

Abstract
Several [FeII 2(N-EtHPTB)(μ-O2X)]2+ complexes (1•O2X) have been synthesized, where N-EtHPTB is the anion of N,N,N′N′-tetrakis(2-benzimidazolylmethyl)-2-hydroxy-1,3-diaminopropane and O2X is an oxyanion bridge. Crystal structures reveal five-coordinate (μ-alkoxo)diiron(II) cores. These diiron(II) complexes react with O2 at low temperatures in CH2Cl2 (−90°) to form blue-green O2 adducts that are best described as triply-bridged (μ-η1:η1-peroxo)diiron(III) species (2•O2X). With one exception, all 2•O2X intermediates convert irreversibly to doubly-bridged, blue (μ-η1:η1-peroxo)diiron(III) species (3•O2X). Where possible, 2•O2X and 3•O2X intermediates were characterized using resonance Raman spectroscopy, showing respective νO-O values of ~850 and ~900 cm−1. How the steric and electronic properties of O2X affect conversion of 2•O2X to 3•O2X was examined. Stopped-flow analysis reveals that oxygenation kinetics of 1•O2X are unaffected by the nature of O2X and for the first time, the benzoate analog of 2•O2X (2•O2CPh) is observed.
Introduction
Nonheme diiron enzymes have attracted great interest because they perform a wide variety of reactions despite having very similar active sites.1–8 For example, soluble methane monooxygenase inserts an oxygen atom into an alkane C-H bond, toluene and o-xylene monooxygenases, as well as phenol hydroxylase insert oxygen atoms into aromatic C-H bonds, the R2 subunit of class I ribonucleotide reductases extracts a hydrogen atom producing a stable radical and, as its name implies, stearoyl acyl carrier protein Δ9-desaturase dehydrogenates a fatty acid hydrocarbon chain. In addition to having similar active site structures, these enzymes all share one other property. The putative catalytic cycles of these enzymes all contain peroxide-bridged diiron(III) complexes formed upon reduction of molecular oxygen. In some cases, these intermediates are stable enough to be trapped and characterized.9–17 While peroxo-diiron(III) species are endemic in these enzymes, their precise role in catalysis is poorly understood, even though they have been well studied. Synthetic attempts to study peroxo-diiron(III) intermediates have resulted in a variety of biomimetic complexes.18–27 These compounds often form peroxide-bridged diiron(III) intermediates, especially when stabilized by carboxylate bridges. They are commonly reported as having a (μ-η1:η1-peroxo)diiron(III) motif.
In a previous publication,28 we discussed how replacing the carboxylate bridges commonly employed in synthesis of these biomimetic complexes affected the behavior of the peroxo intermediates formed. Using the dinucleating ligand N-EtHPTB (anion of N,N,N′,N′-tetrakis(2-benzimidazolylmethyl)-2-hydroxy-1,3-diaminopropane) combined with two equivalents of iron(II) and one equivalent of either benzoate, diphenylphosphinate or dimethylarsinate,29 we demonstrated that two different (μ-η1:η1-peroxo)diiron(III) species can be formed upon reduction of dioxygen. A doubly-bridged (μ-alkoxo)(μ-1,2-peroxo) intermediate was obtained from the benzoate-bridged diiron(II) precursor at −40°, in contrast to the triply-bridged (μ-alkoxo)(μ-1,2-peroxo)(μ-1,3-dimethylarsinato) intermediate produced by the dimethylarsinate-bridged diiron(II) precursor. The diphenylphosphinate-bridged diiron(II) precursor was unique in that it formed a triply-bridged peroxo intermediate as the initial kinetic product, which converted to a metastable doubly-bridged peroxo intermediate by a shift of the phosphinate from a bridging mode to a terminal monodentate binding mode before peroxo decomposition, analogous to the “carboxylate shift” notion described by Lippard twenty years ago.30 We concluded that the nature of the oxyanion bridge (O2X) strongly influenced the nature and stability of any (μ-η1:η1-peroxo)diiron(III) species formed, but we felt further work was required before the effects of bridge differences could be clearly understood. This work is intended to address that issue.
To this end, we focused on three effects: (i) those produced by O2X O···O bite distance differences, (ii) those produced by O2X electronic differences and (iii) those produced by O2X steric differences. Examination required synthesis of several new diiron(II) complexes (1•O2X) using N-EtHPTB and various O2X ligands. We found that all of these new species reacted with O2 in solution to produce blue-green (μ-η1:η1-peroxo)diiron(III) intermediates (2•O2X) at −90°. With one exception, they then converted to deep-blue (μ-η1:η1-peroxo)diiron(III) species (3•O2X) before decaying to yellow products (4•O2X) at higher temperature. In most cases, addition of OPPh3 to solutions of 2•O2X led to conversion to purple-blue species (3′•O2X) similar to 3•O2X. In this paper, we report crystallographic details of three diiron(II) complexes, kinetic data on their reactions with dioxygen, and spectroscopic characterization of a variety of (μ-η1:η1-peroxo)diiron(III) intermediates. Implications of O2X bite distances as well as electronic and steric differences are discussed.
Experimental Section
Materials and Syntheses
All reagents and solvents were purchased from commercial sources and were used as received, unless noted otherwise. The ligand N-EtHPTB was synthesized using a published procedure.31 Solvents were dried according to published procedures and distilled under Ar prior to use.32 The 18O2 (97%) used in resonance Raman experiments was purchased from Cambridge Isotope Laboratories, Inc., Andover, MA. Preparation and handling of air sensitive materials were carried out under an inert atmosphere by using either standard Schlenk and vacuum line techniques or a glovebox. Elemental analyses were performed by Atlantic Microlab, Inc., Norcross, GA.
1•O2CPh, 1•O2PPh2 and 1•O2AsMe2 were synthesized using published procedures.28
1•O2PMe2
N-EtHPTB (157 mg, 0.217 mmol) was dissolved in MeOH (~10 mL) along with Et3N (0.19 mL, 1.4 mmol). Dimethylphosphinic acid (21.6 mg, 0.230 mmol) was added and allowed to dissolve. Fe(OTf)2•2MeCN33 (189 mg, 0.434 mmol) was added, producing a yellow solution. After 5 minutes, NaBPh4 (149 mg, 0.435 mmol) was added, resulting in immediate precipitation of a white powder. The solid was filtered and dried in vacuo. Recrystallization from MeCN and Et2O produced colorless crystals, some suitable for X-ray diffraction structural analysis. Yield: 153 mg (80%). Anal. for [Fe2(N-EtHPTB)( O2PMe2)](BPh4)(OTf) and calcd for C70H75BF3Fe2N10O6PS: C, 60.27; H, 5.42; N, 10.04%. Found: C, 59.82; H, 5.49; N, 10.38%.
1•O2P(OPh)2
N-EtHPTB (176 mg, 0.243 mmol) was dissolved in MeOH (~10 mL) along with Et3N (0.19 mL, 1.4 mmol). Diphenylphosphoric acid (67.7 mg, 0.271 mmol) was added and allowed to dissolve. Fe(OTf)2•2MeCN (222 mg, 0.509 mmol) was added, producing a yellow solution. After 5 minutes, NaBPh4 (173 mg, 0.506 mmol) was added, resulting in immediate precipitation of a white powder. The solid was filtered and dried in vacuo. Recrystallization from MeCN and Et2O produced milky crystals, some suitable for X-ray diffraction structural analysis. Yield: 331 mg (79%). Anal. for [Fe2(N-EtHPTB)( O2P(OPh)2)](BPh4)2 and calcd for C103H99B2Fe2N10O5P: C, 71.87; H, 5.80; N, 8.14%. Found: C, 71.77; H, 5.94; N, 8.41%.
1•O2CCPh3
N-EtHPTB (208 mg, 0.288 mmol) was dissolved in MeOH (~10 mL) along with Et3N (0.19 mL, 1.4 mmol). Triphenylacetic acid (83.3 mg, 0.289 mmol) was added and allowed to dissolve. Fe(OTf)2•2MeCN (257 mg, 0.589 mmol) was added, producing a yellow solution. After 5 minutes, NaBPh4 (205 mg, 0.598 mmol) was added, resulting in immediate precipitation of a white powder. The solid was filtered and dried in vacuo. Recrystallization from MeCN and Et2O produced colorless crystals. Yield: 370 mg (73%). Anal. for [Fe2(N-EtHPTB)( O2CCPh3)](BPh4)2 and calcd for C111H104B2Fe2N10O3: C, 75.78; H, 5.96; N, 7.96%. Found: C, 75.46; H, 5.95; N, 7.96%.
1•O2CCMe3
N-EtHPTB (153 mg, 0.212 mmol) was dissolved in MeOH (~10 mL) along with Et3N (0.19 mL, 1.4 mmol). Trimethylacetic acid (21.7 mg, 0.212 mmol) was added and allowed to dissolve. Fe(OTf)2•2MeCN (186 mg, 0.426 mmol) was added, producing a yellow solution. After 5 minutes, NaBPh4 (152 mg, 0.443 mmol) was added, resulting in immediate precipitation of a white powder. The solid was filtered and dried in vacuo. Recrystallization from MeCN and Et2O produced milky crystals. Yield: 130 mg (50%). Anal. for [Fe2(N-EtHPTB)(O2CCMe3)](OTf)2 and calcd for C50H58F6Fe2N10O9S2: C, 48.71; H, 4.74; N, 11.36%. Found: C, 48.91; H, 4.77; N, 11.39%.
