

Abstract
Nitric oxide synthase (NOS) catalyzes the conversion of L-arginine to L-citrulline through the intermediate Nω-hydroxy-L-arginine (NHA), producing nitric oxide, an important mammalian signaling molecule. Several disease states are associated with improper regulation of nitric oxide production, making NOS a therapeutic target. The first step of the NOS reaction has been well-characterized and is presumed to proceed through a compound I heme species, analogous to the cytochrome P450 mechanism. The second step, however, is enzymatically unprecedented and is thought to occur via a ferric peroxo heme species. To gain insight into the details of this unique second step, we report here the synthesis of NHA analogues bearing guanidinium-methyl or -ethyl substitutions and their investigation as either inhibitors of or alternate substrates for NOS. Radiolabeling studies reveal that Nω-methoxy-L-arginine, an alternative NOS substrate, produces citrulline, nitric oxide, and methanol. On the basis of these results we propose a mechanism for the second step of NOS catalysis in which a methylated nitric oxide species is released and is further metabolized by NOS. Crystal structures of our NHA analogues bound to nNOS have been solved, revealing the presence of an active site water molecule only in the presence of singly methylated analogues. Bulkier analogues displace this active site water molecule; a different mechanism is proposed in the absence of the water molecule. Our results provide new insight into the steric and stereochemical tolerance of the NOS active site and substrate capabilities of NOS.
Nitric oxide synthases (NOSs) catalyze the oxygenation of L-arginine to L-citrulline and nitric oxide (NO) using molecular oxygen (O2) and NADPH (Scheme 1). NO is an important signalling molecule with a wide range of biological functions.(1–3) There are three mammalian NOS isoforms. As products of distinct genes, they maintain highly conserved active sites across all three isoforms and other species. Two are constitutive isoforms, neuronal NOS (nNOS) and endothelial NOS (eNOS), which are involved in neuronal signalling and vascular regulation, respectively.(4, 5) Inducible NOS (iNOS) is expressed in macrophage cells in response to invasion of pathogens.(3) Misregulation of NO has been implicated in various disease states,(1–3) and therefore NOSs are sought-after therapuetic targets. Better understanding of the NOS mechanism will aid in the design of novel NOS inhibitors.
Scheme 1.
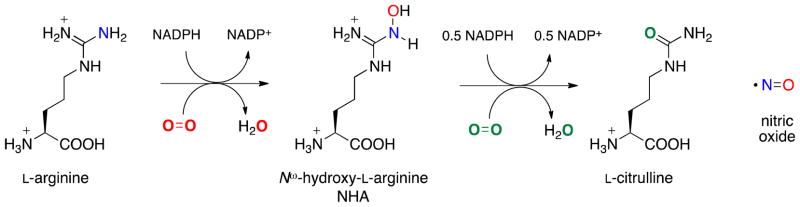
Reaction catalyzed by NOS
NOSs are homodimeric enzymes with a reductase domain that binds NADPH, FAD, and FMN, and an oxygenase domain, which contains heme and binds L-arginine and (6R)-5,6,7,8-tetrahydrobiopterin (H4B). The mechanism catalyzed by NOS occurs in two distinct steps (Scheme 1). In the first step, L-arginine is monooxygenated to Nω-hydroxy-L-arginine (NHA). This is proposed to occur through an oxygen rebound mechanism via the Compound I (CpdI, FeIV•+=O ) heme species, analagous to cytochrome P450 chemistry (Scheme 2).(6) H4B provides the second electron required for oxygen activation.(7, 8) In the second step, NOS converts NHA to citrulline and NO, which requires only one electron. This step is thought to proceed through a ferric peroxo species.(9, 10) Early mechanistic proposals included the nucleophilic addition of the ferric peroxide heme species (FeIII-OO−) to the guanidino carbon of NHA.(11–14) Recent EPR/ENDOR cryoreduction/annealing experiments provide evidence that an active species is ferric hydroperoxide (FeIIIOOH), and in this case, the mechanism could involve a nucleophilic attack of the hydroperoxide on the guanidinium oxime.(15) The source of the proton that forms FeIIIOOH is unknown, but it is speculated to be either an active site water molecule or the substrate itself.(14) The tetrahedral intermediate formed from addition of a ferric peroxo will then collapse, yielding citrulline and nitric oxide. In this step, H4B also serves as a donor of the electron required for oxygen activation. It further acts as an electron acceptor when it is re-reduced either by the tetrahedral intermediate before collapse or by product NO−, since overall this reaction is only a one-electron oxidation.(7) Substrate identity, and perhaps more importantly its pKa (arginine pKa = 12.5; NHA pKa = 8.5),(16) are thought to dictate the formation of these different active heme species in steps one and two.
Scheme 2.

Proposed NOS mechanisms for steps one and two
Although many experiments have been reported, details of the NOS mechanism remain elusive. During NOS turnover, the precise role of H4B and when it is implicated, the true identity of the active heme species, and the source of protons (and how many are required) are still controversial.(17, 18) An active site water molecule has been speculated to play an important role in proton donation – either as a single proton donor or as a shuttle sequestering protons from the bulk solvent to the reaction site.(15, 16, 19) NOS crystal structures of various isoforms show a conserved, specifically oriented water molecule as part of a hydrogen bonding network that includes the substrate, active site residues, and the diatomic heme ligand (O2 for catalysis, but NO and CO are used to form stable crystal structures).(13, 20) A second hypothesis is that the substrate itself acts as a proton donor; ENDOR and X-ray experiments show that NHA is most likely protonated in the active site.(21) Furthermore, Davydov and Hoffman’s cryoreduction EPR experiments find that FeIII-OOH formation is not pH dependent, suggesting the proton is sourced directly from NHA, not bulk solvent.(15)
The first NOS half-reaction closely resembles well-known cytochrome P450 hydroxylation chemistry, but the second-half reaction, the oxidation of an Nω-hydroxyguanidine to a urea and NO, is enzymatically unprecedented. It is therefore poorly understood. Previously we demonstrated that direct hydrogen atom abstraction from the O-H bond of NHA is not necessary for substrate turnover; Nω-tert-butyloxy-L-arginine (tBOA) and Nω-(3-methyl-2-butenyl)oxy-L-arginine are nitric oxide- and citrulline-producing NOS substrates.(22) To further probe the second step of the mechanism of NOS, we have synthesized and investigated a series of methylated (or ethylated) NHA analogues (Figure 1).
Figure 1.
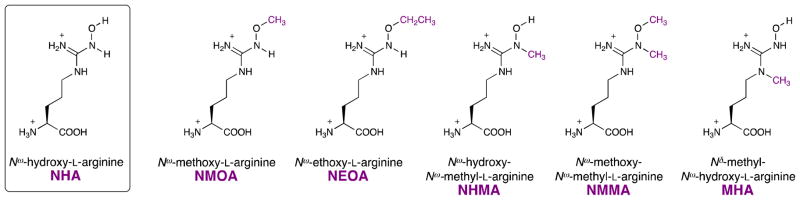
NHA substrate analogues
Nω-Methoxy-L-arginine (NMOA) has been previously synthesized(23, 24) and has been explored as a prodrug inhibitor of arginase,(25) but has not been examined as a NOS substrate. Nω-Methyl-L-arginine (NMA) functions as an inactivator (kinact = 0.07 min−1, KI = 2.7 μM), a competitive inhibitor (Ki = 200 nM), and a slow, alternative substrate for NOS.(26) NOS converts NMA into Nω-hydroxy-Nω-methyl-L-arginine (NHMA), which is subsequently converted into citrulline, NO, and formaldehyde.(26) As substrates, NMA and NHMA both cause significant uncoupling of NADPH oxidation.(26) Nδ-Methyl-L-arginine (dMA) can be converted by NOS to Nδ-methyl-Nω-hydroxy-L-arginine (MHA), but no further.(27, 28) dMA binds weakly to the NOS active site with a Ki of 1.4 mM.(27) Nω-Methoxy-Nω-methyl-L-arginine (NMMA) and Nω-ethoxy-L-arginine (NEOA) have not been previously reported. We previously evaluated tBOA as a NOS substrate,(22) and here we re-examine a co-crystal structure of this compound bound in the nNOS active site. In this report, we present the synthesis, enzymatic evaluation, co-crystal structures, and novel mechanistic insights with respect to these five substrate analogues (Figure 1) as they relate to the second step of the NOS catalytic mechanism (Scheme 2).
