

Abstract
The revolution of epigenetics has revitalized cancer research, shifting focus away from somatic mutation toward a more holistic perspective involving the dynamic states of chromatin. Disruption of chromatin organization can directly and indirectly precipitate genomic instability and transformation. One group of epigenetic mediators, the Polycomb group (PcG) proteins, establishes heritable gene repression through methylation of histone tails. Although classically considered regulators of development and cellular differentiation, PcG proteins engage in a variety of neoplastic processes, including cellular proliferation and invasion. Due to their multifaceted potential, PcG proteins rest at the intersection of transcriptional memory and malignancy. Expression levels of PcG proteins hold enormous diagnostic and prognostic value in breast, prostate, and more recently, gastrointestinal cancers. In this review, we briefly summarize the function of PcG proteins and report the latest developments in understanding their role in pancreatic cancer.
Introduction
Unequivocally, pancreatic cancer carries the worst prognosis of any major malignancy due to its relatively equivalent incidence and mortality rates. In 2010, the American Cancer Society projected 43,410 new cases of the disease and 36,800 related deaths in the USA for both sexes combined [1]. Despite advances in surgical resection and adjuvant chemoradiotherapy treatments, the 5-year survival rate remains below 5% across all stages. The highly aggressive, chemoresistant nature of the disease continues to outpace advances in our understanding of the molecular mechanisms fueling tumorigenesis [2]. Additionally, most patients live asymptomatically until metastasis, a stage at which surgical resection is no longer beneficial. As such, developing early-stage diagnostic tools and identifying novel targets for therapeutic intervention persist as urgent research priorities.
The long-standing model of pancreatic carcinogenesis describes the multistep transformation of ductal cells into invasive pancreatic ductal adenocarcinoma (PDAC) through a series of intermediate microscopic dysplastic precursor lesions known as pancreatic intraepithelial neoplasias (PanINs). PanIN lesions are organized into a linear classification system in which increasing PanIN grade (PanIN-1a, PanIN-1b, PanIN-2, PanIN-3) is concordant with the severity of observed morphological alterations in nuclear organization, epithelial polarity, and mitoses [3]. A similar classification system exists for intraductal papillary mucinous neoplasm (IPMN) and mucinous cystic neoplasm (MCN), macroscopic ductal precursor lesions that may also mature into invasive carcinoma.
To shed light into the potential mechanisms underlying pancreatic cancer development, the past two decades of research have focused heavily on uncovering the genetic basis for the PanIN progression model, resulting in exhaustive catalogs of inherited germline and acquired somatic mutations in cancer-associated genes. The Pancreatic Cancer Genome Project recently published a milestone study that sequenced DNA isolated from 24 advanced pancreatic adenocarcinomas in combination with SNP arrays to detect homozygous deletions and gene amplifications [4]. Jones et al. discovered 1,561 somatic gene mutations across 1,007 genes within the 20,661 protein-coding genes analyzed, yielding an average rate of 63 genetic abnormalities per pancreatic cancer. Clustering of altered genes revealed a core subset of 12 regulatory pathways or processes disrupted in the majority (67-100%) of pancreatic cancers, namely, apoptosis, DNA damage control, regulation of the G1/S phase transition, Hedgehog signaling, homophilic cell adhesion, integrin signaling, KRAS signaling, JNK signaling, regulation of invasion, Wnt/Notch signaling, TGF-β signaling, and other non-KRAS small GTPase-dependent signaling [4]. This study provided independent verification and prevalence rates for a number of well-characterized pancreatic cancer-associated genes, including activating mutations in proto-oncogene KRAS (>95% of tumors) and inactivating mutations in tumor suppressors p16/CDKN2A (75-80%), TP53 (80%), and SMAD4/DPC4 (60%) [5]. Conceptually, these data suggest that pancreatic cancer is fundamentally a disease of pathways. Research into these pathways has also been a matter of intensive investigation in cell biology, rendering clearly that these cascades must ultimately engage the function of epigenetic regulators to silence tumor suppressors and activate oncogenes in a heritable manner. Therefore, studies into epigenetics will supply logical extension to the genetic paradigm.
Epigenetics
Epigenetic mechanisms permit stable and heritable patterns of altered gene expression independent of the DNA sequence, profoundly expanding the functional potential of the human genome beyond its approximately 35,000 protein-coding genes. Two primary types of epigenetic processes dictate chromatin structure in the setting of long-term gene repression: DNA methylation and histone modification.
DNA methylation
The first epigenetic studies into pancreatic cancer sought to link DNA methylation to the inactivation of critical tumor suppressors. DNA methylation involves transfer of a methyl group to the 5′ carbon of the cytosine pyrimidine ring within cytosine-guanine dinucleotides (CpGs) through the activity of DNA methyltransferases (DNMTs). DNMT1 establishes and maintains parental methylation patterns following chromosomal replication and repair while DNMT3A and DNMT3B produce de novo methylation [6], both leading to gene inhibition. While the majority of the genome displays low CpG frequency, a small fraction possesses a high percentage of hypomethylated CpG regions across a minimum of 200bp of DNA (termed CpG islands), which often correspond to transcriptional start sites [7]. Genome imprinting and X-inactivation both require hypermethylation of CpG islands during development, while aberrant hypermethylation appears characteristic in aging and carcinogenesis. Inactivation of the p16 tumor suppressor is observed in the majority of pancreatic carcinomas with hypermethylation of CDKN2A gene accounting for 14-21% of this loss [5, 8, 9]. p16 (CDKN2A) inhibits the cyclin-dependent kinase 4-cyclin D2 complex, regulating cell cycle progression. Using a high-throughput microarray approach, Sato et al. identified 475 genes profoundly upregulated (>fivefold change) in the presence of 5-aza-2′-deoxycytidine, a methyltransferase inhibitor, in pancreatic cancer cell lines compared to normal ductal epithelial cells. Of these genes, the investigators predicted that ~70% will be aberrantly methylated in pancreatic cancer with ~60% of this small subset expected to harbor hypermethylated CpG islands [10]. Subsequently, the same laboratory noted hypermethylation of eight CpG-associated genes in 68% of PanIN-1a samples. One of the markers, NPTX2, exhibited increases in methylation from PanIN-1 to PanIN-2 [11]. Thus, these data imply that aberrant DNA methylation, particularly of tumor suppressors, occurs early in carcinogenesis and increases during neoplastic progression. The full role of DNA methylation in pancreatic cancer development and progression was recently reviewed in depth by Omura et al. [12].
In this review, we will focus exclusively on the role of histone modifications in pancreatic tumorigenesis, specifically those modifications imprinted by Polycomb group (PcG) proteins, a new branch of epigenetics research in pancreatic cancer. PcG proteins display excellent promise as highly sensitive early detection markers, tools that will prove invaluable in lessening the time to diagnosis in this disease. Additionally, as with DNA methylation, histone modifications are theoretically reversible, affording exciting opportunities for treatment.