1•O2CC6H2-3,4,5-(OMe)3
N-EtHPTB (72.9 mg, 0.101 mmol) was dissolved in MeOH (~10 mL) along with Et3N (0.077 mL, 0.56 mmol). 3,4,5-Trimethoxybenzoic acid (21.4 mg, 0.101 mmol) was added and allowed to dissolve. Fe(OTf)2•2MeCN (94.2 mg, 0.216 mmol) was added, producing a yellow solution. After 5 minutes, NaBPh4 (87.2 mg, 0.255 mmol) was added, resulting in immediate precipitation of a pale yellow powder. The solid was filtered and dried in vacuo. Recrystallization from MeCN and Et2O produced yellow crystals, some suitable for X-ray diffraction structural analysis. Yield: 116 mg (74%). Anal. for [Fe2(N-EtHPTB)(O2CCH2-3,4,5-(OMe)3)](BPh4)(OTf) and calcd for C78H80BF3Fe2N10O9S: C, 61.92; H, 5.33; N, 9.26%. Found: C, 62.14; H, 5.55; N, 8.94%.
1•O2CC6H3-3,4-(OMe)2
N-EtHPTB (100.0 mg, 0.138 mmol) was dissolved in MeOH (~10 mL) along with Et3N (0.097 mL, 0.69 mmol). 3,4-Dimethoxybenzoic acid (25.1 mg, 0.138 mmol) was added and allowed to dissolve. Fe(OTf)2•2MeCN (120.3 mg, 0.276 mmol) was added, producing a yellow solution. After 5 minutes, NaBPh4 (196.5 mg, 0.574 mmol) was added, resulting in immediate precipitation of a pale yellow powder. The solid was filtered and dried in vacuo. Recrystallization from MeCN and Et2O produced milky crystals. Yield: 162 mg (89%). Anal. for [Fe2(N-EtHPTB)(O2CC6H3-3,4-(OMe)2)](OTf)2 and calcd for C54H58F6Fe2N10O11S2: C, 49.40; H, 4.45; N, 10.67%. Found: C, 49.65; H, 4.44; N, 10.39%.
1•O2CC6H3-3,5-(OMe)2
N-EtHPTB (146.1 mg, 0.202 mmol) was dissolved in MeOH (~10 mL) along with Et3N (0.142 mL, 1.02 mmol). 3,5-Dimethoxybenzoic acid (37.0 mg, 0.203 mmol) was added and allowed to dissolve. Fe(OTf)2•2MeCN (187.8 mg, 0.431 mmol) was added, producing a yellow solution. After 5 minutes, NaBPh4 (190.6 mg, 0.557 mmol) was added, resulting in immediate precipitation of a pale green-yellow powder. The solid was filtered and dried in vacuo. Recrystallization from MeCN and Et2O produced pale yellow crystals. Yield: 238.5 mg (80%). Anal. for [Fe2(N-EtHPTB)(O2CC6H3-3,5-(OMe)2)](BPh4)(OTf)2 and calcd for C77H78BF3Fe2N10O8S: C, 62.36; H, 5.30; N, 9.44%. Found: C, 62.56; H, 5.11; N, 9.33%.
1•O2CC6H4-4-OMe
N-EtHPTB (149.2 mg, 0.206 mmol) was dissolved in MeOH (~10 mL) along with Et3N (0.144 mL, 1.03 mmol). 4-Methoxybenzoic acid (31.3 mg, 0.206 mmol) was added and allowed to dissolve. Fe(OTf)2•2MeCN (189.2 mg, 0.433 mmol) was added, producing a yellow solution. After 5 minutes, NaBPh4 (205.4 mg, 0.600 mmol) was added, resulting in immediate precipitation of a pale green-yellow powder. The solid was filtered and dried in vacuo. Recrystallization from MeCN and Et2O produced milky crystals. Yield: 245.4 mg (93%). Anal. for [Fe2(N-EtHPTB)(O2CC6H4-4-OMe)](OTf)2 and calcd for C53H56F6Fe2N10O10S2: C, 49.62; H, 4.40; N, 10.92%. Found: C, 49.41; H, 4.51; N, 11.08%.
Physical Methods
UV-Vis spectra were recorded on a Hewlett-Packard 8453 diode array spectrophotometer (2-nm resolution) equipped with an Unisoku Scientific Instruments cryostat (Osaka, Japan). Resonance Raman spectra were collected on an ACTON AM-506M3 monochromator with a Princeton LN/CCD data collection system using a Spectra-Physics Model 2060 krypton laser. Low-temperature spectra of the peroxo intermediates in CH2Cl2 and MeCN were obtained at 77 K using a 135° backscattering geometry. Samples were frozen onto a gold-plated copper cold finger in thermal contact with a Dewar flask containing liquid nitrogen. Raman frequencies were referenced to the features of indene. Slits were set for a band-pass of 4 cm−1 for all spectra.
Time-resolved spectra of rapid oxygenation reactions were acquired with a Hi-Tech Scientific (Salisbury, Wiltshire, UK) SF-43 multi-mixing anaerobic cryogenic stopped-flow instrument combined with either a monochromator (low intensity light irradiation of the sample) or a diode array rapid scanning unit (strong UV-Vis irradiation of the sample), or with a TgK Scientific (formerly HiTech Scientific, Salisbury, Wiltshire, UK) SF-61DX2 cryogenic stopped-flow system equipped with a J&M Diode array (Spectralytics). All manipulations with diiron(II) complexes and their solutions were done using an argon atmosphere glove-box, air-tight syringes, and the anaerobic stopped-flow instrument to avoid contamination with air. Saturated solutions of O2 in CH2Cl2 and CH3CN were prepared by bubbling the dry O2 gas for 20 min in a septum-closed cylinder with the solvent at a constant temperature (20 or 25 °C). The solubility of O2 was accepted to be 5.8 mM in dichloromethane at 20° and 8.1 mM in acetonitrile at 25°.34 Solutions of O2 with smaller concentrations were prepared by diluting the saturated O2 solution with argon-saturated solvent using graduated gas-tight syringes equipped with three-way valves. For the kinetic experiments, dioxygen was always taken in large excess so that its concentration did not change significantly during the reaction with 1•O2X. The solutions of 1•O2X and O2 were cooled to a preset temperature (±0.1°) in the stopped-flow instrument before mixing. Data analysis was performed with the IS-2 Rapid Kinetics Software (Hi-Tech Scientific) for kinetic traces at a single wavelength.
X-ray Crystallography
X-ray diffraction data were collected on a Bruker SMART platform CCD diffractometer at 173(2) K.35 Preliminary sets of cell constants were calculated from reflections harvested from three sets of 20 frames. These initial sets of frames were oriented such that orthogonal wedges of reciprocal space were surveyed. The data collection was carried out using MoKα radiation (graphite monochromator). Randomly oriented regions of reciprocal space were surveyed to the extent of one sphere and to a resolution of 0.84 Å. The intensity data were corrected for absorption and decay using SADABS.36 Final cell constants were calculated after integration with SAINT.37 The structures were solved and refined using SHELXL-97.38 The space groups P21/c, P21/n and P-1 were determined based on systematic absences and intensity statistics. Direct-methods solutions were calculated which provided most non-hydrogen atoms from the E-map. Full-matrix least squares/difference Fourier cycles were performed which located the remaining non-hydrogen atoms. All non-hydrogen atoms were refined with anisotropic displacement parameters. All hydrogen atoms were placed in ideal positions and refined as riding atoms with relative isotropic displacement parameters. The SQUEEZE function of the program PLATON39 was used to remove 143 effective electrons in diffuse scattering from a volume of 1555.2 Å3 per cell of 1•O2P(OPh)2(BPh4)2•MeCN and 120 effective electrons in diffuse scattering from a volume of 1097.4 Å3 per cell of (1•O2CC6H2-3,4,5-(OMe)3)2(BPh4)2(OTf)2•2MeCN. Brief crystal data and intensity collection parameters for the crystalline complexes are shown in Table 1.
Table 1.
Crystal data and structure refinement for 1•O2PMe2(BPh4)(OTf)•MeCN, 1•O2P(OPh)2(BPh4)2•MeCN, and (1•O2CC6H2-3,4,5-(OMe)3)2(BPh4)2(OTf)2•2MeCN
| [1•O2PMe2]-(BPh4)(OTf)•MeCN | [1•O2P(OPh)2]-(BPh4)2•MeCN | [1•O2CC6H2-3,4,5-(OMe)3]2(BPh4)2(OTf)2-•2MeCN | |
|---|---|---|---|
| empirical formula | C72H78BF3Fe2N11O6PS | C105H102B2Fe2N11O5P | C160H166B2F6Fe4N22O18S2 |
| fw | 1435.99 | 1762.27 | 3108.29 |
| T (K) | 173(2) | 173(2) | 173(2) |
| Mo Kα λ, Å | 0.71073 | 0.71073 | 0.71073 |
| space group | P21/c | P21/n | P -1 |
| a (Å) | 16.0709(9) | 23.063(4) | 12.576(4) |
| b (Å) | 15.6360(9) | 18.942(4) | 19.153(7) |
| c (Å) | 28.4703(15) | 23.888(4) | 36.053(12) |
| α (deg) | 90 | 90 | 96.743(5) |
| β (deg) | 103.5800(10) | 99.205(3) | 92.147(5) |
| γ (deg) | 90 | 90 | 103.100(5) |
| V (Å3) | 6954.1(7) | 10301(3) | 8381(5) |
| Z | 4 | 4 | 2 |
| ρ (calc), Mg/m3 | 1.372 | 1.136 | 1.232 |
| abs coeff (mm−1) | 0.539 | 0.352 | 0.437 |
| R1a | 0.0398 | 0.0446 | 0.0713 |
| wR2b | 0.0984 | 0.1271 | 0.1955 |
R1 = Σ||Fo|−|Fc||/Σ|Fo|.
wR2 = [Σ[w(Fo2−Fc2)2]/Σ [w(Fo2)2]]1/2.
