurons, and can prevent defasciculation in motor axons. We speculate that, given that these two processes require the activity of the Abl substrate ena and that Abl-kinase activity is necessary for this gain-of-function effect, Abl acts to inhibit ena function in repulsion and defasciculation through its N-terminal kinase activity (Bashaw et al., 2000; Wills et al., 1999a).
Perhaps the most peculiar result of our study is the failure of AblΔFABDGFP to induce ectopic ap axon crossing whereas AblNGFP appears to be almost as effective as WT-Abl. One possibility is that AblΔFABDGFP is simply unstable and is not expressed at sufficient levels in our experiments. Several observations argue against this interpretation. First, AblΔFABDGFP is expressed at similar levels as AblWTGFP in commissural neurons. Second, and more importantly, AblΔFABDGFP fully rescues ISNb guidance defects when expressed in RP neurons. Given these data, we interpret this to reflect the existence of additional C-terminal motifs that normally antagonize the ability of Abl to inhibit Robo signaling. When either the FABD is present, or the C terminus is deleted, these domains are incapable of inhibiting the role of Abl in the Robo pathway. Taken together, these observations suggest that, in contrast to the role of Abl in commissural guidance and axon growth, antagonism of Slit/Robo signaling occurs in the absence of C-terminal sequences, arguing that the opposing functions of Abl in axon guidance are provided by different structural motifs.
Supplementary Material
Acknowledgments
We would like to thank members of the Bashaw lab for their thoughtful comments and ideas during the development of this manuscript. In particular, we thank Alexandra Neuhaus-Follini and Celine Santiago for their experimental and intellectual contributions. We thank Dr Mark Peifer for providing the Abl-GFP construct.
Footnotes
Funding
This work was supported by National Institutes of Health (NIH) grants [NS-046333and NS054739 to G.J.B.]; March of Dimes grant [#1-FY12-445 to G.J.B.]; and NIH training grants [5-T32-GM07229 and 5-T32-007516 to M.P.O.]. Deposited in PMC for release after 12 months.
Competing interests statement
The authors declare no competing financial interests.
Author contributions
M.P.O. and G.J.B. carried out the experiments and analysed the data. M.P.O. and G.J.B. planned experiments and interpreted results. G.J.B. supervised the research. M.P.O and G.J.B. wrote the paper.
Supplementary material
Supplementary material available online at http://dev.biologists.org/lookup/suppl/doi:10.1242/dev.093831/-/DC1
References
- Antoku S., Saksela K., Rivera G. M., Mayer B. J. (2008). A crucial role in cell spreading for the interaction of Abl PxxP motifs with Crk and Nck adaptors. J. Cell Sci. 121, 3071–3082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bashaw G. J., Goodman C. S. (1999). Chimeric axon guidance receptors: the cytoplasmic domains of slit and netrin receptors specify attraction versus repulsion. Cell 97, 917–926 [DOI] [PubMed] [Google Scholar]
- Bashaw G. J., Kidd T., Murray D., Pawson T., Goodman C. S. (2000). Repulsive axon guidance: Abelson and Enabled play opposing roles downstream of the roundabout receptor. Cell 101, 703–715 [DOI] [PubMed] [Google Scholar]
- Bennett R. L., Hoffmann F. M. (1992). Increased levels of the Drosophila Abelson tyrosine kinase in nerves and muscles: subcellular localization and mutant phenotypes imply a role in cell-cell interactions. Development 116, 953–966 [DOI] [PubMed] [Google Scholar]
- Bradley W. D., Koleske A. J. (2009). Regulation of cell migration and morphogenesis by Abl-family kinases: emerging mechanisms and physiological contexts. J. Cell Sci. 122, 3441–3454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brankatschk M., Dickson B. J. (2006). Netrins guide Drosophila commissural axons at short range. Nat. Neurosci. 9, 188–194 [DOI] [PubMed] [Google Scholar]
- Crowner D., Le Gall M., Gates M. A., Giniger E. (2003). Notch steers Drosophila ISNb motor axons by regulating the Abl signaling pathway. Curr. Biol. 13, 967–972 [DOI] [PubMed] [Google Scholar]
- Elkins T., Zinn K., McAllister L., Hoffmann F. M., Goodman C. S. (1990). Genetic analysis of a Drosophila neural cell adhesion molecule: interaction of fasciclin I and Abelson tyrosine kinase mutations. Cell 60, 565–575 [DOI] [PubMed] [Google Scholar]