MATERIALS AND METHODS
General Methods
All chemicals, unless otherwise noted, were obtained from Sigma Aldrich and used without further purification. Michaelis-Menten kinetics and non-linear regressions were plotted and analyzed using GraphPad Prism5.0c software. Complete procedures for the syntheses, as well as the characterization of NMOA, NEOA, NEOA, NHMA, NMMA, MHA, and [14C]-NMOA, can be found in the Supporting Information.
Measuring NO Production
Murine iNOS(29) and rat nNOS(30) were expressed and purified from E. coli as previously described. NO production was monitored using the hemoglobin capture assay at 22 °C.(31) In addition to various final concentrations of the analogue being evaluated, assay mixtures contained 100 μM NADPH, 3 μM hemoglobin-A0 (Sigma H0267), 10 μM H4B, in 100 mM HEPES with 10% glycerol (pH 7.5). For nNOS assays, 1 mM CaCl2 and 300 U/mL CaM were added. For Ki determination, assays contained 10 μM L-arginine. Assays were initiated by addition of enzyme (approximately 100 nM final concentration), and methemoglobin formation was monitored for 1 min at 401 nm using a Shimadzu UV-1800 spectrophotometer. Km and kcat values were determined from nonlinear regressions (Michaelis-Menten). For Ki determinations, IC50 values were first calculated using nonlinear regressions (dose-response inhibition, four parameter variable slope). Subsequent Ki values were determined using the Cheng-Prushoff relationship Ki = IC50/(1 +[S]/Km), where a Km of 8.3 μM was used for murine iNOS.(32)
When noted, NO production was also measured with the Greiss reagent(31) using the nitrite/nitrate colormetric assay kit from Cayman Chemical (760871). Enzyme incubations contained various concentrations of the compound being evaluated, 100 nM iNOS, 100 μM NADPH, 10 μM H4B, in 100 mM HEPES with 10% glycerol (pH 7.5). Lactate dehydrogenase was added to the reactions to oxidize excess NADPH, and then Griess reagents were added to report nitrate. Absorbance was measured at 540 nm using a Synergy H1 Biotek plate reader.
Determination of the Spectral Binding Constant, Ks
Binding affinities for iNOS were determined using the previously described ferric difference spectral binding assay.(33) Since all compounds were type I heme-coordinating, imidazole, a type II coordinating compound, was used to initially coordinate the heme. The Ks value was then determined from displacement of the imidazole. To a 500 μL quartz cuvette was added 5 μM iNOS, 10 μM H4B, and 100 mM HEPES (pH 7.5) to 200 μL total volume. This cuvette was scanned against a blank containing 100 mM HEPES with 10 μM H4B. Spectra were taken from 380 to 500 nm. To determine the binding affinity of imidazole, spectra were taken after the addition of aliquots of the imidazole (0.5 to 1 mM final concentration). To assay NHA analogues, 300 μM imidazole was used, aliquots of the analogue being examined (0.1 to 1 mM final concentration) were added, and spectra were obtained for each. The total volumes added were kept below 10 μL (5%) to avoid dilution effects. Michaelis-Menton curves were determined for the imidazole as well as for each inhibitor by plotting concentration versus absorbance difference (local maximum – local minimum ). Then Hanes-Wolff plots were used to determine the Ks for imidazole and the apparent Ks for the NHA analogues.(34) The Ks for imidazole with iNOS was found to be 120 – 150 μM over multiple experiments; Ks values of analogues were determined using the following equation:
NDA Derivatization and HPLC Separation
NDA-derivatization reactions contained 25 μL of amino acid standard or sample, 25 μL of 30 mM NaCNω in 100 mM NaB(OH)3 buffer pH 10, and 15 μL of 10 mM NDA in methanol. NDA reactions achieve completion nearly immediately, so all reactions were analyzed after 10 min. Phenylalanine was used as an internal standard to track complete derivatization and assure complete injection. Reactions were analyzed by reversed-phase HPLC (10 μL injection) using an Econosil C18 column with 80% 5 mM sodium acetate pH 6.0: 20% MeOH as Solvent A and 100% acetonitrile as Solvent B. A gradient from 25% B to 75% B was run over 30 min at 0.75 mL/minute. Under these conditions, NDA-derivatized amino acids had the following retention times: citrulline 4.2 min; phenylalanine 9.4 min; NHA 10.5 min; NMOA 11.7 min; NHMA 11.2 min; NMMA 12.8 min. An NDA-citrulline standard curve (R2 = 0.995) was linear from 100 μM to 1 mM.
Measuring Substrate Uncoupling
iNOS-substrate enzyme rections (300 μL total volume in quartz cuvettes, containing 100 μM NADPH, 10 μL H4B, 100 nM iNOS, 3 μM hemoglobin-A0, and 100 mM HEPES pH 7.5) were dually monitored at 401 nM and 340 nM for 1 min. Substrates were evaluated at the following final concentrations: 20 μM arginine, 20 μM NHA, 100 μM NMOA, and 100 μM NHMA. Reactions were initiated by the addition of iNOS. Extinction coefficents used were εHbNO = 38,000 M−1 cm−1 and εNADPH = 6.22 mM−1 cm−1.(35)
DNPH-Derivatization Reactions
DNPH-derivatization reactions were set up by combining 20 μL of aldehyde standard or sample with 10 μL of 20 mM DNPH in 0.4 M H2SO4 in 10% H2O, 90% acetonitrile. DNPH derivatization reactions proceed quickly to completion, so all reactions were injected after 10 min. All DNPH-derivatized reactions were separated by reverse-phase HPLC (10 μL injection) under the following conditions: a Phenomonex Luna C18 column was used with water as Solvent A and acetonitrile as Solvent B. A gradient from 10% B to 90% B was run over 30 min at 1.0 mL/min. DNPH-derivatives were detected at 360 nm. Under these conditions, DNPH eluted at 12.2 min and DNPH-formaldehyde eluted at 13.3 min (DNPH-acetaldehyde 14.2 min, DNPH-acetone 14.9 min). A DNPH-formaldehyde standard curve (R2 = 0.995) was linear from 100 μM to 1.5 mM.
HPLC MS Confirmation of NDA- and DNPH-derivatives
The identities of all NDA- and DNPH-derivatized products of the [14C]-NMOA reactions were confirmed by liquid chromatography-mass spectrometry using an Agilent 1200 series purification system equipped with a diode array detector (SL 1315C) set to 460 (or 360) and 254 nm and an Agilent 6130A Single Quad detector using atmospheric pressure electrospray ionization (API-ES) in positive mode. A Phenomenex Gemini-NX C18 (4.6 x 50 mm, 5 μm, 100 Å) column was used with Solvent A as LCMS grade water + 0.1 % formic acid and Solvent B as LCMS grade ACNω+ 0.1 % formic acid. For NDA-derivatizations, the following gradient was run at a flow rate of 1.5 mL/min: 0–7 minutes (5 – 50 % B), 7–10 minutes (100 % B). Derivatives had the following retention times: NDA-NHA 4.2 min; NDA-citrulline 6.2 min; NDA-NMOA 4.4 min; NDA-NHMA 4.4 min; NDA-NMMA; 4.6 min. For DNPH-derivatizations, the following gradient was used: at a flow rate of 1.5 mL/min, from 0–10 minutes (10 – 90 % B). DNPH-formaldehyde eluted at 3.26 minutes under these conditions.
Crystal Structure Determination
The heme domain of rat nNOS protein sample and crystals were prepared according to the procedures reported previously.(36) Fresh crystals (1–2 days old) were first passed stepwise through cryo-protectant solutions(36) and then soaked with 10 mM NHA analogues for 4–6 h at 4 oC before being mounted on nylon loops and flash cooled by plunging into liquid nitrogen. The cryogenic (100 K) X-ray diffraction data were collected remotely at various beamlines at Stanford Synchrotron Radiation Lightsource or Advanced Light Source through the data collection control software and a crystal mounting robot. Raw data frames were indexed, integrated, and scaled using HKL2000.(37) The binding of NHA analogues was detected by the initial difference Fourier maps calculated with REFMAC.(38) The analogue molecules were then modeled in COOT(39) and refined using REFMAC. Water molecules were added in REFMAC and checked through COOT. The TLS protocol(37, 40) was implemented in the later stage of refinements with each subunit as one TLS group. Finally, an additional round of TLS refinement was carried out with the coordinates of substrate analogue and the water of interest removed from the input model. The map coefficients in the output were used to produce the omit Fo – Fc electron density maps shown in Supplemental Figure S10. The refined structures were validated through the RCSB web server before deposition to the protein data bank. The crystallographic data collection and structure refinement statistics are summarized in Table 1 with PDB accession codes included.