Results
X-ray Crystallography
For this work, we synthesized diiron(II) complexes using a variety of O2X ligands, namely O2PMe2, O2P(OPh)2, and several aliphatic and aromatic carboxylates, to augment a list that includes O2AsMe2, O2PPh2, and benzoate. We were able to obtain crystal structures of three of these 1•O2X compounds (O2X = O2PMe2, O2P(OPh)2 and O2CC6H2-3,4,5-(OMe)3) (Table 2). Like those of previously published complexes,28 the new complexes possess two distorted trigonal bipyramidal iron(II) centers, with N-EtHPTB amine nitrogen atoms and oxygen atoms from the bridging O2X moiety occupying the axial positions. The equatorial sites are occupied by two benzimidazole nitrogen atoms and the alkoxide oxygen atom (Figure 1). With few exceptions, the respective atoms in the first coordination sphere of each complex reflect approximately equal interatomic distances and angles. Differences of note include average τ values,40 O2X bite distances, O-X-O angles and inter-iron distances. The τ values can be grouped into two sets with τave ~ 0.8 (O2X = O2AsMe2, O2PPh2 and O2PMe2) and τave ~ 0.9 (O2X = O2P(OPh)2, O2CC6H2-3,4,5-(OMe)3 and O2CPh). Comparing bite distances of all complexes, we see O···O distances of ~2.55 Å (X = P) and ~2.23 Å (X = C). The lone X = As complex is in a class of its own with a bite distance of ~2.80 Å. It is also apparent that the O-X-O angle varies with the identity of the X atom (X = As, 113.26 deg.; X = P, 115.17 to 119.47 deg.; X = C, 123.4 to 124.2 deg.). The O-X-O angle measurement correlates inversely with O2X bite distance; the species with the longest O···O distance (1•O2AsMe2) has the most acute O-X-O angle, while the X = C complexes have the shortest bite distances and largest O-X-O angles, with the X = P compounds falling in between these extremes. This inverse correlation reflects variations in X-O bond lengths (As-Oave = 1.676 Å; P-Oave = 1.501 Å; C-Oave = 1.261 Å. Even though the O-X-O angle decreases, concomitant lengthening of the X-O bonds results in a greater O···O distance.
Table 2.
Selected interatom distances and bond angles for [Fe2(N-EtHPTB)(O2X)]2+.
| 1•O2AsMe2a | 1•O2PPh2a | 1•O2PMe2 | 1•O2P(OPh)2 | 1•O2CC6H2-3,4,5-(OMe)3b | 1•O2CPha | |
|---|---|---|---|---|---|---|
| τave | 0.81 | 0.77 | 0.82 | 0.91 | 0.89 | 0.93 |
|
| ||||||
| Interatom distances (Å)
| ||||||
| Fe1-O1 | 2.0145(13) | 2.004(2) | 2.0184(18) | 2.0057(16) | 1.967(3) | 1.976(5) |
| Fe2-O1 | 2.0037(14) | 1.992(2) | 1.9995(18) | 2.0031(15) | 1.970(3) | 1.964(5) |
| Fe1-N1 | 2.3366(16) | 2.294(3) | 2.323(2) | 2.3141(18) | 2.328(4) | 2.316(6) |
| Fe2-N2 | 2.3892(16) | 2.352(3) | 2.372(2) | 2.309(2) | 2.314(4) | 2.280(7) |
| Fe1-O2 | 1.9825(14) | 2.008(2) | 2.0037(19) | 2.0562(16) | 2.036(3) | 2.057(5) |
| Fe2-O3 | 1.9863(14) | 2.021(2) | 2.0123(18) | 2.0506(16) | 2.025(3) | 2.019(6) |
| Fe1-N3 | 2.1372(17) | 2.086(3) | 2.105(2) | 2.0668(19) | 2.045(4) | 2.064(6) |
| Fe1-N5 | 2.1023(18) | 2.115(3) | 2.127(2) | 2.0732(19) | 2.046(4) | 2.069(6) |
| Fe2-N7 | 2.1088(17) | 2.075(3) | 2.098(2) | 2.0551(19) | 2.063(4) | 2.080(6) |
| Fe2-N9 | 2.0759(17) | 2.098(3) | 2.080(2) | 2.075(2) | 2.044(4) | 2.064(6) |
| As1/P1/C44-O2 | 1.6740(15) | 1.514(2) | 1.509(2) | 1.4762(17) | 1.254(5) | 1.264(9) |
| As1/P1/C44-O3 | 1.6776(14) | 1.512(2) | 1.517(2) | 1.4784(18) | 1.273(5) | 1.253(9) |
| Fe1···Fe2 | 3.5357(5) | 3.5405(10) | 3.5364(6) | 3.6211(7) | 3.4879(13) | 3.4749(31) |
| O2···O3 | 2.7991(21) | 2.5600(32) | 2.5545(28) | 2.5520(24) | 2.2253(46) | 2.2251(74) |
|
| ||||||
| Bond angles (degrees)
| ||||||
| Fe1-O1-Fe2 | 123.27(6) | 124.76(11) | 123.32(9) | 129.19(8) | 124.73(15) | 123.8(2) |
| O1-Fe1-O2 | 105.77(6) | 105.55(9) | 102.50(8) | 98.99(6) | 98.27(13) | 98.6(2) |
| O1-Fe2-O3 | 107.92(6) | 99.44(9) | 103.98(8) | 98.93(7) | 100.51(13) | 101.5(2) |
| Fe1-O2-As1/P1/C44 | 127.24(8) | 132.01(14) | 131.47(12) | 133.92(11) | 138.1(3) | 136.4(5) |
| Fe2-O3-As1/P1/C44 | 128.15(8) | 138.88(15) | 132.54(12) | 133.24(10) | 134.7(3) | 133.9(5) |
| O2-As1/P1/C44-O3 | 113.26(7) | 115.59(13) | 115.17(12) | 119.47(10) | 123.4(4) | 124.2(7) |
Data from reference 28.
The unit cell of this compound contains virtual twins of 1•O2CC6H2-3,4,5-(OMe)3 produced through a pseudo-inversion center. As there is no substantive difference between the two cations, values listed here are taken from a single cation.
Figure 1.

Crystal structure of the cation 1•O2CC6H2-3,4,5-(OMe)3 (50% ellipsoids) with hydrogen atoms removed. Generic cartoon of 1•O2X: O2X = O2AsMe2 (1•O2AsMe2), O2PPh2 (1•O2PPh2), O2PMe2 (1•O2PMe2), O2P(OPh)2 (1•O2P(OPh)2), O2CC6H2-3,4,5-(OMe)3 (1•O2CC6H2-3,4,5-(OMe)3), O2CPh (1•O2CPh).
In four of the six diferrous complexes crystallographically characterized, increased bite distance correlates to increased Fe···Fe distance. With the other two complexes, we observed that: (i) 1•O2AsMe2 has an O···O distance ~0.24 Å longer than any other complex, although its interiron distance is within 0.01 Å of those found in both 1•O2PPh2 and 1•O2PMe2. (ii) 1•O2P(OPh)2 has an O···O distance on par with 1•O2PPh2 and 1•O2PMe2, yet its interiron distance (3.6211 Å) is the greatest of all the compounds, exceeding the distances found in 1•O2AsMe2 (3.5357 Å) and the two other X = P species (3.5405 and 3.5364 Å) by ~0.08 Å. This is greater than the ~0.06 Å difference observed between the Fe···Fe distances of the X = C complexes (3.4879 and 3.4749 Å) and those with Fe···Fe distances of ~3.54 Å. The unusually long Fe···Fe distance in 1•O2P(OPh)2 is most likely not due to packing effects, because a similar interiron distance of 3.649 Å was reported when 1•O2P(OPh)2 crystallized in a different space group (P -1) with different anions (ClO4).41
There are two crystallographically interesting observations regarding the compounds we report here and those examined in our previous work.28 First, we note that [Fe2(N-EtHPTB)( O2PMe2)](BPh4)(OTf)(MeCN) and [Fe2(N-EtHPTB)(O2AsMe2)](BPh4)(OTf)(MeCN) are crystallographically isostructural. Both fall into the space group P21/c with Z = 4 and share virtually identical unit cells with respective values for a, b and c of 16.0709(9) vs. 16.1151(12), 15.6360(9) vs. 15.7370(12) and 28.4703(15) vs. 28.473(2) Å. The α and γ angles are exactly 90 degrees and the β angles are respectively 103.5800(10) and 103.4900(10) degrees. The respective cations, anions and solvent molecules in each unit cell lie in essentially the same positions with the same orientations. The minor variations in unit cell parameters and atom positions arise from differences around the X atom of the O2X bridging moieties. Small angle changes around X as well as X-C and X-O bond length variations produce minor differences in the cations which slightly alter the position of every other atom in the unit cell due to packing effects. The second observation is that the unit cell containing the cation [Fe2(N-EtHPTB)( O2CC6H2-3,4,5-(OMe)3)]2+ contains a pseudo-inversion center lying between cations (Figure 2). This effectively doubles the number of unique elements per unit cell, thus doubling the size of the unit cell. The cation “inversion” is reasonably true, but the anions are farther away from the pseudo-inversion center, resulting in greater distortion during “inversion”. Because the differences between the cations are not significant, we chose to focus on one cation rather than redundantly discussing what essentially amounts to duplicate cations.