Histone Code Hypothesis
The heart of eukaryotic epigenetics and chromatin is the nucleosome, the fundamental packaging unit comprised of ~146 base pairs of DNA twice coiled around an octamer core of histone proteins (H2A, H2B, H3, H4, two copies each), termed the nucleosome core particle (NCP). Histone H1 resides outside of the NCP, stabilizing the nucleosome and promoting folding of nucleosome arrays into higher-order configurations [13]. The N-terminal tails of histone proteins protrude from the NCP, providing registries upon which the heritable epigenetic profile of each cell is written through post-translational modifications (PTMs) of critical residues. Multiple types of covalent modifications are possible within each tail region, including acetylation, phosphorylation, methylation, ubiquitination, and SUMOylation.
The histone code hypothesis, as coined by David Allis, postulates that unique patterns of histone modifications influence the local configuration of chromatin by altering the binding affinities of non-histone proteins [14]. Recruitment of these chromatin-associated factors influences chromatin structure and the accessibility of the DNA for template-dependent processes (e.g., transcription) [15]. While inscription of the histone code requires specific “writer” proteins, translation requires “reader” proteins to induce chromatin alterations. A number of protein domains interact with specific PTMs, such as the bromodomain (acetylated lysines), chromodomain (methylated lysines), and PHD domain (methylated lysines and arginines) [16]. The chromodomain is one member of the Tudor domain “Royal Family” that also includes Tudor, Agenet, PWWP, and MBT domains evolutionarily conserved from a common ancestor [17]. Furthermore, recent evidence suggests that recognition is not limited to individual PTMs but that groups of adjacent modifications may function as a unit, or “cassette,” foretelling the existence of additional reader modules. Thus, the intrinsic epigenetic potential of a given histone tail is a function of both the number of residues and potential modifications, individually and in combination.
Two systems predominate for instituting a repressive chromatin landscape: polycomb and heterochromatin-associated protein 1 (HP1). A chromodomain protein, HP1 binds mono-, di-, and tri-methyl-lysine 9 of histone 3 (H3-K9) with increasing relative affinities. Bound HP1 recruits histone methyltransferases that methylate adjacent H3 tails at K9 to create additional docking sites for HP1, spreading the heterochromatic (transcriptionally repressed) state in a cyclic fashion [18]. Interestingly, HP-1 exists as three isoforms (HP1α, HP1β, and HP1γ) and undergoes extensive post-translational modifications, suggesting the presence of a histone subcode operating in tandem with the histone code [18]. Investigation of HP1 revealed the presence of “binary switches” in the translation of the histone code. Phosphorylation of serine 10 inhibits HP1 binding and is frequently combined with the acetylation of lysine 9 and/or lysine 14, nurturing a euchromatic (transcriptionally active) state [19]. The role of HP1 in pancreatic cancer is under active investigation, but decreased expression of HP1 may increase malignant potential. MUC1, a transmembrane mucin, is highly overexpressed in metastatic pancreatic cancer. MUC1-negative cell lines display substantially higher levels of H3-K9 methylation and DNA methylation at the MUC1 promoter, indicating loss of HP1 as a critical “hit” in neoplastic progression [20]. Dialynas et al. recently reviewed the role of HP1 in multiple facets of cancer progression, including centromere stability and invasion [21].
Polycomb group (PcG) Proteins
The term “Polycomb” was first coined in the 1940s to describe mutations in Drosophila melanogaster that produced extra sex combs, the bristle-like structures on the forelegs of males. The phenotype arose from loss of homeotic (Hox) gene repression—genes responsible for anterior-posterior axis patterning—yielding transformation of one body segment type to another. Other mutants with Hox de-repression were identified, although the identity of the disrupted genes would not be revealed until decades later. The activity of PcG proteins is critical in development, stem cell maintenance and pluripotency, and cell fate determination [22].
In the mid-1980s, a group of proteins that antagonize the repressive effects of PcG proteins were uncovered, the Trithorax group (TrxG) proteins [23]. While transient fluctuations in the transcriptional state of genes are required during development or cellular processes such as replication and mitosis, the identity of a differentiated cell is preserved throughout its existence. PcG protein complexes induce gene repression via H3-K27 trimethylation while TrxG proteins promote gene activation through H3-K4 trimethylation. Genes in embryonic stem (ES) cells are frequently enriched for both modifications, forming poised “bivalent domains” that are reduced to a stable, “univalent” state during differentiation. The final activated or repressed state of the gene is then inherited throughout subsequent generations, a phenomenon known as long-term transcriptional memory [24].
PcG proteins may be divided into two functional biochemical categories (Table 1): Polycomb Repressive Complex 1 (PRC1) and Polycomb Repressive Complex 2 (PRC2). PRC2 is the ~600kDa complex responsible for initiating gene repression through trimethylation of H3-K27 [25, 26]. The enzymatic engine of PRC2 is EZH2 (enhancer of zeste homologue 2), which possesses the SET domain that confers the complex with its methyltransferase activity [25, 27, 28]. The catalytic function of EZH2 is only active in obligate complex with SUZ12 (suppressor of zeste 12) and EED (embryonic ectoderm development) [29]. The WD40-repeat domains of EED form a seven-bladed β-propeller structure that interacts with EZH2 and the N terminus of H3, linking the methyltransferase to its substrate [30-32]. Curiously, the carboxyl-terminal domain of EED also binds to histone tails enriched for H3-K27me3. Disruption of this binding abolishes the activity of PRC2 [33]. EED may therefore function in the propagation and renewal of the H3-K27me3 mark from parent to progeny during replication and division [34]. SUZ12 also directly interacts with EZH2 to enhance methylation, although its precise function is unclear [29, 35]. A number of other proteins are dispensable for complex function but enhance its activity, such as RBBP4, RBBP7, AEPB2, and PHF1 [29, 36, 37]. PRC2 proteins are extremely well conserved from plants to humans [38], solidifying the fundamental role of H3-K27 methylation in repression. Several alternative PRC2 complexes have been identified. EED undergoes alternative translation to yield four isoforms, resulting in isoform-specific PRC2 complexes with distinct roles in differentiation and cancer [39, 40]. Additionally, homologue EZH1 also forms a PRC2 complex that primarily complements the functions of PRC2-EZH2 and is highly expressed in non-proliferative adult tissues [41].
Table 1.