Table 1.
Crystallographic data collection and refinement statistics
| Data seta | nNOS-NMOA | nNOS-NEOA | nNOS-NHMA | nNOS-NMMA | nNOS-MHA | nNOS-tBOA |
|---|---|---|---|---|---|---|
| Data collection | ||||||
| PDB code | 4FVW | 4FVX | 4FVY | 4FVZ | 4FW0 | 4GQE |
| Space group | P212121 | P212121 | P212121 | P212121 | P212121 | P212121 |
| Cell dimensions | ||||||
| a | 52.1 | 52.0 | 51.9 | 51.9 | 52.1 | 52.0 |
| b | 111.0 | 111.3 | 110.9 | 110.8 | 111.3 | 111.1 |
| c (Å) | 165.3 | 165.1 | 164.5 | 164.5 | 164.1 | 164.3 |
| Resolution (Å) | 1.80 (1.83-1.80) | 2.00 (2.03-2.00) | 1.70 (1.73-1.70) | 2.00 (2.03-2.00) | 1.95 (1.98-1.95) | 1.78 (1.81 -1.78) |
| Rsym or Rmerge | 0.077 (0.626) | 0.071 (0.588) | 0.055 (0.443) | 0.075 (0.636) | 0.064 (0.662) | 0.057 (0.746) |
| I / σI | 29.9 (1.8) | 19.7 (1.7) | 31.8 (2.0) | 23.3 (1.9) | 24.3 (2.0) | 31.6 (2.3) |
| No. unique reflections | 88,431 | 65,282 | 102,661 | 65,397 | 70,555 | 88,147 |
| Completeness (%) | 99.6 (99.9) | 99.6 (97.8) | 97.5 (99.2) | 99.4 (99.9) | 99.6 (99.9) | 99.4 (98.7) |
| Redundancy | 4.2 (3.6) | 4.0 (3.6) | 5.0 (4.6) | 3.8 (3.8) | 4.0 (4.0) | 4.5 (3.8) |
| Refinement | ||||||
| Resolution (Å) | 1.81 | 2.00 | 1.70 | 2.00 | 1.95 | 1.80 |
| No. reflections used | 83,967 | 61,988 | 97,474 | 61,954 | 66,998 | 83,676 |
| Rwork/Rfreeb | 0.181/0.218 | 0.181/0.221 | 0.184/0.212 | 0.202/0.248 | 0.194/0.232 | 0.189/0.222 |
| Mean B value (Å2) | 48.03 | 47.98 | 36.52 | 52.98 | 43.26 | 46.80 |
| No. atoms | ||||||
| Protein | 6687 | 6659 | 6679 | 6667 | 6655 | 6662 |
| Ligand/ion | 171 | 165 | 157 | 160 | 167 | 153 |
| Water | 374 | 388 | 419 | 262 | 322 | 380 |
| R.m.s. deviations | ||||||
| Bond lengths (Å) | 0.013 | 0.013 | 0.012 | 0.015 | 0.013 | 0.013 |
| Bond angles (deg) | 2.019 | 1.424 | 1.303 | 1.522 | 1.352 | 2.051 |
See Figure 1 for nomenclature and chemical formulae of NHA analogues.
Rfree was calculated with the 5% of reflections set aside throughout the refinement. The set of reflections for the Rfree calculation were kept the same for all data sets according to those used in the data of the starting model (1OM4).
MeOX and FDH Reactions
MeOX and FDH were obtained from Sigma Aldrich (A2404 and F8649, respectively). 10 μL of 80 U/mL MeOX in 100 mM HEPES pH 7.5 was reacted with 100 μL of sample (methanol standards, or iNOS-substrate reactions) for one hour at room temperature. MeOX reactions with methanol standards, followed by DNPH derivatization and HPLC separation produced a standard curve (R2 = 0.89) with detection limit of 100 μM. [14C]-NMOA-iNOS reactions incubated with MeOX were DNPH derivatized (see Methods) and analyzed by HPLC and scintillation counted. Reactions longer than one hour did not show stoichiometric turnover.
In a 20 mL glass scintillation vial the following were combined: 100 uL of [14C]-NMOA-iNOS reaction, 10 μL 80 U/mL MeOX and 10 μL 80 U/mL FDH. The vial was sealed with a septum containing a suspended 1 mL plastic well. The vial was reacted for 12 hours at room temperature. Using a syringe, 200 μL of an 8 % (v/v) aqueous solution of NaOH was carefully added to the plastic well and 200 μL of a 20 % (v/v) aqueous solution of TCA was added to the multi-enzyme reaction in the bottom of the vial. The vial was further incubated at 37 °C with gentle shaking for 2 hours. The suspended reaction well was carefully separated from the vial and each were scintillation counted. Supplementary Figure S10 shows the chromatograms for those experiments.
RESULTS
Synthesis of the NHA Analogues
NMOA, NEOA, NHMA, and NMMA were synthesized through nucleophilic addition of appropriate amines to a protected ornithine thiourea (see Supplemental Scheme S1).(24, 41) MHA was synthesized by the procedure of Clement and coworkers.(42) tBOA was synthesized as previously described and was purified by HPLC.(22)
Kinetic Evaluation of NHA Analogues
NO production from iNOS and nNOS was evaluated using the hemoglobin capture assay, which monitors the absorbance increase at 401 nm as the hemoglobin-NO complex is produced.(31) Figure 2 shows Michaelis-Menten curves for NHA analogues that behave as substrates, Table 2 reports kinetic values for the compounds measured, Supplementary Figure S1 provides the individual Michaelis-Menten curves used for the determination. The mechanism is speculated to be highly conserved among NOS isoforms. In many cases we examine both isoforms, while in other experiments (such as crystal structure determination), we use only nNOS. Subtle mechanistic differences between NOS isoforms are not taken into account. Of the five NHA analogues studied, only NMOA and NHMA were found to produce NO. Overall, the substrates have a slightly greater affinity for nNOS than iNOS. Alternative substrates, NMOA and NHMA, produce NO with similar Km values to that for NHA, but with lower kcat (turnover) values; the kcat for NMOA is about 4 and 7 times lower and that for NHMA is about 18 and 13 times lower with iNOS and nNOS, respectively, when compared to NHA. Kinetic trends are the same for both NOS isoforms; for nNOS and iNOS, NMOA has a weaker binding affinity for the active site (higher Km), but greater turnover (higher kcat) than NHMA. An enzyme would likely be evolutionarily optimized for its native substrate, so the lower enzyme efficencies (kcat/Km) for these analogues are expected. The absence of NO production was confirmed for NEOA, NMMA, and MHA by longer incubations with iNOS (1, 2, 6, 12, and 24 hours) using the Griess reagent.(31)
Figure 2.

Michaelis-Menten curves of NHA, NMOA, and NHMA with (A) nNOS and (B) iNOS. (C) Michaelis-Menten curve of tBOA with iNOS, note the different units along both axes. The corresponding Michaelis-Menten values are reported in Table 2.
Table 2.
Kinetic values determined for NHA analoguesa
| NHA | NMOA | NHMA | NEOA | NMMA | MHA | tBOAc | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| iNOS | nNOS | iNOS | nNOS | iNOS | nNOS | iNOS | iNOS | iNOS | iNOS | |
| Km (μM) | 22 ± 5 | 5.7±2.1 | 88 ± 8 | 11 ± 2 | 34 ± 6 | 2.0 ± 0.4 | - | - | - | 2.7 ± 0.8 mM |
| kcat (min−1) | 16 ± 2 | 13 ± 4 | 4.7±1.3 | 1.8 ±0.5 | 0.9±0.5 | 1.0 ± 0.2 | - | - | - | 0.3 ± 0.1 |
| kcat/Km (μM min−1) | 0.5 | 2.8 | 0.05 | 0.16 | 0.03 | 0.03 | - | - | - | 0.11 |
| Ks (μM) | 29 ± 4 | - | 122 ±20 | - | 34 ± 4 | - | 120±13 | 70 ± 5 | >10mM | - |
| Ki (μM)b | n/a | n/a | n/a | 26 ± 3 | 28 ± 3 | >10mM | 7.4 ± 4.5 mM | |||
See Supplementary Information for Michaelis-Menten plots (Figure S1) and inhibition curves and difference spectra (Figures S2–9).