Figure 2.
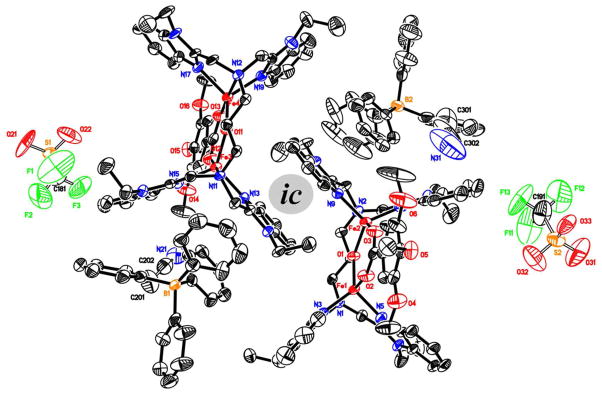
ORTEP diagram (50% ellipsoids) of the unit cell containing two molecules of [Fe2(N-EtHPTB)( O2CC6H2-3,4,5-(OMe)3)]2+ and accompanying anions and solvent molecules (hydrogen atoms removed for clarity). There is a pseudo-inversion center (ic) between the cations. Comparing the cations, we find that the “inversion” is almost true, whereas comparing the BPh4 ions reveals differences in thermal ellipsoid size and minor differences in atom locations, indicating that the inversion center is not real.
Observation of Two Peroxo Intermediates
Upon reaction with O2, the virtually colorless solutions of 1•O2X produce blue-green O2 adducts (2•O2X), which in all cases except two convert to deep blue species (3•O2X) before decaying to yellow final products (4•O2X). Previously, we reported the formation of 2•O2PPh2 and its conversion to 3•O2PPh2 at −40°.28 Corresponding studies with O2X = benzoate afforded only evidence for the formation of 3•O2CPh under these conditions.29,42,43 However, visible spectral evidence was obtained for the formation of both 2•O2CR and 3•O2CR by going to −90° in CH2Cl2, as shown in Figure 3 for O2X = O2CCPh3 with respective λmax values at 708 and 630 nm. In general, the visible chromophores of 2•O2X and 3•O2X fall in the ranges of 630–710 and 580–620 nm, respectively (Table 3), which have been previously assigned to peroxo-to-iron( III) charge transfer transitions for O2X = benzoate, Ph2PO2, and Me2AsO2 by resonance Raman spectroscopy.28,42
Figure 3.
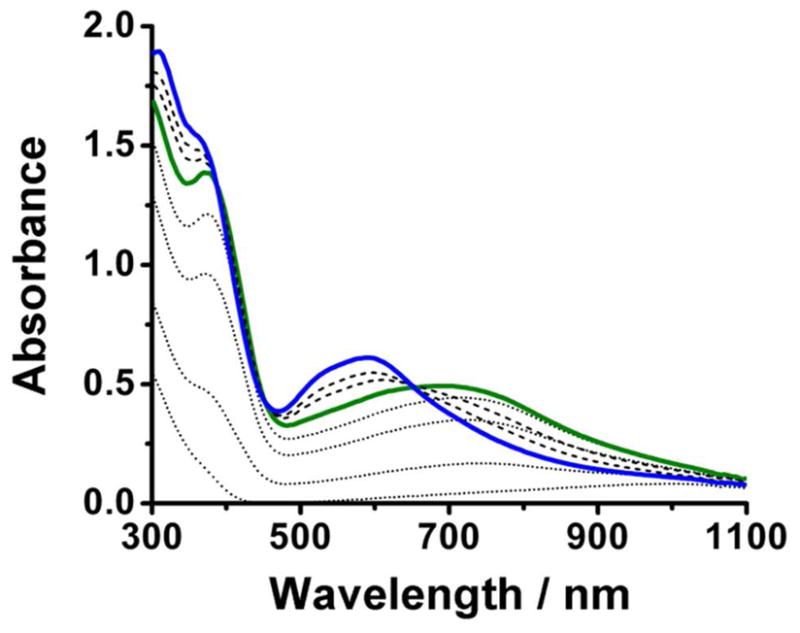
Selected UV-Vis spectra obtained in the reaction of O2 and 1•O2CCPh3 in CH2Cl2 at −90° showing initial formation of 2•O2CCPh3 (solid green line) and subsequent conversion to 3•O2CCPh3 (solid blue line). Dotted lines correspond to spectra leading to the appearance of 2•O2CCPh3 within the first 40 seconds, while dashed lines correspond to spectra associated with the conversion of 2•O2CCPh3 to 3•O2CCPh3 over the course of the following hour.
Table 3.
UV-Vis properties of 2•O2X, 3•O2X and 3′•O2X in CH2Cl2.
| O2X | λmax (nm), [ε] (M−1 cm−1)
|
||
|---|---|---|---|
| 2•O2X | 3•O2X | 3′•O2X | |
| O2AsMe2 | 632 [2100] | -- a | -- b |
| O2PMe2 | 674 [1800] | -- a | 616 [2400] |
| O2PPh2 | 678 [2100] | 621 [1800] | 592 [2600] |
| O2P(OPh)2 | 680 [2800] | 576 [3100] | 594 [3600] |
| O2CCPh3 | 708 [2500] | 630 [2200] | 595 [3300] |
| O2CCMe3 | 706 [--] c | 580 [3100] | 577 [3800] |
| O2CPh | 704 [--]c | 588 [1500]d | 592 [2300] |
| O2CC6H2-3,4,5-(OMe)3 | 707 [2600] | 590 [3200] | 592 [4300] |
| O2CC6H3-3,4-(OMe)2 | 704 [--]c | 578 [6200] | 590 [5300] |
| O2CC6H3-3,5-(OMe)2 | 706 [--]c | 588 [5400] | 592 [5600] |
| O2CC6H4-4-OMe | 705 [--]c | 580 [3000] | 590 [4000] |
Conversion to 3•O2X did not take place at any temperature.
Conversion to 3′•O2X did not take place at any temperature even with addition of 100 equivalents of OPPh3.
Conversion of 2•O2X to 3•O2X began before complete formation of 2•O2X, so ε was not determined.
Values from reference 42.
Resonance Raman spectra for 2•O2CCPh3, 3•O2CCPh3, 2•O2P(OPh)2, and 3•O2P(OPh)2 shown in Figure 4 are representative of the species observed in this study. 16O2 and 18O2 labeling experiments reveal O-O stretching frequencies (Table 4) that by comparison to those previously published28,42 allow us to sort them into two clusters. The 2•O2X cluster exhibits νO-O values ranging from 839 to 851 cm−1 that downshift to 791–807 cm−1 upon 18O substitution, while the 3•O2X cluster has νO-O values ranging from 897 to ~910 cm−1 that downshift to 845–857 cm−1 with 18O incorporation. On the other hand, no systematic difference is observed for the νFe-O values of the 2•O2X and 3•O2X complexes, all of which range from 457 to 479 cm−1 (Table 4). These features can be assigned to the symmetric Fe-O stretch of the Fe–O2-Fe moiety and often appear as Fermi doublets. The difference in the νO-O values of 2•O2X and 3•O2X complexes has previously been attributed to a change in the Fe–Fe distance in the O2 adducts on the basis of a systematic study of 1,2-peroxo-bridged diiron(III) complexes by Fiedler et al.27 For the 2•O2X and 3•O2X complexes, the change in the Fe-Fe distance indicated by the difference in the νO-O values is postulated to derive from the conversion of the O2X moiety from a bidentate bridging ligand in 2•O2X to a terminal monodentate ligand in 3•O2X (Scheme 1).
Figure 4.

Resonance Raman spectra of 2•O2CCPh3 (A), 3•O2CCPh3 (B), 2•O2P(OPh)2 (C) and 3•O2P(OPh)2 (D). Solid red lines (16O2) and dotted blue lines (18O2); T = 77 K, λex = 647 nm.
Table 4.