PcG proteins and their implication in the pathogenesis of pancreatic cancer.
| PRC2: INITIATION COMPLEX | |||
|
| |||
| PROTEIN | DOMAIN | FUNCTION | ROLE IN PaCa |
| EZH1/EZH2 | SET | Histone methyltransferase activity (HMT), establishes H3- K37me3 mark |
Oncogene verexpression associated with increased proliferation and invasion with reduction in cell cycle regulators; decreased E- cadherin expression associated with migration and invasion |
| SUZ12 | Zinc finger | Required forHMT activity of PRC2 |
|
| EED | WD40 repeat | Stabilization of N-terminal histone tail; required for HMT activity of PRC2 |
|
| RBBP4/RBBP7 | Histone binding | Histone binding | |
|
| |||
| PRC1: MAINTENANCE COMPLEX | |||
|
| |||
| PROTEIN | DOMAIN | FUNCTION | ROLE IN PaCa |
| CBX2/4/8 | Chromodomain | H3-K27me3 binding | |
| CBX7 | Chromodomain | Nucleosome targeting | Low levels associated with poor differentiation; decreased E-cadherin expression associated with migration and invasion |
| PHC1/2/3 | Zinc finger SPM | ||
| RING1A/1B and RNF2 |
RING finger | Ubiquitin ligase | Oncogenes; overexpression associated with poor differentiation and invasion. |
| BMI1 and MEL18 |
Zinc finger, RING-type |
Protein-protein interactions | Oncogenes; overexpression associated with poor differentiation, increased proliferation, EMT, invasion and “stemness” |
The H3-K27me3 mark imprinted by PRC2 subsequently serves to recruit PRC1, a poorly conserved ~1-2 MDa complex [38, 42]. The complex typically contains CBX2, 4, or 8 (chromobox homologue 2/4/8), PHC1, 2, or 3 (polyhomeotic homologue 1/2/3), BMI1 (B-cell-specific Moloney murine leukemia virus insertion site 1), and RING1A/B or RNF2 (RING finger domain protein). CBX proteins harbor the chromodomain responsible for recognition of trimethylated H3-K27 [43]. Depletion studies of PRC1 and PRC2 complex components have established a primarily hierarchical relationship between the two complexes [44]. While knockdown of PRC1 proteins BMI1 and RING1A fails to affect H3-K27me3 levels, loss of SUZ12 significantly reduces occupancy of BMI1 and RING1A at gene promoters [45]. PRC1 functions to maintain the repressive state initiated by PRC2, although the mechanisms by which PRC1 accomplishes this task are unclear. In addition to direct chromatin compaction, PRC1 acts as an E3 ubiquitin ligase complex that catalyzes monoubiquitination of histone H2A through the catalytic activity of RING1 proteins, likely inhibiting transcriptional initiation by RNA polymerase II [45-47]. Repression may also be mediated through the recruitment of other chromatin-remodeling enzymes, specifically DNMTs and histone deacetylases (HDACs) [48]. PcG complexes are capable of “pre-marking” genes for de novo methylation in cancer, linking the two major mechanisms of epigenetic repression [49, 50]. However, gene repression also occurs in the presence of the H3-K27me3 mark alone, independent of DNMT-mediated promoter methylation [51]. Finally, PRC1 can restrict access of chromatin remodeling or transcriptional machinery to the DNA template, as evidenced by the ability of PRC1 to hinder ATP-dependent SWI/SNF remodeling [52, 53]. Together, these data substantiate the diversity of PRC proteins and the multitude of varying interactions with other chromatin remodeling mechanisms. The flexibility in composition and function of PcG complexes enables exquisitely subtle, content-dependent regulation of repression. Such precision is an absolute necessity in processes with strict spatial-temporal gene expression requirements (e.g., development).
As the core PRC1and PRC2 proteins lack inherent DNA binding capabilities, a pressing question remains how complexes are initially recruited to the promoters of genes. In Drosophila, PRC complexes bind to specific DNA sequences flanking promoters known as polycomb response elements (PREs). Mammalian PREs have largely eluded detection although a few laboratories report progress [54, 55]. The Drosophila protein pleiohomeotic (PHO) and its mammalian ortholog, transcription factor YinYang1 (YY1), may bind specific DNA sequences and cooperate with other cofactors to recruit PRCs to target loci [56, 57]. Therefore, the proposed working model of PRC-mediated repression (Figure 1A) begins with the recruitment of the PRC2 initiation complex to target loci. PRC2 marks the gene target for repression through trimethylation of H3-K27. The methylated mark then serves as a docking site for the PRC1 maintenance complex, leading to stabilization of gene repression through a multitude of downstream pathways. Recent discoveries have added additional complexity to the step-wise recruitment model, such as the simultaneous occupancy of PRC1 and PRC2 at certain bivalent promoters [24].
Figure 1A. Mechanism of polycomb-mediated gene silencing.
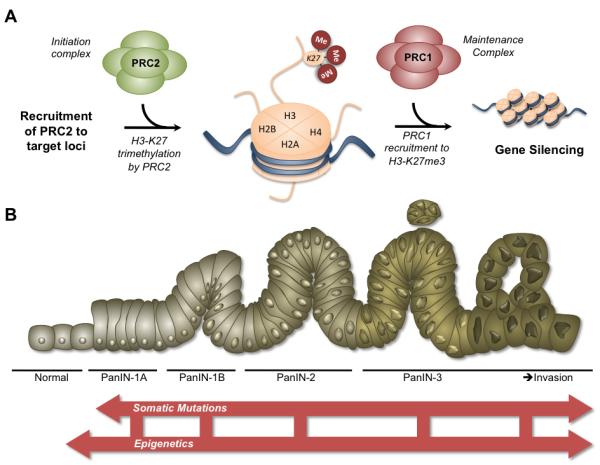
The working model of PRC repression proposes that recruitment of the initiation complex, PRC2, results in H3-K27 trimethylation of gene promoters. The H3-K27 mark subsequently functions to recruit PRC1, which maintains the repressed state through a variety of potential mechanisms. Aberrant overexpression of PcG proteins may lead to malignancy by the repression of vital tumor suppressors. 1B: Revised model of pancreatic carcinogenesis. The current model of pancreatic carcinogenesis proposes the graded transformation of ductal cells from dysplastic precursor lesions, deemed pancreatic intraepithelial neoplasias (PanINs) toward invasive pancreatic ductal adenocarcinomas (PDACs) fueled by inactivating mutations in tumor suppressors and activating mutations in oncogenes. The model requires expansion to incorporate the role of epigenetics in tumorigenesis as well the interaction of epigenetic mechanisms with somatic mutations in progression of the disease.