- Forsthoefel D. J., Liebl E. C., Kolodziej P. A., Seeger M. A. (2005). The Abelson tyrosine kinase, the Trio GEF and Enabled interact with the Netrin receptor Frazzled in Drosophila. Development 132, 1983–1994 [DOI] [PubMed] [Google Scholar]
- Fox D. T., Peifer M. (2007). Abelson kinase (Abl) and RhoGEF2 regulate actin organization during cell constriction in Drosophila. Development 134, 567–578 [DOI] [PubMed] [Google Scholar]
- Garbe D. S., Bashaw G. J. (2007). Independent functions of Slit-Robo repulsion and Netrin-Frazzled attraction regulate axon crossing at the midline in Drosophila. J. Neurosci. 27, 3584–3592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garbe D. S., O’Donnell M., Bashaw G. J. (2007). Cytoplasmic domain requirements for Frazzled-mediated attractive axon turning at the Drosophila midline. Development 134, 4325–4334 [DOI] [PubMed] [Google Scholar]
- Gertler F. B., Bennett R. L., Clark M. J., Hoffmann F. M. (1989). Drosophila abl tyrosine kinase in embryonic CNS axons: a role in axonogenesis is revealed through dosage-sensitive interactions with disabled. Cell 58, 103–113 [DOI] [PubMed] [Google Scholar]
- Grevengoed E. E., Loureiro J. J., Jesse T. L., Peifer M. (2001). Abelson kinase regulates epithelial morphogenesis in Drosophila. J. Cell Biol. 155, 1185–1198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halekoh U., Højsgaard S., Yan J. (2006). The R package geepack for generalized estimating equations. J. Stat. Softw. 15, 1–11 [Google Scholar]
- Henkemeyer M. J., West S. R., Gertler F. B., Hoffmann F. M. (1990). A novel tyrosine kinase-independent function of Drosophila abl correlates with proper subcellular localization. Cell 63, 949–960 [DOI] [PubMed] [Google Scholar]
- Hsouna A., VanBerkum M. F. A. (2008). Abelson tyrosine kinase and Calmodulin interact synergistically to transduce midline guidance cues in the Drosophila embryonic CNS. Int. J. Dev. Neurosci. 26, 345–354 [DOI] [PubMed] [Google Scholar]
- Hsouna A., Kim Y.-S., VanBerkum M. F. A. (2003). Abelson tyrosine kinase is required to transduce midline repulsive cues. J. Neurobiol. 57, 15–30 [DOI] [PubMed] [Google Scholar]
- Kolodziej P. A., Timpe L. C., Mitchell K. J., Fried S. R., Goodman C. S., Jan L. Y., Jan Y.-N. (1996). frazzled encodes a Drosophila member of the DCC immunoglobulin subfamily and is required for CNS and motor axon guidance. Cell 87, 197–204 [DOI] [PubMed] [Google Scholar]
- Lai Wing Sun K., Correia J. P., Kennedy T. E. (2011). Netrins: versatile extracellular cues with diverse functions. Development 138, 2153–2169 [DOI] [PubMed] [Google Scholar]
- Lapetina S., Mader C. C., Machida K., Mayer B. J., Koleske A. J. (2009). Arg interacts with cortactin to promote adhesion-dependent cell edge protrusion. J. Cell Biol. 185, 503–519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H., Engel U., Rusch J., Scherrer S., Sheard K., Van Vactor D. L. (2004). The microtubule plus end tracking protein Orbit/MAST/CLASP acts downstream of the tyrosine kinase Abl in mediating axon guidance. Neuron 42, 913–926 [DOI] [PubMed] [Google Scholar]
- Li X., Meriane M., Triki I., Shekarabi M., Kennedy T. E., Larose L., Lamarche-Vane N. (2002). The adaptor protein Nck-1 couples the netrin-1 receptor DCC (deleted in colorectal cancer) to the activation of the small GTPase Rac1 through an atypical mechanism. J. Biol. Chem. 277, 37788–37797 [DOI] [PubMed] [Google Scholar]
- Liebl E. C., Forsthoefel D. J., Franco L. S., Sample S. H., Hess J. E., Cowger J. A., Chandler M. P., Shupert A. M., Seeger M. A. (2000). Dosage-sensitive, reciprocal genetic interactions between the Abl tyrosine kinase and the putative GEF trio reveal trio’s role in axon pathfinding. Neuron 26, 107–118 [DOI] [PubMed] [Google Scholar]
- Mayer B. J., Jackson P. K., Van Etten R. A., Baltimore D. (1992). Point mutations in the abl SH2 domain coordinately impair phosphotyrosine binding in vitro and transforming activity in vivo. Mol. Cell. Biol. 12, 609–618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller A. L., Wang Y., Mooseker M. S., Koleske A. J. (2004). The Abl-related gene (Arg) requires its F-actin-microtubule cross-linking activity to regulate lamellipodial dynamics during fibroblast adhesion. J. Cell Biol. 165, 407–419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitchell K. J., Doyle J. L., Serafini T., Kennedy T. E., Tessier-Lavigne M., Goodman C. S., Dickson B. J. (1996). Genetic analysis of Netrin genes in Drosophila: Netrins guide CNS commissural axons and peripheral motor axons. Neuron 17, 203–215 [DOI] [PubMed] [Google Scholar]