Fe–O and O–O stretching frequencies of 2•O2X and 3•O2X complexes determined using resonance Raman spectroscopy.
| O2X | 2•O2X | 3•O2X | ||
|---|---|---|---|---|
| νFe-O (cm−1) [18O] |
νO-O (cm−1) [18O2] |
νFe-O (cm−1) [18O] |
νO-O (cm−1) [18O2] |
|
| O2AsMe2 | 464 [433] | 845 [796] | -- | -- |
| O2PMe2 | 467, 479 [449, 460] | 839 [791] | -- | -- |
| O2PPh2 | 465, 476 [455] | 845, 853 [807] | 477 [458] | 897 [848] |
| O2P(OPh)2 | 457, 469 [436, 446] | 851 [806] | 479 [460] | 897 [845] |
| O2CCPh3 | 466, 474 [447, 454] | 841 [792] | 466, 479 [448, 456] | 903, 917 [857] |
| O2CCMe3 | -- | -- | 466, 475 [447, 457] | 897, 912 [846] |
| O2CPha | -- | -- | 476 [460] | 900 [850] |
| O2CC6H2-3,4,5-(OMe)3 | -- | -- | 465, 476 [449, 457] | 900, 914 [853] |
| O2CC6H3-3,4-(OMe)2 | -- | -- | 466, 474 [449, 456] | 898, 912 [847] |
| O2CC6H3-3,5-(OMe)2 | -- | -- | 465, 476 [449, 458] | 899, 913 [848] |
| O2CC6H4-4-OMe | -- | -- | 466, 474 [448, 457] | 899, 913 [847] |
Values from reference 42.
Scheme 1.

Conversion of 2•O2X to 3•O2X or 3′•O2X.
Attempts to obtain resonance Raman spectra for either 2•O2CC6H2-3,4,5-(OMe)3 or 2•O2CC6H3-3,4-(OMe)2 were unsuccessful, even though they exhibited a longer half life than 2•O2CCPh3. A color change was observed during the freezing process. Their respective Raman spectra were comparable to those of 3•O2CC6H2-3,4,5-(OMe)3 and 3•O2CC6H3-3,4-(OMe)2, indicating conversion took place during the phase change (Figure S1). On the other hand, the longer lifetimes of the 3•O2X intermediates allowed us to collect spectra of every one (Figures 4 and S1), with the exception of 3•O2PMe2, which did not form in quantities sufficient for characterization. The resonance Raman spectrum of 3′•O2PPh2, formed by treatment of 2•O2PPh2 with an excess of OPPh3 (Figure 5) was also acquired. As it was nearly identical to the previously reported spectrum of 3′•O2CPh,27 we did not carry out the corresponding experiments with other 3′•O2X intermediates.
Figure 5.
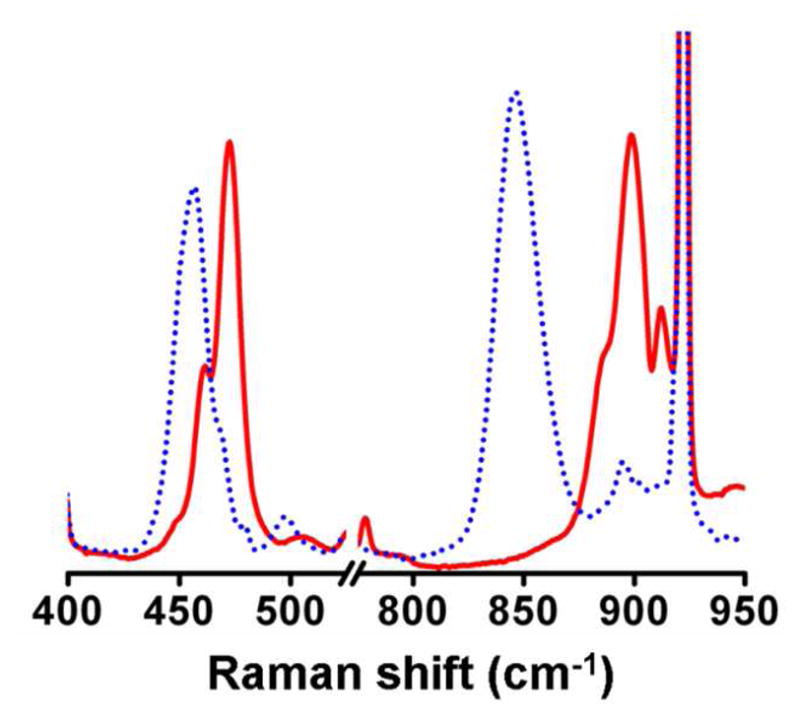
Resonance Raman spectra of 3′•O2PPh2 (16O2 = solid red line, 18O2 = dotted blue line).
Conversion of 2•O2X to 3•O2X
The spectral differences between 2•O2X and 3•O2X make UV-Vis absorption spectroscopy an excellent tool for following the transition from the first peroxo intermediate to the second. Based on the data shown in Figure 3, analysis of the spectral changes observed upon oxygenation of 1•O2CCPh3 at −90° afforded a first-order rate constant of 1.7 × 10−3 s−1 for the conversion of 2•O2CCPh3 to 3•O2CCPh3. This value is comparable to that previously reported for the conversion of 2•O2PPh2 to 3•O2PPh2 at −40°, thus implicating a more facile conversion for the carboxylate-bridged species. Figure 6 provides evidence for the formation of two peroxo species at −90° as well even for the well studied complex 1•O2CPh,29,42,44 although they are not as spectroscopically distinct as those of 2•O2CCPh3 and 3•O2CCPh3. Stopped-flow kinetic analysis at −80°C revealed the conversion from 2•O2CPh to 3•O2CPh with a kobs of 5.3 × 10−2 s−1, much faster than that observed for 2•O2CCPh3 to 3•O2CCPh3 at −90°. This comparison provides a rationale for why 2•O2CPh was not observed previously.29,42,44 The rate of conversion of 2•O2X to 3•O2X decreased as electron donating substituents were introduced onto the benzoate ring or as the steric bulk of the carboxylate bridge was increased (Table 5). The rate of conversion was further decreased by replacing the trigonal carboxylate moiety with tetrahedral anions such as (PhO)2PO2−, Ph2PO2−, Me2PO2−, and Me2AsO2− (Table 5).
Figure 6.

Selected UV-Vis spectra obtained in the reaction of O2 with 1•O2CPh at −90° showing evidence for the formation of two peroxo intermediates. The solid green line represents the spectrum with the largest fraction of 2•O2CPh formed, while the solid blue line corresponds to the subsequently formed 3•O2CPh. Dotted lines correspond to spectra leading to the maximum amount of 2•O2CPh formed within the first 90 seconds after oxygenation, while dashed lines correspond to spectra associated with the conversion of 2•O2CPh to 3•O2CPh over the course of the following 30 minutes.
Table 5.
pKa values of HO2X and first order kobs values for conversion of 2•O2X to 3•O2X and 2•O2X to 3′•′ O2X.
| O2X | pKa of HO2X | 2 → 3 a Kobs (s−1) | 2 → 3′b Kobs (s−1) |
|---|---|---|---|
| O2AsMe2 | 6.27 | -- c | -- d |
| O2PMe2 | 3.08 e | -- c | 1.8(4) × 10−4 (−40 °C) |
| O2PPh2 | 2.32 e | 3.1 × 10−4 f (−40 °C) |
4.3(4) × 10−3 (−40 °C) |
| O2P(OPh)2 | 1.85 e | 1.2(1) × 10−4 | 3.1(1) × 10−2 |
| O2CC6H2-3,4,5-(OMe)3 | 4.24 | 4.2(6) × 10−4 | 0.13(5) |
| O2CC6H3-3,4-(OMe)2 | 4.36 | 6.6(9) × 10−4 | 0.12(1) |
| O2CCPh3 | 3.96 | 1.7(2) × 10−3 | 2.6(3) × 10−2 |
| O2CC6H3-3,5-(OMe)2 | 3.97 | 4.8(2) × 10−3 | 0.20(1) |
| O2CC6H4-4-OMe | 4.50 | 4.7(4) × 10−3 | 0.12(1) |
| O2CCMe3 | 5.03 | 1.8(1) × 10−2 | 8.0(3) × 10−2 |
| O2CPh | 4.19 | 5.3(1) × 10−2 g (−80 °C) |
-- h |
All rates measured at −90 °C except where noted.
All rates measured after addition of 20 equivalents of OPPh3 at −90 °C except where noted.
Conversion to 3•O2X did not take place at any temperature.
Conversion to 3′•O2X did not take place at any temperature even with addition of 100 equivalents of OPPh3.
Measured in 7% EtOH (Reference 53).
t −40 °C in CH2Cl2, 3•O2PPh2 starts to decay before complete conversion from 2•O2PPh2 occurs. For this reason, kobs was calculated from the y-intercept of the OPPh3 concentration dependence plot for the conversion of 2•O2PPh2 to 3′•O2PPh2 (Figure S2).
Rate measured at −80 °C using stopped-flow techniques.
By the time enough 2•O2CPh had formed to allow for addition of OPPh3, significant conversion to 3•O2CPh had occurred, preventing accurate rate determination for the conversion of 2•O2CPh to 3′•O2CPh.