PcG Proteins: Epigenetic Oncogenes
BMI1 was the first PcG protein linked with neoplastic development and remains the best characterized. The initial discovery of BMI1 as a proto-oncogene found that the protein cooperates with Myc to significantly accelerate B and T-cell lymphomagenesis [58] by repressing the CDKN2A tumor repressor locus to control cellular proliferation and senescence [59]. Overexpression of BMI1 has become a significant prognostic indicator in a wide-variety of cancers, including ovarian [60], bladder [61], and breast [62]. PRC2 proteins have only recently been implicated in cancer development. Levels of EZH2 strongly associate with severity of malignant progression and poor prognosis in breast and prostate cancer [63, 64]. Experimentally, EZH2 overexpression promotes cellular proliferation [48, 65-69], migration [70-72], angiogenesis [73], and survival [74, 75]. For example, EZH2 competes with HDAC1 and growth repressors pRb2/p130 at the cyclin A promoter, leading to disruption of cell cycle progression while promoting genomic instability and transformation [48, 76]. Additionally, EZH2 overexpression leads to repression of E-cadherin, a protein that maintains epithelial cell morphology, directly implicating the protein in cellular adhesion, migration, and metastasis [71]. The multiplicity of downstream functions disrupted by PcG overexpression points to the pervasiveness of PcG-mediated repression in the maintenance of identity throughout the lifetime of a cell.
The precise etiology of PcG protein overexpression appears to be context and tissue-type-dependent. A variety of mechanisms for aberrant expression of PcG proteins have been observed in cancer, including copy number variation, regulatory failure via post-translational modifications or micro-RNA, loss of nuclear localization, and somatic mutations affecting activity. The nascent understanding of PcG proteins in malignant development, therefore, appears to rest on the inactivation of critical tumor suppressors and cell cycle regulators due to an imbalance of PcG-mediated function, culminating in transformation. The mechanisms driving PcG overexpression and its effects on cell cycle, DNA repair, apoptosis, senescence, and other cellular processes were recently reviewed in detail by Sauvageau et al. [77].
Additionally, PcG proteins play a critical role in the maintenance and proliferation of pluripotent progenitor cells in a variety of tissues under physiological conditions [78-80]. Overexpression of these proteins may promote tumorigenesis by fostering a self-renewing population of cells with stem cell-like chromatin patterns capable of rampant proliferation, lending credence to the hypothesis of cancer stem cells. In this scenario, repressive priming of differentiation and tumor suppressors to maintain plasticity leaves these areas of the genome vulnerable to DNA hypermethylation and permanent stabilization of inappropriate gene silencing [81].
PcG Proteins: New Frontier in Pancreatic Cancer Research
The precise role of PcG proteins in pancreatic organogenesis remains unclear. In mice, BMI1 and RING1B are detected in acinar cell nuclei at stages E14.5-15.5 in regions positive for key pancreatic enzymes Elastase 3B, Amylase 2, and Carboxypeptidase A1 [82]. When endocrine cells expand at E17.5 to form islets, BMI1 expression is localized to the developing islets and, by the adult stage, is expressed in the ducts and islets but only weakly so in acinar cells. A similar pattern is observed for RING1B [82]. EZH2 expression is also observed at high levels of expression in the developing pancreas (Grzenda, unpublished data). Therefore, PcG-mediated repression appears pivotal to pancreatic development although virtually no data exists regarding the mechanisms governing these processes.
Recent studies have also uncovered a role for PcG proteins in the programming of β cells during development and regeneration. β cell proliferation and mass display an age-dependent decline, resulting in a reduced capacity for response to metabolic change [83]. A corresponding age-dependent decline in BMI1 expression in the islets is also observed [84]. BMI1 depleted mice (BMI-/-) demonstrate significantly decreased β-cell mass and proliferation with an observed increase in p16 expression, decreased H2A ubiquitination, and increased H3-K4me3 levels at the CDKN2A locus [84]. In the exocrine pancreas, BMI1 nurtures a subset of differentiated acinar cells capable of self-renewal for >1 year [85]. Such a population bolsters evidence of certain carcinomas that develop from non-ductal cell origins observed in mice [86]. Furthermore, EZH2 levels similarly decrease in aging β cells. EZH2 depletion in juvenile mice yields a similar phenotype as BMI1 loss with decreased levels of H3-K27me3 mark at the CDKN2A locus, increases in p16 and p19 expression, and decreased β-cell proliferation [87]. Interestingly, lineage mapping of key histone modifications in β-cell development reveals that differentiation of β cells from progenitors occurs in two stages: first, a wave of cell-specific de novo H3-K27 trimethylation in non-CpG island genes followed by selective temporal removal of the repressive marks in a core β-cell-specific gene program during differentiation [88]. As such, residual PcG expression in the pancreas following development bestows the organ with regenerative potential for tissue homeostasis that is gradually restricted during aging but may be expropriated during neoplastic progression.
According to the classification established by Gunster et al., the adult pancreas is a Class I Polycomb tissue as determined by Northern blot of 23 human tissue samples, on par with the testis and heart. Class I tissues express a minimum of five of the six representative PcG transcripts probed. Levels of RING1, CBX4, BMI1, and EZH2 exhibited intermediate expression while CBX2 demonstrated high levels and EDR1 could not be detected [89]. Immunohistochemistry detected BMI1 and EDR1 primarily in the islet cells, CBX2 and EZH2 primarily in the exocrine compartment, and RING1 and CBX4 in both compartments [89]. Expression of MEL18 is also observed in normal pancreatic tissues [90]. Variable levels of PcG protein expression indicate the potential for complex-independent roles for PcG proteins.
BMI1 overexpression is oncogenic in pancreatic tumorigenesis and strongly linked to poor patient survival. In a conditional knock-in mouse model of selective overexpression of the KRAS oncogene in acinar and centroacinar cells to induce PanIN formation, BMI1 expression was found strongly upregulated in PanIN lesions and PDAC. RING1B, on the other hand, was weakly expressed in PanIN lesions but strongly expressed in PDAC. Correlation of murine findings to patient samples demonstrated similar expression patterns. As such, RING1B may serve to overcome the barrier between the progression from PanIN-1 and PanIN-2/3 lesions [91]. BMI1 depletion in pancreatic cancer cells suppresses growth and delays the G1/S transition with resultant inhibition of cellular proliferation, foci formation, and tumor volume [92]. Furthermore, BMI1 knockdown increases p21 and apoptosis-mediator Bax while downregulating cyclin D1, CDK2, Bcl-2, and phosphorylation of Akt, yielding an overall increased susceptibility to apoptosis [92]. Recently, epithelial-to-mesenchymal (EMT)-activator ZEB1 (zinc finger E-box binding homeobox 1) was found significantly overexpressed in poorly differentiated PDAC samples with correlation to increased BMI1 expression [93]. Knockdown of ZEB1 reduced tumorigenicity of pancreatic cancer cells in a xenograft model as well as their stem cell-like properties such as sphere formation and resistance to gemcitabine, a deoxycytidine nucleoside analog commonly used as adjuvant therapy for pancreatic cancer. The microRNA-200 (miR-200) family, as part of a reciprocal feedback loop, regulates ZEB1 expression. Reduced BMI1 expression and “stemness” were observed with overexpression of miR-200 family members in pancreatic cancer cells, hinting at one putative mechanism of BMI1 deregulation in pancreatic cancer [93]. Downregulation of miR-200 family members has been observed in gemcitabine-resistant pancreatic cancer cell lines that manifest increased EMT characteristics [94]. Collectively, these results indicate that BMI1 disrupts cell cycle and apoptotic pathways, stimulating increased proliferation and survival. Additionally, BMI1 may regulate the critical balance point between regenerative potential and development of the malignant stemness that contributes significantly to PDAC chemoresistance.