All compounds were also evaluated as inactivators, but none was found to cause time-dependent inhibition (see Supplementary Information Figure S9).
The kcat value differs from that previously reported;(22) the same enzyme concentration was used here for all of the compounds, which is lower than that used in the earlier publication for tBOA, so that all data in this table could be meaningfully compared.
Spectral binding affinities (Ks) for iNOS were also determined (Table 2, Figures S2–S8) using the previously described spectral binding assay.(33) In this assay, imidizole, which coordinates directly to the heme iron causing a Soret shift, is first bound to NOS and then NHA analogues are titrated into the cuvette while monitoring the Soret shift as these compounds displace the imidazole (see Methods). Therefore, these values directly reflect the affinity the compound has for the active site. For the substrates, Ks values are similar to Km values, as expected. For the compounds that are not substrates, the Ks values may rationalize why turnover cannot occur. MHA, for example, was found to have a very high Ks value, indicating its very poor binding affinity. This was confirmed upon analysis of the crystal structure (see below). NEOA and NMMA are not substrates, but have good binding affinities, so other factors must be preventing these compounds from being substrates.
Inhibition constants (Ki) with iNOS were determined for those compounds that were not substrates (Table 2, Figure S9). Slow substrates will also be competitive inhibitors, but measuring a Ki value is not meaningful since slower, alternate substrate turnover is also occurring. For the four compounds for which Ki values are meaningful (NEOA, NMMA, MHA, and tBOA), and therefore reported, these values agree with binding affinity (Ks) values. The Ki and the Ks for MHA are both very high, over 10 mM, while the Ki values for NEOA and NMMA are mid-micromolar. Ki values also indicate competition with arginine, thereby confirming that the analogues are inhibiting NOS at its active site.
We also re-evaluated tBOA, a previously reported weak NOS substrate. It has a poor binding affinity for iNOS, evidenced by its 7.4 mM Ki value and 2.7 mM Km value. tBOA has the slowest kcat, about one-third the rate of NHMA, the slowest of our NHA analogues for iNOS.
The NHA analogues were also evaluated as time-dependent inactivators of iNOS. In all cases, NOS activity was fully restored by addition of L-arginine to pre-incubations of each compound with iNOS under turnover conditions, indicating that time-dependent inactivation was not occurring (Figure S9C).
Citrulline Production
For substrates NMOA and NHMA, the amino acid product was confirmed to be solely citrulline (chromatographs are shown in Supplementary Figure S13). This was done by naphthalene-2,3-dicarboxyaldehyde-(NDA)-derivatization of the products of NOS (both iNOS and nNOS were used and performed in at least triplicate) reactions with subsequent HPLC separation and spectral detection at 460 nm (see Methods). An authentic standard of Nω-cyanoornithine (CN-Orn) was prepared to confirm its absence in all of the analogue-NOS reactions.
NADPH Consumption and NO Production
The rates of production of NO and consumption of NADPH were compared for all substrates (Table 3) with iNOS. NO production was measured using the hemoglobin capture assay, monitoring methemoglobin formation at 401 nm, while simultaneously measuring the conversion of NADPH to NADP+ at 340 nm. Table 3 shows that arginine and NHA consume approximately 1.5 and 0.5 equivalents, respectively, of NADPH for each NO molecule released. NMOA and NHMA, however, consume many more equivalents of NADPH (8 and 15, respectively) than NO is produced. This result is consistent with the slower kcat values (Table 2) for these two analogues when compared to that of NHA; NMOA and NHMA are not efficent substrates for NOS.
Table 3.
Uncoupling of NO production from NADPH consumption
| Substrate: | arginine | NHA | NMOA | NHMA |
|---|---|---|---|---|
| NADPH:NOa | 1.7 ± 0.2 | 0.7 ± 0.3 | 8.0 ± 1.5 | 15.0 ± 2.7 |
Ratios are expressed as moles of NADPH consumed per mole of NO formed with iNOS, averaged over five or more experiments.
Determination of the One-Carbon Metabolite of NMOA
NOS turnover of NMOA produces citrulline and NO, leaving the methyl of the Nω-methoxyl group unaccounted for. To address this issue, Nω-[14C]-methoxy-L-arginine ([14C]-NMOA) was synthesized using the chemistry shown in Supplementary Scheme S1,(24, 41) with the exception that [14C]-methoxylamine (34 mCi/mmol) was used as the amine added to the activated thiourea (2, Scheme S1).
iNOS reactions with [14C]-NMOA, NMOA, NHA, or substrate-free were 1) analyzed by NDA-derivatization and HPLC separation to quantify amino acids, 2) analyzed by 2,4-dinitrophenyl hydrazine (DNPH)-derivatization and HPLC separation to identify aldehydes and ketones, and 3) allowed to react with methanol oxidase and/or formate dehydrogenase to convert methanol into formaldehyde and formate and to convert formate into bicarbonate, respectively (Scheme 3). Before HPLC analysis, reactions were filtered through a 10,000 MWC filter. Filters were found to contain no [14C], indicating that covalent modification of the enzyme is not occurring.
Scheme 3.
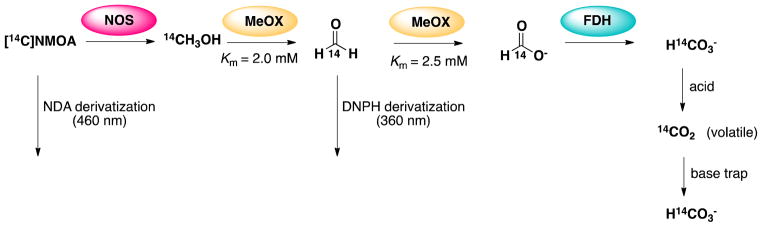
Flowchart of enzyme reactions and detection methods used in determination of the one-carbon metabolite of NMOA
NDA-derivatization of the [14C]-NMOA-iNOS reaction was separated by HPLC and scintillation counted (see Supplementary Figure S10). These spectra show that the only [14C] species to elute, in addition to [14C]-NMOA, is located in the early fractions (2–4 minutes), suggesting that this metabolite is highly polar. We hypothesize that this is a one-carbon metabolite from the [14C]-NMOA-iNOS reaction. DNPH was next used to detect aldehydes and ketones from the iNOS reactions in search of formaldehyde as a potential one-carbon biproduct of [14C]-NMOA-NOS metabolism. With an excess of DNPH no significant amount of 14C eluted with DNPH-formaldehyde standards, indicating that the one-carbon metabolite from these reactions is not formaldehyde.
Numerous attempts at detecting methanol directly by chromatography or mass spectrometry were unsuccessful, which excluded experiments run in isotopic water or oxygen. Consequently, enzymatic conversion was employed to convert any methanol produced to formaldehyde, then to formic acid, then to carbon dioxide, as depicted in Scheme 3. Methanol oxidase (MeOX) converts methanol to formaldehyde and H2O2 with a Km of approximately 2 mM (depending on the O2 concentration), but also catalyzes the conversion of formaldehyde to formate with a Km of 2.5 mM.(43) Following reaction with MeOX, [14C]-NMOA-iNOS reactions produced small amounts of [14C]-DNPH-formaldehyde, but stoichiometrically less than the amount of citrulline produced by the same reactions in the absence of MeOX, because of the conversion of some of the produced formaldehyde to formate by MeOX. To better quantify the methanol formation in the iNOS-catalyzed reaction, [14C]-NMOA was incubated with a mixture of iNOS, MeOX, and formate dehydrogenase (FDH); FDH converts the formate produced in the MeOX reaction to bicarbonate, which, after acidification, produces carbon dioxide, which can be trapped in base, allowing for quantitative detection of all of the [14C] metabolites as DNPH-formaldehyde and CO2. Experiments repeated with MeOX but with FDH omitted did not produce [14C]-CO2, confirming the identity of the one-carbon metabolite of the [14C]-NMOA-iNOS reaction as [14C]-methanol exclusively.