In our previous publication,28 we postulated that the conversion of 2•O2X to 3•O2X in acetonitrile involves a change in O2X binding mode from bridging to terminal and coordination of a solvent molecule in position L (Scheme 1). For this work we added OPPh3 to solutions of 2•O2X in hopes of substituting it into the L position. Upon OPPh3 addition, purple-blue species (3′•O2X) indeed form with λmax values at wavelengths near each respective 3•O2X, although the extinction coefficients of the new intermediates are higher than those observed for each respective 3•O2X (Table 3) with the exception of the O2CC6H3-3,4-(OMe)2-based species. While facile conversion to 3′•O2X from most 2•O2X intermediates was accomplished by adding 20 equivalents of OPPh3 at −90°, 2•O2PMe2 and 2•O2PPh2 had to be warmed to −40° to attain reasonable rates of conversion. In contrast, 2•O2AsMe2 appears to be unaffected by addition of up to 100 equivalents of OPPh3, even at temperatures as high as 20°.
After finding evidence indicating the formation of 2•O2CPh, we opted to further examine the oxygenation kinetics of both 1•O2PPh2 and 1•O2CPh using stopped-flow techniques. 1•O2PPh2 offers an excellent opportunity to characterize the initial oxygen coordination with the formation of 2•O2PPh2, since the subsequent rearrangement of 2•O2PPh2 to 3•O2PPh2 is relatively slow. The reaction between 1•O2PPh2 and O2 in either MeCN or CH2Cl2, which is accompanied by rapid growth of the absorption band with λmax ≈ 680 nm, was studied. At low temperatures (down to −80 °C in CH2Cl2), the initially formed diiron-O2 intermediate 2•O2PPh2 is stable for at least an hour. However, noticeable decay of this intermediate was observed under intense illumination with polychromatic, UV-rich light of the arc lamp. In order to avoid complications from undesirable photodecomposition, quantitative kinetic measurements were performed in a single-wavelength mode, with low-intensity monochromatic light and a sensitive photomultiplier detector. Under these conditions, photobleaching was not observed. In order to determine the rate law, kinetic traces were acquired under large excess of O2, showing single-exponential growth of 2•O2PPh2 and yielding the values of observed pseudo-first-order rate constants, kobs. The observed rate constants did not depend on the initial concentration of 1•O2PPh2, in agreement with a rate law that is first-order in the diiron precursor. The observed rate constants increased linearly with an increase in the concentration of O2 (Figure S3), indicating that the reaction was first order in dioxygen. It can be concluded that the formation of 2•O2PPh2 is a second-order process (first-order in diiron complex and first-order in O2): v = k2[1•O2PPh2][O2]
As temperature was raised, the oxygenation rate of 1•O2PPh2 increased modestly, and the conversion of 2•O2PPh2 to 3•O2PPh2 became pronounced. The Eyring plot (Figure 7) for the oxygen binding step was linear over a broad temperature range (from −80 to −20° in CH2Cl2), yielding the activation parameters that are summarized in Table 6. Kinetics of oxygen binding to 1•O2PPh2 in MeCN were also examined, and very similar activation parameters were extracted (Table 6). Subsequent disappearance of 2•O2PPh2 was readily observed in MeCN at temperatures above −40 °C (Figure S4). The spectral changes agreed well with the conversion of 2•O2PPh2 into 3•O2PPh2 described in detail by Frisch et al.28 Low activation enthalpies and large negative activation entropies for the oxygenation of 1•O2PPh2 leading to 2•O2PPh2 are typical of associative O2 coordination at vacant or labile iron sites,34,45 which are present in the starting diiron(II) complex. The rates and activation parameters for the oxygenation of 1•O2PPh2 in CH2Cl2 or in acetonitrile are very similar to previously published kinetic parameters for the oxygenation of 1•O2CPh in propionitrile.43 The kinetics of oxygenation of 5-coordinate diiron(II) complexes with a related dinucleating ligand HPTP are also very similar (Table 6).46
Figure 7.
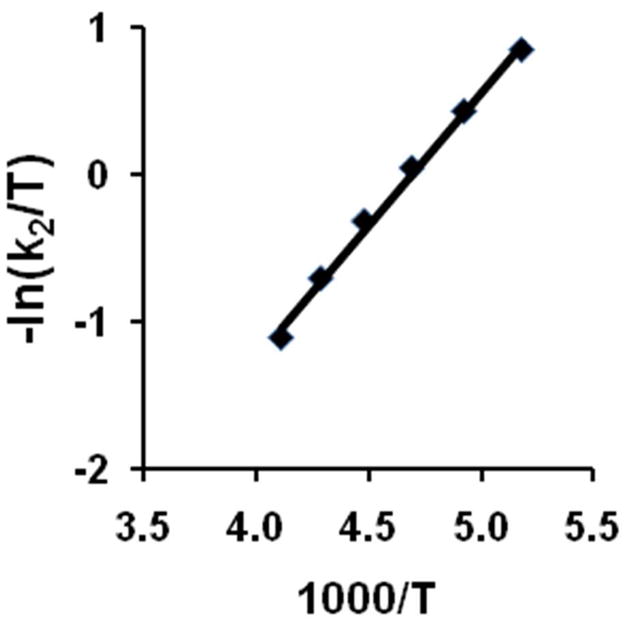
Eyring plot for O2 binding to 1•O2PPh2 in CH2Cl2.
Table 6.
Kinetic parameters for reactions of diiron(II) complexes with dioxygen
| Complex | Solvent | T range (°C) | K (−40 °C) (M−1 s−1) | ΔH‡ (kJ/mol) | ΔS‡ (J/K·mol) | Reference |
|---|---|---|---|---|---|---|
| 1•O2PPh2 | CH2Cl2 | −80 to −20 | 4.7×102 | 15.7(4) | −124(6) | This work |
| 1•O2PPh2 | MeCN | −40 to 0 | 2.55×103 | 13.7(4) | −119(10) | This work |
| 1•O2CPh | EtCN | −75 to −15 | 1.03(12)×102 | 15.4(6) | −121(3) | 43 |
| [Fe2(HPTP)(O2CPh)]2+ | MeCN | −40 to 0 | 7.3 × 103 | 15.8(4) | −101(10) | 46 |
| [Fe2(HPTP)(O2CPh)]2+ | CH2Cl2 | −80 to 0 | 67 | 16.7(2) | −132(8) | 46 |
A closer examination of dioxygen binding to 1•O2CPh identified the individual reaction steps of the overall reaction. Stopped-flow observation of the reaction of 1•O2CPh with dioxygen (2.9 mM) in CH2Cl2 at −80° reveals a two-step process (Figure S5), in agreement with sequential formation of intermediates 2•O2CPh and 3•O2CPh. At longer wavelengths (greater than ca. 650 nm), the initial absorbance increase takes place within 15 seconds and is followed by decay that is complete within 100 seconds. At 640 nm, the initial absorbance increase was also seen, but after 15 seconds the kinetic trace becomes flat. At shorter wavelengths, a biexponential absorbance increase was seen. Two-exponential fit of the variable-wavelengths data gave the following value of the observed rate constant: k1 = 0.22 s−1 at −80 °C (76 M−1s−1 under an assumption of a second-order process). This rate constant corresponds to the first reaction step, formation of 2•O2CPh. Interestingly, it is in excellent agreement with the reported value of the rate constant for the reaction of 1•O2CPh with O2 (135 M−1s−1 at −75 C in propionitrile, with the activation parameters of ΔH‡ = 15.4 kJ/mol ΔS‡ = −121 J K−1 mol−1).43 In that work, 3•O2CPh was characterized spectroscopically, and it was assumed that the kinetics of oxygenation corresponded to the formation of 3•O2CPh. Based on our studies, the kinetic parameters reported by Feig et al.43 correspond to the first oxygenation step, the formation of 2•O2CPh; the next step, conversion of 2•O2CPh to 3•O2CPh, is very rapid at most temperatures.
Discussion
One step in the catalytic activation of dioxygen by biological diiron(II) systems often produces (μ-η1:η1-peroxo) diiron(III) moieties. Some of these intermediates exhibit a level of stability that allows for them to be trapped and characterized.9–16 The same is true for many synthetic (μ-η1:η1-peroxo) diiron(III) complexes,18,25,27,41,47 some of which are so stable they have been crystallographically characterized.19–22 In previously published work,28 we used the dinucleating ligand N-EtHPTB and different oxyanions (O2X) to synthesize a set of three dioxygen binding diiron(II) complexes (1•O2X). Our efforts revealed that the (μ-η1:η1-peroxo) diiron(III) intermediates produced upon oxygenation of the diiron(II) precursors come in two forms, green-blue 2•O2X and deep blue 3•O2X (Scheme 1). In the initial form, the oxyanion acts as a three-atom bridge between the iron centers and the pendant benzimidazoles are cis to each other on each iron. In many cases, this intermediate converts to a second form, wherein the O2X ligand has moved to a terminal position, allowing the pendant benzimidazoles of the N-EtHPTB ligand to rearrange from a cis to a trans disposition on each iron. We found that the stability of 2•O2X is influenced by the identity of O2X (O2X = O2AsMe2, O2PPh2 and O2CPh) and concluded that the dominant factor governing 2•O2X stability is the bite distance (O···O) of the O2X moiety in 1•O2X as determined by X-ray crystallography. Anions with greater bite distances are better able to accommodate the >3 Å Fe···Fe distance in 2•O2X and gave rise to more stable 2•O2X intermediates; for X = O2AsMe2, the 2•O2X intermediate was so stable that no observable conversion to 3•O2X was observed prior to decomposition.