At least one PRC1 protein may act as a tumor suppressor in pancreatic cancer progression. Tissue microarray (TMA) analysis of 210 cases of PDAC as well 40 PanIN-3 cases with 40 normal controls demonstrated a step-wise reduction in CBX7 protein expression from normal tissue to cancer [95]. CBX is a chromodomain-containing PRC1 protein that interacts with RING1B, targeting PRC1 to H2A [96]. CBX7 expression levels were inversely correlated with tumor grade, remaining highest in very well differentiated PDAC and decreasing in conjunction with increasing dedifferentiation and poor prognosis. Tumors with depleted CBX7 levels also possessed depleted E-cadherin expression, contributing to their aggressiveness. CBX7 has previously been shown to occupy and repress the CDKN2A promoter in primary human fibroblasts along with CBX8, MEL18, and BMI1, indicating an additional role in cell cycle regulation. Depletion of any of the four proteins results in increased p16 expression and proliferative arrest [97]. At this time, no other PRC1 proteins have been examined in PDAC progression, although one study noted increases in RNF2-positive cells in a small number of PDAC samples compared to normal controls [90]. In other cancerous tissues, CBX7 participates in oncogenesis. These seemingly contradictory results arise from the multifaceted potential inherent to the complexes that may be differentially channeled depending on the tissue-level microenvironment.
EZH2 is the only PRC2 protein thus far studied in pancreatic cancer. In normal tissues, EZH2 protein expression in ductal and acinar cells appears relatively weak. In a comprehensive immunohistochemical analysis of a spectrum of PDACs, strong nuclear accumulation of EZH2 was observed in 11/20 (55%) of well differentiated, 29/50 (58%) of moderately differentiated, and 31/34 (98%) of poorly differentiated samples [98]. The degree of nuclear accumulation significantly correlated with the degree of dedifferentiation of the tumors. An independent study examined 60 cases of primary PDAC against 39 cases of non-cancerous pancreatic tissue by TMA that revealed that the majority (67.5%) of advanced tumors (T3/T4) demonstrated intense EZH2 expression that corresponded significantly with dedifferentiation [99].
EZH2, like BMI1, appears critical in cell cycle regulation and proliferation, processes prominently deregulated in early carcinogenesis. In one study, depletion of EZH2 from pancreatic cancer cell lines decreased proliferation, although examination of common cyclin-dependent kinase inhibitors p15, p16, p21, p27, and p57 revealed that only p27 was uniformly re-expressed [98]. Overexpression of EZH2 significantly parallels the loss of p27 in PDAC samples (61-67%) [98]. However, a subset of pancreatic cancer cell lines have been identified that also re-express p57 in the presence of methyltransferase inhibitor [100]. Knockdown of EZH2 by lentivirus in pancreatic cancer cell lines yields decreased proliferation as well as reduced tumor formation by xenograft [99]. Furthermore, intrasplenic injection of EZH2-depleted pancreatic cancer cells results in reduced liver metastasis [99]. EZH2 depletion also significant decreases H3-K27me3 levels at the RUNX3 promoter, instigating increases in RUNX3 expression that inhibit cellular proliferation [101]. A study examining RUNX3 expression in pancreatic cancer cell lines found that 9/12 of the lines studied possessed absent RUNX3 expression. 100% of these RUNX3-absent lines were hypermethylated at the RUNX3 promoter [102]. RUNX3 plays a critical role in regulating p21 in cell cycle control, perhaps explaining the pro-proliferative effect of EZH2 overexpression in carcinomas where RUNX3 is aberrantly methylated. EZH2 expression is required for normal cell cycle function at a multitude of transitions points, perhaps explaining the wide range of effects on cell cycle regulators and the ease at which EZH2 overexpression disables the normal cellular clock to enhance proliferation.
EZH2 overexpression also appears to participate in late stage invasion and EMT through downregulation of epithelial proteins. Increased EZH2 expression in PDAC samples correlates with decreased E-cadherin expression (70% of cases examined), node positivity (86% of cases), and large tumor size [103]. The degree of differentiation strongly predicts the level of E-cadherin expression with well differentiated PDACs displaying almost normal expression. EZH2 expression is also strongly related to the degree of dysplasia in IPMN cases [103]. Silencing of E-cadherin in breast and prostate cancer cell lines occurs through EZH2-mediated methylation of the E-cadherin promoter [71]. Additionally, decreased E-cadherin is observed in non-cohesive PDAC with associated lack of β-catenin expression, indicating possible zonula adherens defects [104]. As such, EZH2 functions as an epigenetic oncogene throughout neoplastic progression in the pancreas, promoting both the early expansion of the cancerous population and the later aggressive evolution of this population toward invasion. Treatment of pancreatic cancer cells with gemcitabine or doxorubicin, an anthracycline antibiotic, actually increases EZH2 levels while EZH2 depletion sensitizes cells to both treatments [98]. This troubling observation underscores the need to fully characterize the epigenetic mechanisms at play in pancreatic tumors to prevent ineffective or deleterious treatment.
Overexpression of EZH2, the catalytic component of PRC2, would predict increased levels of H3-K27me3 on target genes, a prediction confirmed in a number of cancers. Surprisingly, a study in pancreatic cancer demonstrated H3-K27me3 expression in only 26% (43/165) of pancreatic cancer samples compared to 89% (64/72) of normal samples [105]. Low H3-K27me3 expression samples corresponded to an 11% 5-year survival rate compared with 23% in high H3-K27me3 expression samples. Destabilization of PRC2 in the presence of EZH2 overexpression may explain the contradiction by shifting activity away from H3-K27 trimethylation [39, 40]. EZH2 overexpression in prostate cancer cells leads to formation of PRC4, a complex containing EED isoform 2, which associates with HDAC SirT1 and preferentially methylates H1 [39]. Alternatively, post-translational modifications of EZH2 that inhibit its activity may also be responsible for reduced H3-K27me3 levels. Akt-mediated phosphorylation of EZH2 represses its methyltransferase activity, disrupting gene silencing, which could contribute to oncogenesis in certain contexts [106].