Crystal Structures of the Substrate Analogues
Crystal structures of substrate analogues bound to rat nNOS oxygenase domain were obtained (Figure 3 and omit electron densities in Figure S11). Similar to NHA and arginine, all analogues retain the four hydrogen bonds conserved for an L-amino acid moiety: AA-COO− to Tyr588-OH; AA-COO− to Asp597-COO− ; AA-NH3+ to Glu592-COO−; and AA-NH3+ to heme propionate A. The planarity of the guanidino group is, more or less, maintained for all compounds but MHA. This planarity allows the two guanidino nitrogens, Nδ and Nω′, to hydrogen bond to Glu592. These key interactions place the guanidino head over the heme for potential catalysis. However, with MHA the presence of an Nδ-methyl destroys the planarity of guanidine, resulting in only one hydrogen bond from the Nω′ nitrogen to Glu592. The close distance (~ 3.9 Å) from the extra Nδ-methyl to heme could be sterically preventing O2 from binding to the heme iron, thus preventing turnover. Steric blockage of productive oxygen binding was hypothesized to be the reason that arginine and NHA analogues bearing a C5 methyl substitution at the pro-R position are not substrates.(19) The electron density for MHA is the poorest among the five analogue structures, which reflects unfavorable interactions between the distorted guanidino group of MHA and the NOS active site and is consistent with the poor binding affinity determined for this compound (see Table 2).
Figure 3.

Crystal structures of analogues complexed with nNOS. Heme is shown in light pink and H4B in dark blue; nNOS active site residues are in green; active site water is shown as a sphere. (A) NHA (green, PDB 1LZX) (B) NMOA (cyan) which was modeled with the methyl group in two alternate positions but with only major one shown. (C) NHMA (magenta) (D) NMMA (peach) (E) NEOA (grey) (F) MHA (yellow), which showed poorer density quality indicating partial disordering. Hydrogen bonding interactions are depicted by black dashed lines; distances are reported in Å. See Figure S12 for crystal structure of tBOA.
Similar to NHA, the Nω-OH of NHMA and MHA forms a weak hydrogen bond to the backbone nitrogen of Gly586 (Figure 3). Any substituent on this hydroxyl group would eliminate this hydrogen bond by either dragging the oxygen atom away from Gly586 as in NEOA, totally swinging away as in NMMA, or being blocked by a methyl group as in NMOA (Figure 3). The orientation and bulkiness of this substituted hydroxyl will, in turn, influence whether or not an active site water molecule can bind next to the analogue. The active site water usually hydrogen bonds to the substrate Nω atom. This hydrogen bonding interaction is maintained in NHMA, and is at least partially retained in MHA and NMOA via the oxygen atom from the analogue. In MHA the water molecule is not fully occupied because of the closeness of the hydroxyl group; while in NMOA the methyl of the methoxyl moiety adopts two conformations, the predominant conformation, shown in Figure 3B, allows for a partially occupied water molecule that shares the same space with the methyl group in its minor conformation (Figure S11A). However, in NEOA and NMMA either an ethyl or a methyl group, respectively, occupies the space of the active site water. In the nitric oxide ferrous complex of nNOS, this same water molecule is within hydrogen-bonding distance of the O atom of NO, and thus is in an ideal position to serve as a proton donor.(20)
We also have obtained a crystal structure of tBOA in the active site of nNOS (see Figure S12). There is no water molecule present in the active site in this co-crystal structure. The tert-butyl group is apparently too bulky to fit in the site and is, therefore, disordered; the three tert-butyl methyl groups exchange their positions but are populated more in the space that a water molecule normally occupies. Therefore, there is no active site water molecule present even though this compound is a weak substrate.
DISCUSSION
The products of NOS turnover with the various NHA analogues was investigated. Citrulline is the only amino acid product formed from NMOA and NHMA. In addition to citrulline, NOS produces CN-Orn when NHA is the substrate and when H2O2 is used in place of NADPH.(44) It is speculated that citrulline is the product of native NOS-NHA chemistry (through the ferric peroxo intermediate), while CN-Orn is the product of non-native NOS-NHA chemistry, when NOS is forced to go through CpdI.(10) Since citrulline is the only amino acid product of NMOA and NHMA, this suggests NOS is performing native chemistry through FeIII-OO−/FeIII-OOH, not through CpdI, on these substrates.
There is significantly higher consumption of NADPH per NO released for substrates NMOA and NHMA (8 and 15 equiv, respectively) in comparison to L-arginine and NHA (1.5 and 0.5 equiv, respectively). These ratios were measured at substrate concentrations approximately equal to their Km values. Uncoupling of substrate turnover from electron consumption happens when superoxide (O2•−) is released from the ferric superoxide complex (FeIIIOO•; see Scheme 2) before substrate modification. This unproductive consumption of electrons by NOS occurs at subsaturating concentrations of L-arginine or H4B.(6, 35) Uncoupling also occurs in the presence of alternate substrates such as homoarginine(45) or inactivators such as N5-(1-iminooethyl)-L-ornithine,(46) suggesting substrate identity affects the feasibility of oxidation. B. subtilis NOS Trp66 (the Trp residue that hydrogen bonds to H4B) mutants also show uncoupling.(47) This demonstrates the importance of the entire NOS enzyme structure in implementing efficiently coupled substrate turnover and that even seemingly small, single residue, changes can largely affect enzymatic outcomes. Both substrates, NMOA and NHMA, show some uncoupling during turnover, which means NADPH reducing equivalents may be used for the production of other products, such as superoxide, rather than NO (Table 3). A highly coupled system requires precise proton transfer steps, and therefore those substrates that are uncoupled, very likely, perturb the local proton transfer mechanism. It should be noted that substrate oxidation is a multi-step process and that alternative substrates may potentially affect other steps, for example, electron transfer from the reductase. Furthermore, the consistency between low kcat values (Table 2) and higher uncoupling for NMOA and NHMA suggests that oxidation of these alternative subsrtates represents only 5 to 10% of iNOS catalysis. The decrease in kcat (approximately 5 and 10% the kcat of NHA) could be the result of this dramatic degree of uncoupling.
The one-carbon metabolite of [14C]-NMOA was found to be methanol. This result differs from the one-carbon metabolite determined to be present in NOS-NHMA reactions; Olken and Marletta found that NHMA produces formaldehyde, first going through NHA as an intermediate.(26). The higher uncoupling (Table 3) of NHMA compared to NMOA is consistent with the fact that NHMA processing requires an additional oxidation step to yield the observed formaldehyde. This suggests that different substrates are metabolized by NOS through different mechanisms.
We have previously reported(22) that both Nω-tert-butyloxy-L-arginine and Nω-(3-methyl-2-butenyl)oxy-L-arginine are NOS substrates; therefore, a direct hydrogen atom abstraction from the Nω-OH hydroxyl does not appear to be required in the second step of the NOS reaction for these two alternative substrates. It, therefore, may seem surprising that NHMA is a NOS substrate since NHMA lacks the Nω-H proton. In light of this, we propose that at least one of the two protons, either the Nω-H or the NωO-H proton, is necessary for turnover. The positions of these protons are seemingly interchangeable because of the conformational isomers that exist, and either may be functioning as a proton source during oxygen activation (see Figure 4A). Density functional theory calculations suggest that both are comparable in energy for hydrogen atom donation.(21) Crystal structures typically reflect the most stable binding conformation, but do not reveal other possible conformations that may exist under the dynamic conditions of an enzyme active site. By comparison of the crystal structures for the three substrates, along with their turnover rates, we hypothesize that deprotonation at the Nω-H is favored on the basis of its physical proximity to the heme-iron and on the slower kcat for NHMA than NMOA (Table 2). While even the native substrate (NHA) may go through many pathways, this suggests that the Nω-H, positioned down towards the heme-iron (Figures 4B and 4C), is more easily removed, but that the NωO-H, positioned farther away from the heme-iron (Figures 4B and 4D), can also serve as a viable proton source, but less efficiently. The substrate might rotate around the C-Nω bond in order for the Nω O-H to be aligned for deprotonation, but since this is not the conformation depicted in the crystal structure, it is likely not the most energetically favorable conformation. The fact that NMMA, a compound in which both the Nω-H and the Nω O-H protons are replaced with methyl groups, has good binding affinity for the NOS active sites but is not a NOS substrate, is consistent with our hypothesis that the presence of at least one Nω proton is essential for turnover.