For this work, we synthesized several more 1•O2X complexes to assess additional factors that may affect the stability of 2•O2X. Specifically, we were interested in examining effects produced by electronic and steric changes in O2X and how those results relate to effects produced by differences in O2X bite distances. We also investigated how OPPh3 could be used to destabilize some 2•O2X intermediates. In addition, we used low-temperature stopped-flow techniques to demonstrate that the oxygenation of 1•O2CPh was in fact a two-step process like those we have described for the other 1•O2X complexes. Indeed, 2•O2CPh is short-lived even at −90°, so the adduct we originally observed at −40° and assigned to be 2•O2CPh in 199029,42 is in actuality the more stable 3•O2CPh isomer. Finally, we determined the activation parameters for the oxygenation of 1•O2PPh2 in CH2Cl2 and in MeCN and found them to be very similar to those reported earlier for 1•O2CPh in EtCN (Table 6),43 strongly suggesting a common rate determining step for these reactions corresponding to the formation of 2•O2X.
The focus of the experiments reported in this paper has been to gain further insight into the factors that affect the conversion of 2•O2X to 3•O2X, in which irreversible conversion to 3•O2X is preceded by movement of the O2X moiety from a bridging to a terminal position (Scheme 1). We postulate that there is a rapid equilibrium between bridging and terminal coordination modes of the O2X ligand in 2•O2X and only the isomer with a terminal O2X ligand can undergo conversion to 3•O2X during which the benzimidazole arms of the N-EtHPTB ligand shift from a cis relationship to each other to a trans configuration on both iron centers. Thus the initial preequilibrium should be affected by the basicity of O2X ligands, with the more basic ligand favoring the bridging mode and thereby decreasing the fraction of monodentate O2X isomer available to undergo conversion to 3•O2X. On the other hand, as the transformation from 2•O2X to 3•O2X entails a significant rearrangement of the coordination spheres about each iron center, the conversion should be slowed down by an increase in the steric bulk of O2X.
The trend in the stability of the 2•O2X complexes is best discerned by an examination of the kinetic data for the large subset of complexes with carboxylate bridges. A perusal of Table 5 suggests that, while it may be possible to observe an effect of ligand basicity in comparisons of select pairs (e.g. O2CPh vs. O2CC6H4-4-OMe and O2CPh vs. O2CCMe3), for the most part steric considerations supersede ligand basicity arguments. For example, the stability of 2•O2CR is enhanced 30-fold in the series, R = Ph < CMe3 < CPh3, commensurate with the increase in steric bulk on the carbon atom adjacent to the carboxylate function. Although the higher pKa (5.03) of HO2CCMe348 relative to benzoic acid (pKa = 4.19)48 could be used to rationalize the threefold longer lifetime of 2•O2CCMe3, the 30-fold greater stability of 2•O2CCPh3 than 2•O2CPh cannot be explained by the slightly lower pKa of HO2CCPh3 (3.96)49 but can easily be rationalized by the much greater bulk of the triphenylmethyl group relative to phenyl.
Complexes with methoxy-substituted benzoate bridges support the above arguments. The conversion of 2•O2CC6H2-3,4,5-(OMe)3 to its 3•O2X form is 100-fold slower that for 2•O2CPh, despite the fact that the two carboxylic acids have essentially identical pKa values (4.24 and 4.19, respectively).48,50 Therefore the considerable difference in the stabilities of 2•O2CC6H2-3,4,5-(OMe)3 and 2•O2CPh (Table 5) must arise from steric considerations. Other methoxy-substituted benzoate complexes exhibit intermediate rates of conversion; 2•O2CC6H3-3,4-(OMe)2 converts just slightly faster than 2•O2CC6H2-3,4,5-(OMe)3, while 2•O2CC6H3-3,5-(OMe)2 and 2•O2CC6H4-4-(OMe) are 10-fold faster. From these results, it seems clear that additional steric bulk in the meta and/or para positions of benzoate-based oxyanions is the primary factor that stabilizes the resultant 2•O2X complexes.
The lifetimes of 2•O2CR complexes are shortened upon addition of OPPh3. We previously found that OPPh3 had a good binding affinity for the parent FeIII 2(N-Et-HPTB)-peroxo complex and exerted such a significant stabilizing effect that its presence led to the crystallization and structural characterization of [FeIII 3(μ-1,2-O2)(N-Et-HPTB)(OPPh3)2]3+.20 Addition of 20 equivalents of OPPh3 to 2•O2CR at −90° in fact accelerated the conversion to 3′•O2CR with rates of 0.03–0.2 s−1, representing a much smaller range of values than for the conversions in the absence of OPPh3 (Table 5). These results suggest that the binding of OPPh3 to the diiron center facilitates the ligand rearrangement required to convert from 2•O2X to 3•O2X (Scheme 1).
Tetrahedral O2X bridges also increase the stability of 2•O2X. In this series, we compared complexes with O2AsMe2, O2PMe2, O2PPh2 and O2P(OPh)2 bridges that differ in steric properties and basicity (pKa = 6.27, 3.08, 2.32 and 1.85, respectively).51–53 The order of stability in this subset is O2AsMe2 > O2PMe2 > O2PPh2 > O2P(OPh)2 (Table 4), following the trend of decreasing pKa values. Only 2•O2P(OPh)2 undergoes conversion to the corresponding 3•O2X form at −90°, with kobs = 1.2(1) × 10−4 s−1, a rate that is slower than observed for five of the six 2•O2CR complexes at −90° (Table 5). While 2•O2PPh2 is indefinitely stable at −90°, it converts to 3•O2PPh2 when warmed to −40°, at a rate comparable to that of 2•O2P(OPh)2 at −90°. Neither 2•O2PMe2 nor 2•O2AsMe2 generate an observable 3•O2X form; instead they appear to decay directly to 4•O2X. However, 2•O2PMe2 can be converted to 3′•O2PMe2 at −40° by addition of OPPh3, whereas 2•O2AsMe2 does not convert to 3′•O2AsMe2, even when reacted with 100 equivalents of OPPh3 at room temperature. The increasing stability of 2•O2X with the bite distance of the oxyanion bridge (X = C, 2.23 Å; X = P, 2.55 Å; X = As, 2.80 Å) reflects the ability of the O2X bridge to span the Fe•••Fe distance required by the dinucleating HPTB ligand framework without imposing a strain on the six-member ring formed by the three atoms of the oxyanion bridge, the two iron atoms and the alkoxide oxygen on the N-EtHPTB ligand. The O2AsMe2 complex, having the longest O•••O bite distance, represents the most stable of the 2•O2X intermediates.
In this paper, we have investigated the various factors that influence the conversion of 2•O2X O2 adducts to corresponding 3•O2X species. This conversion entails the shift of the O2X ligand from a bidentate bridging mode to a terminal monodentate mode, providing another example of a diiron complex involved in a mechanistically important “carboxylate shift” first recognized by Lippard and coworkers twenty years ago.30 An analogous carboxylate shift has been suggested by Do et al54 to occur upon protonation of O2 adducts of [FeII 2(N-EtHPTB)(μ-O2CR)]2+ complexes (R = Ph or C6F5) at −30°. However, although unequivocal evidence was provided for the protonation of the carboxylate ligand, it is clear from a comparison with the spectroscopic data we have presented in this paper that the O2 adducts studied by Do et al54 must be assigned structures we now associate with 3•O2CR complexes, which have terminal monodentate carboxylates. On the other hand, the corresponding 2•O2CR complexes (with bridging carboxylates) are in fact formed upon oxygenation of the diiron(II) precursors (as previously assumed29,42,54) but can be observed only at −90°, as they readily undergo the “carboxylate shift” at that temperature to isomerize to corresponding 3•O2CR species. However, the key take-home message of the study by Do et al54 is that protonation of a carboxylate ligand on a diiron complex can occur and give rise to a complex with a carboxylic acid ligand. Indeed a carboxylate ligand may serve as a convenient conduit for delivering a proton to a bound peroxide in order to facilitate O-O bond cleavage, for example, in the conversion of intermediate P to Q in the case of sMMO for which a proton is clearly implicated.13,55
Supplementary Material
Acknowledgments
This work was supported by the National Institutes of Health (GM-38767 to L.Q.) and the Department of Energy (DE-FG02-06ER15799 to E.R.-A.); stopped-flow instrumentation at Tufts was supported by the NSF (CHE 0639138). We thank Dr. Victor Young, Jr. of the X-ray Crystallographic Laboratory at the University of Minnesota for his invaluable assistance.