Recently, Li et al. identified a subset of so-called pancreatic cancer stem cells that possess self-renewal properties and the ability to produce differentiated progeny. The cells comprise <5% of total cancer cells. Xenograft of these CD44+ CD24+ ESA+ stem cells resulted in a 100-fold increase in tumorigenic potential and produced tumors that were histological quite similar to PDAC samples [107]. Upregulation of Hedgehog signaling, a pathway previously implicated in PanIN formation, was also observed. Furthermore, treatment with gemcitabine resulted in increased enrichment of these stem cells in pancreatic tumors. CD133+ pancreatic cancer stem cells have also been isolated which display similar increases in tumorigenic potential and resistance to gemcitabine [108]. The highly metastatic nature of these cells depends on expression of the CXCR4 receptor, a marker of stem cell migration. Depletion of CXCR4 in CD133+ stem cells resulted in decreased liver metastasis. High expression of CXCR4 has been observed with poor prognosis in PDAC and the CXCR4 signaling axis may contribute to chemoresistance [109, 110]. The role of PcG proteins in stem cell pluripotency anticipated the existence of self-renewing pancreatic cancer cells. These cells, although a minor population, further explain the chemoresistant nature of pancreatic neoplasms as well as high rates of reoccurrence following treatment.
PcG Proteins: Gateway to Improved Adjuvant Therapy
Classically, surgical resection has been the only curative treatment for PDAC patients. Unfortunately, at presentation, only 15-20% of PDAC patients are eligible for the resection procedure. Gemcitabine has emerged in the past 10-15 years as a moderately effective palliative treatment in the metastatic setting, particularly in combination with other cytotoxic agents, while the efficacy of radiotherapy in combination with chemotherapy remains unclear [111, 112]. Progressive chemoresistance remains the most significant barrier in the adjunctive therapy of PDAC.
Unraveling the precise function of PcG proteins may explain the mechanisms driving the inherent chemoresistant nature of pancreatic neoplasms as well as provide effective targets for stand-alone or combination therapy. In pancreatic cancer cell lines, depletion of EZH2 sensitizes cancer cells to the effects of gemcitabine (as well as doxorubicin) with a significant increases in apoptosis [98]. Recent investigations have focused on targeting the PRC2 complex through small molecule inhibitors. One compound, 3-deazaneplanocin A (DZNep), an S-adenosylhomocysteine hydrolase inhibitor, depletes levels of PRC2 proteins (EZH2, SUZ12, and EED), inhibits H3-K27 methylation, and preferentially induces apoptosis in cancer cells [113]. Treatment of breast cancer cells with DZNep re-activates a large number of PRC2-repressed gene targets [114]. Additionally, chromatin immunoprecipitation analysis demonstrates increased recruitment of RNA Polymerase II following DZNep treatment with decreased occupancy of SUZ12 at the promoters of eight target genes, indicative of transcriptional re-activation of these previously repressed targets. Two other drugs, sinefungin and adenosine dialdehyde (AdOx), reduce H3-K27me3 levels in a similar fashion [114, 115]. Reduced cellular proliferation and migration in pancreatic cancer cells is also observed with AdOx treatment (Grzenda, unpublished data).
Evidence suggests that PcG proteins are downstream targets of a number of pathways linked to cancer development, such as E2F [66], β-catenin [116], Wnt [117], Hedgehog [118], and ERK [119]. Designing therapeutic strategies to disrupt PcG complex formation or pathways downstream of PcG-mediated repression, such as the CXCR4 axis described above, holds great potential. Thorough elucidation of PcG proteins at the molecular and functional level is required to identify multiple points for treatment, thus maximizing the potential for more beneficial adjuvant therapies.
Conclusion
Epigenetics represents an exciting new chapter in the understanding of pancreatic cancer. The current model of pancreatic carcinogenesis requires expansion to include the contribution of epigenetics, not in isolation from somatic processes but in conjunction throughout neoplastic transformation and progression (Figure 1B). Evidence indicates that certain epigenetic alterations may precede somatic mutations, serving as viable first “hits” or by establishing pro-tumorigenic foundations for fostering neoplasia. In addition to providing valuable information regarding the progressive development of adenocarcinomas from pre-neoplastic lesions, molecular targeting of PcG complexes presents a promising new strategy in the management of these aggressive neoplasias. The recent identification of pancreatic cancer “stem cells” opens yet another branch of epigenetics-focused research. As demonstrated from the paucity of data regarding the function of PcG proteins and complexes in the normal pancreatic development and malignancy, the field is clearly fertile ground for multiple avenues of investigation.
Acknowledgements
This (National review of Health Grants T32CA148073 by Institutes was supported Institute), National and DK52913, DK058185. Cancer Institute), DK52913, and DK058185.
References
- 1.Jemal A, et al. Cancer statistics. CA Cancer J Clin. 2010;60(5):277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 2.Wang Z, et al. Pancreatic cancer: understanding and overcoming chemoresistance. Nat Rev Gastroenterol Hepatol. 8(1):27–33. doi: 10.1038/nrgastro.2010.188. [DOI] [PubMed] [Google Scholar]
- 3.Hruban RH, et al. Pancreatic intraepithelial neoplasia: a new nomenclature and classification system for pancreatic duct lesions. Am J Surg Pathol. 2001;25(5):579–86. doi: 10.1097/00000478-200105000-00003. [DOI] [PubMed] [Google Scholar]
- 4.Jones S, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321(5897):1801–6. doi: 10.1126/science.1164368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hruban RH, Adsay NV. Molecular classification of neoplasms of the pancreas. Hum Pathol. 2009;40(5):612–23. doi: 10.1016/j.humpath.2009.01.008. [DOI] [PubMed] [Google Scholar]
- 6.Cheng X, Blumenthal RM. Mammalian DNA methyltransferases: a structural perspective. Structure. 2008;16(3):341–50. doi: 10.1016/j.str.2008.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Antequera F, Bird A. CpG islands as genomic footprints of promoters that are associated with replication origins. Curr Biol. 1999;9(17):R661–7. doi: 10.1016/s0960-9822(99)80418-7. [DOI] [PubMed] [Google Scholar]