- Musacchio A., Saraste M., Wilmanns M. (1994). High-resolution crystal structures of tyrosine kinase SH3 domains complexed with proline-rich peptides. Nat. Struct. Biol. 1, 546–551 [DOI] [PubMed] [Google Scholar]
- O’Donnell M. P., Bashaw G. J. (2013). Src inhibits midline axon crossing independent of Frazzled/Deleted in Colorectal Carcinoma (DCC) receptor tyrosine phosphorylation. J. Neurosci. 33, 305–314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan W. (2001). Akaike’s information criterion in generalized estimating equations. Biometrics 57, 120–125 [DOI] [PubMed] [Google Scholar]
- Ren R., Ye Z. S., Baltimore D. (1994). Abl protein-tyrosine kinase selects the Crk adapter as a substrate using SH3-binding sites. Genes Dev. 8, 783–795 [DOI] [PubMed] [Google Scholar]
- Round J., Stein E. (2007). Netrin signaling leading to directed growth cone steering. Curr. Opin. Neurobiol. 17, 15–21 [DOI] [PubMed] [Google Scholar]
- Shekarabi M., Moore S. W., Tritsch N. X., Morris S. J., Bouchard J.-F., Kennedy T. E. (2005). Deleted in colorectal cancer binding netrin-1 mediates cell substrate adhesion and recruits Cdc42, Rac1, Pak1, and N-WASP into an intracellular signaling complex that promotes growth cone expansion. J. Neurosci. 25, 3132–3141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sink H., Whitington P. M. (1991). Pathfinding in the central nervous system and periphery by identified embryonic Drosophila motor axons. Development 112, 307–316 [DOI] [PubMed] [Google Scholar]
- Van Etten R. A., Jackson P. K., Baltimore D., Sanders M. C., Matsudaira P. T., Janmey P. A. (1994). The COOH terminus of the c-Abl tyrosine kinase contains distinct F- and G-actin binding domains with bundling activity. J. Cell Biol. 124, 325–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waksman G., Kominos D., Robertson S. C., Pant N., Baltimore D., Birge R. B., Cowburn D., Hanafusa H., Mayer B. J., Overduin M., et al. (1992). Crystal structure of the phosphotyrosine recognition domain SH2 of v-src complexed with tyrosine-phosphorylated peptides. Nature 358, 646–653 [DOI] [PubMed] [Google Scholar]
- Waksman G., Shoelson S. E., Pant N., Cowburn D., Kuriyan J. (1993). Binding of a high affinity phosphotyrosyl peptide to the Src SH2 domain: crystal structures of the complexed and peptide-free forms. Cell 72, 779–790 [DOI] [PubMed] [Google Scholar]
- Wang Y., Miller A. L., Mooseker M. S., Koleske A. J. (2001). The Abl-related gene (Arg) nonreceptor tyrosine kinase uses two F-actin-binding domains to bundle F-actin. Proc. Natl. Acad. Sci. USA 98, 14865–14870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wills Z., Bateman J., Korey C. A., Comer A., Van Vactor D. L. (1999a). The tyrosine kinase Abl and its substrate enabled collaborate with the receptor phosphatase Dlar to control motor axon guidance. Neuron 22, 301–312 [DOI] [PubMed] [Google Scholar]
- Wills Z., Marr L., Zinn K., Goodman C. S., Van Vactor D. L. (1999b). Profilin and the Abl tyrosine kinase are required for motor axon outgrowth in the Drosophila embryo. Neuron 22, 291–299 [DOI] [PubMed] [Google Scholar]
- Wills Z., Emerson M., Rusch J., Bikoff J., Baum B., Perrimon N., Van Vactor D. L. (2002). A Drosophila homolog of cyclase-associated proteins collaborates with the Abl tyrosine kinase to control midline axon pathfinding. Neuron 36, 611–622 [DOI] [PubMed] [Google Scholar]
- Woodring P. J., Hunter T., Wang J. Y. (2001). Inhibition of c-Abl tyrosine kinase activity by filamentous actin. J. Biol. Chem. 276, 27104–27110 [DOI] [PubMed] [Google Scholar]
- Yan J., Fine J. (2004). Estimating equations for association structures. Stat Med 23, 859–74 [DOI] [PubMed] [Google Scholar]
- Yang L., Garbe D. S., Bashaw G. J. (2009). A frazzled/DCC-dependent transcriptional switch regulates midline axon guidance. Science 324, 944–947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H., Chen J. K., Feng S., Dalgarno D. C., Brauer A. W., Schreiber S. L. (1994). Structural basis for the binding of proline-rich peptides to SH3 domains. Cell 76, 933–945 [DOI] [PubMed] [Google Scholar]
- Zhu G., Decker S. J., Mayer B. J., Saltiel A. R. (1993). Direct analysis of the binding of the abl Src homology 2 domain to the activated epidermal growth factor receptor. J. Biol. Chem. 268, 1775–1779 [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.