Figure 4.

(A) Conformational isomers of NHA analogues. (B) Crystal structure of NHA (green) with Nω -H and Nω -OH protons (white) rendered. (C) Crystal structure of NMOA (cyan) with Nω -H proton (white) rendered. The methyl of the methoxyl group occupies two alternate positions, but only one is shown here. (D) Crystal structure of NHMA (magenta) with Nω -OH proton (white) rendered. In all panels heme (light pink) is shown as lines; NHA and analogues are shown as sticks and active site water molecules as spheres. Rendered protons are white; nitrogens atoms are blue; oxygen atoms are red; carbon atom colors are specified for each substrate.
Previous ENDOR and X-ray experiments, as well as DFT calculations, show that NHA is most likely protonated in the active site.(21) The existence of a water molecule next to the guanidino group of the substrate could play a role in maintaining its protonated state, thus affecting the rate of catalysis. On the basis of the substrate activities of NMOA and NHMA, and the lack of activity with NMMA, we propose a possible mechanism for the reaction of NOS with NHA and NMOA (Scheme 4). The turnover and one carbon metabolite of NHMA has been previously examined in detail.(26) As shown in Scheme 4, the ferric superoxide heme species is activated upon receiving an electron from H4B. The resulting ferric peroxide species is then protonated by the substrate to form the reactive ferric hydroperoxide species. On the basis of experimental evidence supporting FeIIIOOH as the active species, (15) and our results suggesting the necessity of the active site proton, we hypothesize that FeIIIOOH needs to be formed to create the proper “push-pull” dynamics and electronics for the reaction to occur. The ferric hydroperoxide then undergoes attack on the electrophilic guanidinium carbon. Driven by the formation of citrulline, this tetrahedral intermediate collapses, producing a protonated or methylated nitric oxide species, which is subsequently processed, aided via appropriate proton transfers by the active site water molecule. A heme-NO complex forms, which donates an electron back to the radical cation of H4B to produce H4B, NO, and ferric heme. This proposed mechanism invokes the active site water in the processing of the released NO-containing product; however, additional roles, such as other proton transfers, structural stability, or pKa maintenance, are also possible.
Scheme 4.

NOS turnover of NHA and NMOA.
Because both NMOA and NHMA are NOS substrates, but NMMA is not, it is reasonable to conclude that NOS can process a singly-methylated NO species, but not a doubly-methylated species. However, although the proton is necessary, it is not sufficient, as NEOA has the Nω-H proton available, but is not a substrate. This can be accounted for by considering two mechanistic pathways: 1) in a nucleophilic pathway, as shown in Scheme 4, an NωO-ethyl species may be too sterically hindered to undergo reaction, while methyl-NωO and NωO-methyl species are more reactive and can be processed by NOS, or 2) in an electrophilic cleavage of the NωO-R bond it would be expected that none of the small alkyl-substituted analogues are likely to be cleaved because of the high energy of the resulting cation. Nω-tert-Butyloxy-L-arginine and Nω-(3-methyl-2-butenyl)oxy-L-arginine(22), however, are substrates, which argues in favor of the second mechanism and against the first. According to the second mechanism, the stability of the resulting cations (tert-butyl and dimethylallyl, respectively) might suggest that these compounds should be excellent substrates, but they are only weak substrates. (22) The crystal structures of NEOA (Figure 3E) and tBOA (Supplemenary Figure S12) show that there is no active site water molecule bound; the bulky hydrophobic substituents apparently displace it. However, whereas NEOA is not a substrate, tBOA is a substrate, suggesting that the water molecule may not be essential for activity, and, perhaps more importantly, that there is likely more than one mechanism by which substrates can be turned over. Perhaps, substrates with larger substituents that displace the water molecule but can form stabilized carbocations are metabolized through a mechanism that does not require a water molecule, a mechanism in which electron transfer from the H4B to ferric superoxide occurs initially followed by ferric peroxide attack on the substrate (Scheme 5). Because of the stability of the resulting carbocation, spontaneous breakdown of the tetrahedral intermediate gives citrulline, HNO, and ferrous oxo radical. Abstraction of a hydrogen atom from HNO by FeIIO• with loss of water produces the heme-NO complex. As in Scheme 4, the radical cation of H4B accepts an electron to give NO and ferric heme. In the case of NEOA, the water molecule is displaced but because an ethyl cation is not sufficiently stable, breakdown of the second intermediate does not occur, and the equilbrium favors substrate.
Scheme 5.
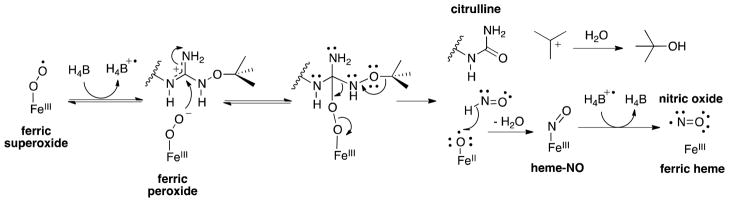
Pathway by which tBOA could be acting as a substrate despite the lack of an active site water molecule
In the unique case of MHA bound to nNOS, an active site water, at least partially, exists, and both Nω protons are present, but this compound is not a substrate. In the crystal structure (Figure 3F) the Nδ-methyl distorts the planarity of the guanidino group, thereby weakening its crucial interactions with Glu592, which contributes to its poor binding affinity. The close positioning of this Nδ-methyl to the heme-iron (~3.9 Å) could prevent productive binding of molecular oxygen.
The role of the active site water in the second step of NOS catalysis is still quite controversial. Martin and coworkers cleverly examined C5-methylated arginine and NHA analogues, finding that Nω-hydroxy-(5S)-methyl-L-arginine is a NOS substrate while (5S)-methyl-L-arginine is not.(19) They speculated that both analogues bearing a (5S)-methyl substituent displace the active site water molecule. On the basis of their modeling results, they suggested that the active site water is required for the first step of NOS catalysis, but is not required for the second step. The NHA analogue crystal structures presented here (Figure 2) show that, when present, the active site water molecule does not bind near C5, but interacts with Nω near Ser585, and MHA, which has an Nδ-methyl substituent, still binds with an active site water molecule (Figure 3F). On the basis of our crystal structures, it is possible that (5S)-methyl compounds also would allow active site water binding; further structural data is needed to confirm this hypothesis. However, as supported by the example of tBOA, this water molecule cannot be deemed essential for catalysis with all NHA analogues; our results suggest it does play a role in the turnover of NHA, NMOA, and NHMA.
Our findings suggest that substrate identity, especially its steric bulkiness, dictates the ability of a water molecule to bind in the active site; if the water is involved in the predominant turnover pathway, its binding could determine the ability of NOS to catalyze a reaction on a substrate and determine the rate of catalysis. A caveat to this hypothesis arises because our crystal structures do not contain a heme-oxo species; substrates would be repositioned when O2 binds and/or the O2 ligand must bend in a different direction than we have observed in the NO complexes of nNOS and eNOS. When NO coordinates to the heme iron in nNOS, the substrate, L-arginine, must move about 0.7 Å.(20) The substrates under investigation in this study are larger, causing greater steric restriction for O2 binding and hence must move to enable O2 to bind. Even so, there still should be sufficient room for the “catalytic” water to remain in place and provide a potential proton source for catalysis in the cases of NMOA and NHMA.
Compared to their substrate-promiscuous cytochrome P450 relatives, NOSs are very specific enzymes because of their small, highly conserved active sites. An intricate set of hydrogen bond interactions holds substrates in the active site. The research described here demonstrates that NOS can metabolize several different substrates and may proceed through different mechanisms during metabolism of these various substrates.