References
- 1.Wallar BJ, Lipscomb JD. Chem Rev. 1996;96:2625. doi: 10.1021/cr9500489. [DOI] [PubMed] [Google Scholar]
- 2.Que L., Jr . In: Bioinorganic Catalysis. 2. Reedijk J, Bouwman E, editors. Marcel Dekker; New York: 1999. p. 269. [Google Scholar]
- 3.Solomon EI, Brunold TC, Davis MI, Kemsley JN, Lee SK, Lehnert N, Neese F, Skulan AJ, Yang YS, Zhou J. Chem Rev. 2000;100:235. doi: 10.1021/cr9900275. [DOI] [PubMed] [Google Scholar]
- 4.Merkx M, Kopp DA, Sazinsky MH, Blazyk JL, Müller J, Lippard SJ. Angew Chem Int Ed. 2001;40:2782. doi: 10.1002/1521-3773(20010803)40:15<2782::AID-ANIE2782>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- 5.Fox BG, Lyle KS, Rogge CE. Acc Chem Res. 2004;37:421. doi: 10.1021/ar030186h. [DOI] [PubMed] [Google Scholar]
- 6.Bollinger JM, Jr, Diao Y, Matthews ML, Xing G, Krebs C. Dalton Trans. 2009:905. doi: 10.1039/b811885j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sazinsky MH, Lippard SJ. Acc Chem Res. 2006;39:558. doi: 10.1021/ar030204v. [DOI] [PubMed] [Google Scholar]
- 8.Murray LJ, Lippard SJ. Acc Chem Res. 2007;40:466. doi: 10.1021/ar600040e. [DOI] [PubMed] [Google Scholar]
- 9.Liu KE, Valentine AM, Qiu D, Edmondson DE, Appelman EH, Spiro TG, Lippard SJ. J Am Chem Soc. 1995;117:4997. [Google Scholar]
- 10.Bollinger JM, Jr, Krebs C, Vicol A, Chen S, Ley BA, Edmondson DE, Huynh BH. J Am Chem Soc. 1998;120:1094. [Google Scholar]
- 11.Broadwater JA, Ai J, Loehr TM, Sanders-Loehr J, Fox BG. Biochemistry. 1998;37:14664. doi: 10.1021/bi981839i. [DOI] [PubMed] [Google Scholar]
- 12.Broadwater JA, Achim C, Münck E, Fox BG. Biochemistry. 1999:12197. doi: 10.1021/bi9914199. [DOI] [PubMed] [Google Scholar]
- 13.Lee SK, Lipscomb JD. Biochemistry. 1999;38:4423. doi: 10.1021/bi982712w. [DOI] [PubMed] [Google Scholar]
- 14.Saleh L, Krebs C, Ley BA, Naik S, Huynh BH, Bollinger JM. Biochemistry. 2004;43:5953. doi: 10.1021/bi036099e. [DOI] [PubMed] [Google Scholar]
- 15.Murray LJ, Garcia-Serres R, Naik S, Huynh BH, Lippard SJ. J Am Chem Soc. 2006;128:7458. doi: 10.1021/ja062762l. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yun D, Garcia-Serres R, Chicalese BM, An YH, Huynh BH, Bollinger JM., Jr Biochemistry. 2007;46:1925. doi: 10.1021/bi061717n. [DOI] [PubMed] [Google Scholar]
- 17.Vu VV, Emerson JP, Martinho M, Kim YS, Münck E, Park MH, Que JL. Proc Nat Acad Sci. 2009;106:14814. doi: 10.1073/pnas.0904553106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tshuva EY, Lippard SJ. Chem Rev. 2004;104:987. doi: 10.1021/cr020622y. [DOI] [PubMed] [Google Scholar]
- 19.Ookubo T, Sugimoto H, Nagayama T, Masuda H, Sato T, Tanaka K, Maeda Y, Okawa H, Hayashi Y, Uehara A, Suzuki M. J Am Chem Soc. 1996;118:701. [Google Scholar]
- 20.Dong Y, Yan S, Young VG, Jr, Que L., Jr Angew Chem Int Ed Engl. 1996;35:618. [Google Scholar]
- 21.Kim K, Lippard SJ. J Am Chem Soc. 1996;118:4914. [Google Scholar]
- 22.Zhang X, Furutachi H, Fujinami S, Nagatomo S, Maeda Y, Watanabe Y, Kitagawa T, Suzuki M. J Am Chem Soc. 2005;127:826. doi: 10.1021/ja045594a. [DOI] [PubMed] [Google Scholar]
- 23.Kryatov SV, Chavez FA, Reynolds AM, Rybak-Akimova EV, Que L, Jr, Tolman WB. Inorg Chem. 2004;43:2141. doi: 10.1021/ic049976t. [DOI] [PubMed] [Google Scholar]
- 24.Korendovych IV, Kryatov SV, Reiff WM, Rybak-Akimova EV. Inorg Chem. 2005;44:8656. doi: 10.1021/ic051739i. [DOI] [PubMed] [Google Scholar]
- 25.Kryatov SV, Taktak S, Korendovych IV, Rybak-Akimova EV, Kaizer J, Torelli S, Shan X, Mandal S, MacMurdo V, Mairata i Payeras A, Que L., Jr Inorg Chem. 2005;44:85. doi: 10.1021/ic0485312. [DOI] [PubMed] [Google Scholar]
- 26.Yoon S, Lippard SJ. Inorg Chem. 2006;45:5438. doi: 10.1021/ic060307k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fiedler AT, Shan X, Mehn MP, Kaizer J, Torelli S, Frisch JR, Kodera M, Que L., Jr J Phys Chem A. 2008;112:13037. doi: 10.1021/jp8038225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Frisch JR, Vu VV, Martinho M, Münck E, Que L., Jr Inorg Chem. 2009;48:8325. doi: 10.1021/ic900961k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ménage S, Brennan BA, Juarez-Garcia C, Münck E, Que L., Jr J Am Chem Soc. 1990;112:6423. [Google Scholar]
- 30.Rardin RL, Tolman WB, Lippard SJ. New J Chem. 1991;15:417. [Google Scholar]
- 31.McKee V, Zvagulis M, Dagdigian JV, Patch MG, Reed CA. J Am Chem Soc. 1984;106:4765. [Google Scholar]
- 32.Armarego WLF, Perrin DD. Purification of Laboratory Chemicals. Butterworth-Heinemann; Oxford: 1997. [Google Scholar]
- 33.Hagen KS. Inorg Chem. 2000;39:5867. doi: 10.1021/ic000444w. [DOI] [PubMed] [Google Scholar]
- 34.Kryatov SV, Rybak-Akimova EV, Schindler S. Chem Rev. 2005;105:2175. doi: 10.1021/cr030709z. [DOI] [PubMed] [Google Scholar]
- 35.Bruker. SMART V5.054. Bruker Analytical X-Ray Systems; Madison, WI: 2001. [Google Scholar]
- 36.Blessing RH. Acta Cryst. 1995;A51:33. [Google Scholar]
- 37.Bruker. SAINT+ V6.45. Bruker Analytical X-Ray Systems; Madison, WI: 2003. [Google Scholar]
- 38.Bruker. SHELXTL V6.14. Bruker Analytical X-Ray Systems; Madison, WI: 2000. [Google Scholar]
- 39.Spek AL. PLATON A multipurpose crystallographic tool. Utrecht University; Utrecht, The Netherlands: 2002. [Google Scholar]
- 40.Addison AW, Rao TN, Reedijk J, Rijn Jv, Verschoor GC. J Chem Soc, Dalton Trans. 1984:1349. [Google Scholar]
- 41.Yan S, Cheng P, Wang Q, Liao D, Jiang Z, Wang G. Science in China, Series B: Chemistry. 2000;43:405. [Google Scholar]
- 42.Dong Y, Ménage S, Brennan BA, Elgren TE, Jang HG, Pearce LL, Que L., Jr J Am Chem Soc. 1993;115:1851. [Google Scholar]
- 43.Feig AL, Becker M, Schindler S, van Eldik R, Lippard SJ. Inorg Chem. 1996;35:2590. doi: 10.1021/ic951242g. [DOI] [PubMed] [Google Scholar]
- 44.Feig AL, Lippard SJ. J Am Chem Soc. 1994;116:8410. [Google Scholar]
- 45.Korendovych IV, Kryatov SV, Rybak-Akimova EV. Acc Chem Res. 2007;40:510. doi: 10.1021/ar600041x. [DOI] [PubMed] [Google Scholar]
- 46.Costas M, Cady CW, Kryatov SV, Ray M, Ryan MJ, Rybak-Akimova EV, Que L., Jr Inorg Chem. 2003;42:7519. doi: 10.1021/ic034359a. [DOI] [PubMed] [Google Scholar]
- 47.Than R, Schrodt A, Westerheide L, Eldik Rv, Krebs B. Eur J Inorg Chem. 1999:1537. [Google Scholar]
- 48.Lide DR. CRC Handbook of Chemistry and Physics. 72. CRC Press; Boston: 1991. [Google Scholar]
- 49.Kilpatrick ML, Fackenthal E. Journal of the Electrochemical Society. 1953;100:185. [Google Scholar]
- 50.Sergeant EP, Dempsey B. Ionisation Constants of Organic Acids. Pergamon Press; New York: 1979. [Google Scholar]
- 51.Kilpatrick ML. J Am Chem Soc. 1949;71:2607. [Google Scholar]
- 52.Shin TW, Kim K, Lee IJ. J Sol Chem. 1997;26:379. [Google Scholar]
- 53.Edmundson RS. Dictionary of Organophosphorus Compounds. Chapman and Hall; New York: 1988. [Google Scholar]
- 54.Do LH, Hayashi T, Moënne-Loccoz P, Lippard SJ. J Am Chem Soc. 2010;132:1273. doi: 10.1021/ja909718f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tinberg CE, Lippard SJ. Biochemistry. 2009;48:12145. doi: 10.1021/bi901672n. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.