- 8.Schutte M, et al. Abrogation of the Rb/p16 tumor-suppressive pathway in virtually all pancreatic carcinomas. Cancer Res. 1997;57(15):3126–30. [PubMed] [Google Scholar]
- 9.Rosty C, et al. p16 Inactivation in pancreatic intraepithelial neoplasias (PanINs) arising in patients with chronic pancreatitis. Am J Surg Pathol. 2003;27(12):1495–501. doi: 10.1097/00000478-200312000-00001. [DOI] [PubMed] [Google Scholar]
- 10.Sato N, et al. Discovery of novel targets for aberrant methylation in pancreatic carcinoma using high-throughput microarrays. Cancer Res. 2003;63(13):3735–42. [PubMed] [Google Scholar]
- 11.Sato N, et al. CpG island methylation profile of pancreatic intraepithelial neoplasia. Mod Pathol. 2008;21(3):238–44. doi: 10.1038/modpathol.3800991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Omura N, Goggins M. Epigenetics and epigenetic alterations in pancreatic cancer. Int J Clin Exp Pathol. 2009;2(4):310–26. [PMC free article] [PubMed] [Google Scholar]
- 13.Luger K. Structure and dynamic behavior of nucleosomes. Curr Opin Genet Dev. 2003;13(2):127–35. doi: 10.1016/s0959-437x(03)00026-1. [DOI] [PubMed] [Google Scholar]
- 14.Jenuwein T, Allis CD. Translating the histone code. Science. 2001;293(5532):1074–80. doi: 10.1126/science.1063127. [DOI] [PubMed] [Google Scholar]
- 15.Chi P, Allis CD, Wang GG. Covalent histone modifications--miswritten, misinterpreted and mis-erased in human cancers. Nat Rev Cancer. 10(7):457–69. doi: 10.1038/nrc2876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bottomley MJ. Structures of protein domains that create or recognize histone modifications. EMBO Rep. 2004;5(5):464–9. doi: 10.1038/sj.embor.7400146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Maurer-Stroh S, et al. The Tudor domain ‘Royal Family’: Tudor, plant Agenet, Chromo, PWWP and MBT domains. Trends Biochem Sci. 2003;28(2):69–74. doi: 10.1016/S0968-0004(03)00004-5. [DOI] [PubMed] [Google Scholar]
- 18.Lomberk G, et al. Evidence for the existence of an HP1-mediated subcode within the histone code. Nat Cell Biol. 2006;8(4):407–15. doi: 10.1038/ncb1383. [DOI] [PubMed] [Google Scholar]
- 19.Mateescu B, et al. Tethering of HP1 proteins to chromatin is relieved by phosphoacetylation of histone H3. EMBO Rep. 2004;5(5):490–6. doi: 10.1038/sj.embor.7400139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yamada N, et al. MUC1 expression is regulated by DNA methylation and histone H3 lysine 9 modification in cancer cells. Cancer Res. 2008;68(8):2708–16. doi: 10.1158/0008-5472.CAN-07-6844. [DOI] [PubMed] [Google Scholar]
- 21.Dialynas GK, Vitalini MW, Wallrath LL. Linking Heterochromatin Protein 1 (HP1) to cancer progression. Mutat Res. 2008;647(1-2):13–20. doi: 10.1016/j.mrfmmm.2008.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sparmann A, van Lohuizen M. Polycomb silencers control cell fate, development and cancer. Nat Rev Cancer. 2006;6(11):846–56. doi: 10.1038/nrc1991. [DOI] [PubMed] [Google Scholar]
- 23.Mills AA. Throwing the cancer switch: reciprocal roles of polycomb and trithorax proteins. Nat Rev Cancer. 10(10):669–82. doi: 10.1038/nrc2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ku M, et al. Genomewide analysis of PRC1 and PRC2 occupancy identifies two classes of bivalent domains. PLoS Genet. 2008;4(10):e1000242. doi: 10.1371/journal.pgen.1000242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cao R, et al. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science. 2002;298(5595):1039–43. doi: 10.1126/science.1076997. [DOI] [PubMed] [Google Scholar]
- 26.Czermin B, et al. Drosophila enhancer of Zeste/ESC complexes have a histone H3 methyltransferase activity that marks chromosomal Polycomb sites. Cell. 2002;111(2):185–96. doi: 10.1016/s0092-8674(02)00975-3. [DOI] [PubMed] [Google Scholar]
- 27.Jenuwein T, et al. SET domain proteins modulate chromatin domains in euand heterochromatin. Cell Mol Life Sci. 1998;54(1):80–93. doi: 10.1007/s000180050127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kuzmichev A, et al. Histone methyltransferase activity associated with a human multiprotein complex containing the Enhancer of Zeste protein. Genes Dev. 2002;16(22):2893–905. doi: 10.1101/gad.1035902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cao R, Zhang Y. SUZ12 is required for both the histone methyltransferase activity and the silencing function of the EED-EZH2 complex. Mol Cell. 2004;15(1):57–67. doi: 10.1016/j.molcel.2004.06.020. [DOI] [PubMed] [Google Scholar]
- 30.Han Z, et al. Structural basis of EZH2 recognition by EED. Structure. 2007;15(10):1306–15. doi: 10.1016/j.str.2007.08.007. [DOI] [PubMed] [Google Scholar]
- 31.Tie F, et al. The N terminus of Drosophila ESC binds directly to histone H3 and is required for E(Z)-dependent trimethylation of H3 lysine 27. Mol Cell Biol. 2007;27(6):2014–26. doi: 10.1128/MCB.01822-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Denisenko O, et al. Point mutations in the WD40 domain of Eed block its interaction with Ezh2. Mol Cell Biol. 1998;18(10):5634–42. doi: 10.1128/mcb.18.10.5634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Margueron R, et al. Role of the polycomb protein EED in the propagation of repressive histone marks. Nature. 2009;461(7265):762–7. doi: 10.1038/nature08398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Xu C, et al. Binding of different histone marks differentially regulates the activity and specificity of polycomb repressive complex 2 (PRC2) Proc Natl Acad Sci U S A. 107(45):19266–71. doi: 10.1073/pnas.1008937107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yamamoto K, et al. Polycomb group suppressor of zeste 12 links heterochromatin protein 1alpha and enhancer of zeste 2. J Biol Chem. 2004;279(1):401–6. doi: 10.1074/jbc.M307344200. [DOI] [PubMed] [Google Scholar]
- 36.Sarma K, et al. Ezh2 requires PHF1 to efficiently catalyze H3 lysine 27 trimethylation in vivo. Mol Cell Biol. 2008;28(8):2718–31. doi: 10.1128/MCB.02017-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tie F, et al. The Drosophila Polycomb Group proteins ESC and E(Z) are present in a complex containing the histone-binding protein p55 and the histone deacetylase RPD3. Development. 2001;128(2):275–86. doi: 10.1242/dev.128.2.275. [DOI] [PubMed] [Google Scholar]
- 38.Schuettengruber B, et al. Genome regulation by polycomb and trithorax proteins. Cell. 2007;128(4):735–45. doi: 10.1016/j.cell.2007.02.009. [DOI] [PubMed] [Google Scholar]