In summary, we demonstrate that NOS can metabolize NHA analogues having a methyl substituted for either the Nω-H proton (NHMA) or the Nω-OH hydroxyl proton (NMOA), but not both (NMMA), and this is consistent with the importance of either the Nω-H proton or the Nω-OH proton in catalysis. Crystal structures reveal the presence of an active site water molecule that could also serve as a proton donor during substrate turnover, but tert-butoxy-L-arginine acts as a substrate, even though the tert-butyl group displaces the active site water, as shown in the crystal structure. We propose potential, alternative pathways (Schemes 4 and 5) consistent with our findings for these analogues as NOS substrates. Our crystal structures demonstrate that substrate identity dictates the presence or absence of the active site water molecule, but this does not always dictate substrate turnover. As a unique and complex enzyme, NOS not only is able to achieve two different oxygenation chemistries (step one and step two) within its active site, but is also flexible enough to oxidize various substituted NHA analogues, possibly by more than one mechanism.
Supplementary Material
Acknowledgments
Funding
This work was funded by National Institutes of Health, Grants GM049725 to RBS, and GM057353 to TLP. We thank Dr. Bettie Sue Siler Masters (NIH grant GM52419, with whose laboratory P.M. and L.J.R. are affiliated). B.S.S.M. also acknowledges the Welch Foundation for a Robert A. Welch Distinguished Professorship in Chemistry (AQ0012). P.M. is supported by grant 0021620849 from MSMT of the Czech Republic. Support for KJL was provided by Reach for the Stars, a GK-12 program supported by the National Science Foundation under grant DGE-0948017.
We thank the SSRL and ALS beamline staff for their support during remote X-ray diffraction data collection. We thank Dr. Jinwen Huang for synthesizing the NEOA.
Abbreviations
- NOS
nitric oxide synthase
- NO
nitric oxide
- NHA
Nω-hydroxy-L-arginine
- nNOS
neuronal nitric oxide synthase
- eNOS
endothelial nitric oxide synthase
- iNOS
inducible nitric oxide synthase
- H4B
(6R)-5,6,7,8-tetrahydrobiopterin
- CYP450
cytochrome P450
- CpdI
compound I
- tBOA
Nω-tert-butyloxy-L-arginine
- NMOA
Nω-methoxy-L-arginine
- NEOA
Nω-ethoxy-L-arginine
- NHMA
Nω-hydroxy-Nω-methyl-L-arginine
- MHA
Nδ-methyl- Nω-hydroxy-L-arginine
- NMMA
Nω-methoxy-Nω-methyl-L-arginine
- δMA
Nδ-methyl-L-arginine
- [14C]-NMOA
Nω-[14C]-methoxy-L-arginine
- CN-Orn
Nδ-cyanoornithine
- NDA
naphthalene-2,3-dicarboxyaldehyde
- DNPH
2,4-dinitrophenyl hydrazine
- MeOX
methanol oxidase
- FDH
formate dehydrogenase
Footnotes
Coordinates have been deposited with the Protein Data Bank (codes: 4FVW, 4FVX, 4FVY, 4FVZ, 4FW0, and 4GQE.
PDB Accession Codes. The PDB accession codes for NHA analogues with nNOS, shown in Figures 3 and S10 are as follows: nNOS-NMOA, 4FVW; nNOS-NEOA, 4FVX; nNOS-NHMA, 4FVY; nNOS-NMMA, 4FVZ; nNOS-MHA, 4FW0; nNOS-tBOA, 4GQE.
Supporting Information Available
Supporting information is available, including: synthetic methods, substrate and inhibitor plots, 14C-labeling methods, crystal structure information, MeOX and FDH reactions, HPLC-MS confirmation of NDA- and DNPH derivatives, and NMR spectra of final products. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Griffith OW, Stuehr DJ. Nitric oxide synthases: properties and catalytic mechanism. Annu Rev Physiol. 1995;57:707–736. doi: 10.1146/annurev.ph.57.030195.003423. [DOI] [PubMed] [Google Scholar]
- 2.Calabrese V, Mancuso C, Calvani M, Rizzarelli E, Butterfield DA, Stella AMG. Nitric oxide in the central nervous system: neuroprotection versus neurotoxicity. Nat Rev Neurosci. 2007;8:766–775. doi: 10.1038/nrn2214. [DOI] [PubMed] [Google Scholar]
- 3.Alderton WK, Cooper CE, Knowles RG. Nitric oxide synthases: structure, function and inhibition. Biochem J. 2001;357:593–615. doi: 10.1042/0264-6021:3570593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhou L, Zhu D-Y. Neuronal nitric oxide synthase: Structure, subcellular localization, regulation, and clinical implications. Nitric Oxide. 2009;20:223–230. doi: 10.1016/j.niox.2009.03.001. [DOI] [PubMed] [Google Scholar]
- 5.Förstermann U, Pollock JS, Schmidt HH, Heller M, Murad F. Calmodulin-dependent endothelium-derived relaxing factor/nitric oxide synthase activity is present in the particulate and cytosolic fractions of bovine aortic endothelial cells. Proc Natl Acad Sci USA. 1991;88:1788–1792. doi: 10.1073/pnas.88.5.1788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gorren ACF, Mayer B. Nitric-oxide synthase: a cytochrome P450 family foster child. Biochim Biophys Acta. 2007;1770:432–445. doi: 10.1016/j.bbagen.2006.08.019. [DOI] [PubMed] [Google Scholar]
- 7.Stuehr DJ, Wei C-C, Wang Z, Hille R. Exploring the redox reactions between heme and tetrahydrobiopterin in the nitric oxide synthases. Dalton Trans. 2005:3427–3435. doi: 10.1039/b506355h. [DOI] [PubMed] [Google Scholar]
- 8.Stoll S, NejatyJahromy Y, Woodward JJ, Ozarowski A, Marletta MA, Britt RD. Nitric oxide synthase stabilizes the tetrahydrobiopterin cofactor radical by controlling its protonation state. J Am Chem Soc. 2010;132:11812–11823. doi: 10.1021/ja105372s. [DOI] [PubMed] [Google Scholar]
- 9.Hurshman AR, Marletta MA. Reactions catalyzed by the heme domain of inducible nitric oxide synthase: evidence for the involvement of tetrahydrobiopterin in electron transfer. Biochemistry. 2002;41:3439–3456. doi: 10.1021/bi012002h. [DOI] [PubMed] [Google Scholar]
- 10.Woodward JJ, Chang MM, Martin NI, Marletta MA. The second step of the nitric oxide synthase reaction: evidence for ferric-peroxo as the active oxidant. J Am Chem Soc. 2009;131:297–305. doi: 10.1021/ja807299t. [DOI] [PubMed] [Google Scholar]
- 11.Feldman PL, Griffith OW, Stuehr DJ. The surprising life of nitric-oxide. Chem Eng News. 1993;71:26–38. [Google Scholar]
- 12.Korth HG, Sustmann R, Thater C, Butler AR, Ingold KU. On the mechanism of the nitric oxide synthase-catalyzed conversion of Nω -hydroxyl-L-arginine to citrulline and nitric oxide. J Biol Chem. 1994;269:17776–17779. [PubMed] [Google Scholar]
- 13.Pant K, Crane BR. Nitrosyl-heme structures of Bacillus subtilis nitric oxide synthase have implications for understanding substrate oxidation. Biochemistry. 2006;45:2537–2544. doi: 10.1021/bi0518848. [DOI] [PubMed] [Google Scholar]
- 14.Crane BR, Sudhamsu J, Patel BA. Bacterial nitric oxide synthases. Annu Rev Biochem. 2010;79:445–470. doi: 10.1146/annurev-biochem-062608-103436. [DOI] [PubMed] [Google Scholar]
- 15.Davydov R, Sudhamsu J, Lees NS, Crane BR, Hoffman BM. EPR and ENDOR characterization of the reactive intermediates in the generation of NO by cryoreduced oxy-nitric oxide synthase from Geobacillus stearothermophilus. J Am Chem Soc. 2009;131:14493–14507. doi: 10.1021/ja906133h. [DOI] [PubMed] [Google Scholar]
- 16.Giroud C, Moreau MM, Mattioli TA, Balland V, Boucher JL, Xu-Li Y, Stuehr DJ, Santolini J. Role of arginine guanidinium moiety in nitric-oxide synthase mechanism of oxygen activation. J Biol Chem. 2010;285:7233–7245. doi: 10.1074/jbc.M109.038240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Santolini J. The molecular mechanism of mammalian NO-synthases: a story of electrons and protons. J Inorg Biochem. 2011;105:127–141. doi: 10.1016/j.jinorgbio.2010.10.011. [DOI] [PubMed] [Google Scholar]
- 18.Daff S. NO synthase: structures and mechanisms. Nitric Oxide. 2010;23:1–11. doi: 10.1016/j.niox.2010.03.001. [DOI] [PubMed] [Google Scholar]
- 19.Martin NI, Woodward JJ, Winter MB, Beeson WT, Marletta MA. Design and synthesis of C5 methylated L-arginine analogues as active site probes for nitric oxide synthase. J Am Chem Soc. 2007;129:12563–12570. doi: 10.1021/ja0746159. [DOI] [PubMed] [Google Scholar]
- 20.Li H, Igarashi J, Jamal J, Yang W, Poulos TL. Structural studies of constitutive nitric oxide synthases with diatomic ligands bound. J Biol Inorg Chem. 2006;11:753–768. doi: 10.1007/s00775-006-0123-8. [DOI] [PubMed] [Google Scholar]
- 21.Tantillo DJ, Fukuto JM, Hoffman BM, Silverman RB, Houk KN. Theoretical studies on NωG-hydroxy-l-arginine and derived radicals: Implications for the mechanism of nitric oxide synthase. J Am Chem Soc. 2000;122:536–537. [Google Scholar]
- 22.Huang H, Hah JM, Silverman RB. Mechanism of nitric oxide synthase. Evidence that direct hydrogen atom abstraction from the OH bond of NG-hydroxyarginine is not relevant to the mechanism. J Am Chem Soc. 2001;123:2674–2676. doi: 10.1021/ja005900u. [DOI] [PubMed] [Google Scholar]
- 23.Komori Y, Wallace GC, Fukuto JM. Inhibition of purified nitric oxide synthase from rat cerebellum and macrophage by L-arginine analogs. Arch Biochem Biophys. 1994;315:213–218. doi: 10.1006/abbi.1994.1492. [DOI] [PubMed] [Google Scholar]
- 24.Schade D, Kotthaus J, Clement B. Efficient synthesis of optically pure Nω-alkylated l-arginines. Synthesis. 2008;2008:2391–2397. [Google Scholar]
- 25.Schade D, Kotthaus J, Klein N, Kotthaus J, Clement B. Prodrug design for the potent cardiovascular agent Nω-hydroxy-L-arginine (NOHA): synthetic approaches and physicochemical characterization. Org Biomol Chem. 2011;9:5249–5259. doi: 10.1039/c0ob01117g. [DOI] [PubMed] [Google Scholar]
- 26.Olken NM, Marletta MA. NG-Methyl-L-arginine functions as an alternate substrate and mechanism-based inhibitor of nitric oxide synthase. Biochemistry. 1993;32:9677–9685. doi: 10.1021/bi00088a020. [DOI] [PubMed] [Google Scholar]
- 27.Luzzi SD, Marletta MA. L-arginine analogs as alternate substrates for nitric oxide synthase. Bioorg Med Chem Lett. 2005;15:3934–3941. doi: 10.1016/j.bmcl.2005.05.088. [DOI] [PubMed] [Google Scholar]
- 28.Kotthaus J, Schade D, Töpker-Lehmann K, Beitz E, Clement B. N(delta)-Methylated L-arginine derivatives and their effects on the nitric oxide generating system. Bioorg Med Chem. 2008;16:2305–2312. doi: 10.1016/j.bmc.2007.11.066. [DOI] [PubMed] [Google Scholar]
- 29.Hevel JM, White KA, Marletta MA. Purification of the inducible murine macrophage nitric oxide syntase: Identification as a falvoprotein. J Biol Chem. 1991;266:22789–22791. [PubMed] [Google Scholar]
- 30.Roman LJ, Sheta EA, Martásek P, Gross SS, Liu Q, Masters BSS. High-level expression of functional rat neuronal nitric oxide synthase in Escherichia coli. Proc Natl Acad Sci USA. 1995;92:8428–8432. doi: 10.1073/pnas.92.18.8428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hevel JM, Marletta MA. Nitric-oxide synthase assays. Methods Enzymol. 1994;233:250–258. doi: 10.1016/s0076-6879(94)33028-x. [DOI] [PubMed] [Google Scholar]
- 32.Cheng YC, Prusoff WH. Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 33.Martell JD, Li H, Doukov T, Martásek P, Roman LJ, Soltis M, Poulos TL, Silverman RB. Heme-coordinating inhibitors of neuronal nitric oxide synthase. Iron-thioether coordination is stabilized by hydrophobic contacts without increased inhibitor potency. J Am Chem Soc. 2010;132:798–806. doi: 10.1021/ja908544f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Silverman RB. The Organic Chemistry of Enzyme-Catalyzed Reactions. 2. Academic Press; San Diego: 2002. pp. 563–596. [Google Scholar]
- 35.Adak S, Wang Q, Stuehr DJ. Arginine conversion to nitroxide by tetrahydrobiopterin-free neuronal nitric-oxide synthase. Implications for mechanism. J Biol Chem. 2000;275:33554–33561. doi: 10.1074/jbc.M004337200. [DOI] [PubMed] [Google Scholar]
- 36.Li H, Shimizu H, Flinspach ML, Jamal J, Yang W, Xian M, Cai T, Wen EZ, Jia Q, Wang PG, Poulos TL. The novel binding mode of N-alkyl-N′-hydroxyguanidine to neuronal nitric oxide synthase provides mechanistic insights into NO biosynthesis. Biochemistry. 2002;41:13868–13875. doi: 10.1021/bi020417c. [DOI] [PubMed] [Google Scholar]
- 37.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- 38.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 39.Emsley P, Lohkamp B, Scott WG. Features and development of Coot. Acta Crystallogr. 2010;D66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Winn MD, Isupov MN, Murshudov GN. Use of TLS parameters to model anisotropic displacements in macromolecular refinement. Acta Crystallogr. 2001;D57:122–133. doi: 10.1107/s0907444900014736. [DOI] [PubMed] [Google Scholar]
- 41.Martin NI, Woodward JJ, Marletta MA. NG-hydroxyguanidines from primary amines. Org Lett. 2006;8:4035–4038. doi: 10.1021/ol061454p. [DOI] [PubMed] [Google Scholar]
- 42.Schade D, Töpker-Lehmann K, Kotthaus J, Clement B. Synthetic approaches to N(delta)-methylated L-arginine, N(omega)-hydroxy-L-arginine, L-citrulline, and N(delta)-cyano-L-ornithine. J Org Chem. 2008;73:1025–1030. doi: 10.1021/jo702150d. [DOI] [PubMed] [Google Scholar]
- 43.van der Klei IJ, Bystrykh LV, Harder W. Alcohol oxidase from Hansenula polymorpha CBS 4732. Methods Enzymol. 1990;188:420–427. doi: 10.1016/0076-6879(90)88067-k. [DOI] [PubMed] [Google Scholar]
- 44.Clague MJ, Wishnok JS, Marletta MA. Formation of Nδ-cyanoornithine from NG-hydroxy-L-arginine and hydrogen peroxide by neuronal nitric oxide synthase: implications for mechanism. Biochemistry. 1997;36:14465–14473. doi: 10.1021/bi971024u. [DOI] [PubMed] [Google Scholar]
- 45.Moali C, Boucher J-L, Sari M-A, Stuehr DJ, Mansuy D. Substrate specificity of NO synthases: detailed comparison of L-arginine, homo-L-arginine, their Nω-hydroxy derivatives, and Nω-hydroxynor-L-arginine. Biochemistry. 1998;37:10453–10460. doi: 10.1021/bi980742t. [DOI] [PubMed] [Google Scholar]
- 46.Fast W, Nikolic D, van Breemen RB, Silverman RB. Mechanistic studies of the inactivation of inducible nitric oxide synthase by N5-(1-Iminoethyl)-l-ornithine (L-NIO) J Am Chem Soc. 1999;121:903–916. [Google Scholar]
- 47.Brunel A, Wilson A, Henry L, Dorlet P, Santolini J. The proximal hydrogen bond network modulates bacillus subtilis nitric-oxide synthase electronic and structural properties. J Biol Chem. 2011;286:11997–12005. doi: 10.1074/jbc.M110.195446. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.