- 39.Kuzmichev A, et al. Composition and histone substrates of polycomb repressive group complexes change during cellular differentiation. Proc Natl Acad Sci U S A. 2005;102(6):1859–64. doi: 10.1073/pnas.0409875102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kuzmichev A, et al. Different EZH2-containing complexes target methylation of histone H1 or nucleosomal histone H3. Mol Cell. 2004;14(2):183–93. doi: 10.1016/s1097-2765(04)00185-6. [DOI] [PubMed] [Google Scholar]
- 41.Margueron R, et al. Ezh1 and Ezh2 maintain repressive chromatin through different mechanisms. Mol Cell. 2008;32(4):503–18. doi: 10.1016/j.molcel.2008.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shao Z, et al. Stabilization of chromatin structure by PRC1, a Polycomb complex. Cell. 1999;98(1):37–46. doi: 10.1016/S0092-8674(00)80604-2. [DOI] [PubMed] [Google Scholar]
- 43.Vincenz C, Kerppola TK. Different polycomb group CBX family proteins associate with distinct regions of chromatin using nonhomologous protein sequences. Proc Natl Acad Sci U S A. 2008;105(43):16572–7. doi: 10.1073/pnas.0805317105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang L, et al. Hierarchical recruitment of polycomb group silencing complexes. Mol Cell. 2004;14(5):637–46. doi: 10.1016/j.molcel.2004.05.009. [DOI] [PubMed] [Google Scholar]
- 45.Cao R, Tsukada Y, Zhang Y. Role of Bmi-1 and Ring1A in H2A ubiquitylation and Hox gene silencing. Mol Cell. 2005;20(6):845–54. doi: 10.1016/j.molcel.2005.12.002. [DOI] [PubMed] [Google Scholar]
- 46.Wang H, et al. Role of histone H2A ubiquitination in Polycomb silencing. Nature. 2004;431(7010):873–8. doi: 10.1038/nature02985. [DOI] [PubMed] [Google Scholar]
- 47.Zhou W, et al. Histone H2A monoubiquitination represses transcription by inhibiting RNA polymerase II transcriptional elongation. Mol Cell. 2008;29(1):69–80. doi: 10.1016/j.molcel.2007.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tonini T, et al. Ezh2 reduces the ability of HDAC1-dependent pRb2/p130 transcriptional repression of cyclin A. Oncogene. 2004;23(28):4930–7. doi: 10.1038/sj.onc.1207608. [DOI] [PubMed] [Google Scholar]
- 49.Schlesinger Y, et al. Polycomb-mediated methylation on Lys27 of histone H3 pre-marks genes for de novo methylation in cancer. Nat Genet. 2007;39(2):232–6. doi: 10.1038/ng1950. [DOI] [PubMed] [Google Scholar]
- 50.Vire E, et al. The Polycomb group protein EZH2 directly controls DNA methylation. Nature. 2006;439(7078):871–4. doi: 10.1038/nature04431. [DOI] [PubMed] [Google Scholar]
- 51.Kondo Y, et al. Gene silencing in cancer by histone H3 lysine 27 trimethylation independent of promoter DNA methylation. Nat Genet. 2008;40(6):741–50. doi: 10.1038/ng.159. [DOI] [PubMed] [Google Scholar]
- 52.Francis NJ, et al. Reconstitution of a functional core polycomb repressive complex. Mol Cell. 2001;8(3):545–56. doi: 10.1016/s1097-2765(01)00316-1. [DOI] [PubMed] [Google Scholar]
- 53.Dellino GI, et al. Polycomb silencing blocks transcription initiation. Mol Cell. 2004;13(6):887–93. doi: 10.1016/s1097-2765(04)00128-5. [DOI] [PubMed] [Google Scholar]
- 54.Sing A, et al. A vertebrate Polycomb response element governs segmentation of the posterior hindbrain. Cell. 2009;138(5):885–97. doi: 10.1016/j.cell.2009.08.020. [DOI] [PubMed] [Google Scholar]
- 55.Woo CJ, et al. A region of the human HOXD cluster that confers polycombgroup responsiveness. Cell. 140(1):99–110. doi: 10.1016/j.cell.2009.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Brown JL, et al. The Drosophila pho-like gene encodes a YY1-related DNA binding protein that is redundant with pleiohomeotic in homeotic gene silencing. Development. 2003;130(2):285–94. doi: 10.1242/dev.00204. [DOI] [PubMed] [Google Scholar]
- 57.Wilkinson F, Pratt H, Atchison ML. PcG recruitment by the YY1 REPO domain can be mediated by Yaf2. J Cell Biochem. 109(3):478–86. doi: 10.1002/jcb.22424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Haupt Y, et al. Novel zinc finger gene implicated as myc collaborator by retrovirally accelerated lymphomagenesis in E mu-myc transgenic mice. Cell. 1991;65(5):753–63. doi: 10.1016/0092-8674(91)90383-a. [DOI] [PubMed] [Google Scholar]
- 59.Jacobs JJ, et al. Bmi-1 collaborates with c-Myc in tumorigenesis by inhibiting c-Myc-induced apoptosis via INK4a/ARF. Genes Dev. 1999;13(20):2678–90. doi: 10.1101/gad.13.20.2678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang FB, Sui LH, Xin T. Correlation of Bmi-1 expression and telomerase activity in human ovarian cancer. Br J Biomed Sci. 2008;65(4):172–7. doi: 10.1080/09674845.2008.11732824. [DOI] [PubMed] [Google Scholar]
- 61.Qin ZK, et al. Expression of Bmi-1 is a prognostic marker in bladder cancer. BMC Cancer. 2009;9:61. doi: 10.1186/1471-2407-9-61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Saeki M, et al. Diagnostic importance of overexpression of Bmi-1 mRNA in early breast cancers. Int J Oncol. 2009;35(3):511–5. doi: 10.3892/ijo_00000362. [DOI] [PubMed] [Google Scholar]
- 63.Kleer CG, et al. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc Natl Acad Sci U S A. 2003;100(20):11606–11. doi: 10.1073/pnas.1933744100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Varambally S, et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature. 2002;419(6907):624–9. doi: 10.1038/nature01075. [DOI] [PubMed] [Google Scholar]
- 65.Bohrer LR, et al. Androgens suppress EZH2 expression via retinoblastoma (RB) and p130-dependent pathways: a potential mechanism of androgenrefractory progression of prostate cancer. Endocrinology. 151(11):5136–45. doi: 10.1210/en.2010-0436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bracken AP, et al. EZH2 is downstream of the pRB-E2F pathway, essential for proliferation and amplified in cancer. EMBO J. 2003;22(20):5323–35. doi: 10.1093/emboj/cdg542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kotake Y, et al. pRB family proteins are required for H3K27 trimethylation and Polycomb repression complexes binding to and silencing p16INK4alpha tumor suppressor gene. Genes Dev. 2007;21(1):49–54. doi: 10.1101/gad.1499407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shi B, et al. Integration of estrogen and Wnt signaling circuits by the polycomb group protein EZH2 in breast cancer cells. Mol Cell Biol. 2007;27(14):5105–19. doi: 10.1128/MCB.00162-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tang X, et al. Activated p53 suppresses the histone methyltransferase EZH2 gene. Oncogene. 2004;23(34):5759–69. doi: 10.1038/sj.onc.1207706. [DOI] [PubMed] [Google Scholar]
- 70.Bryant RJ, et al. EZH2 promotes proliferation and invasiveness of prostate cancer cells. Prostate. 2007;67(5):547–56. doi: 10.1002/pros.20550. [DOI] [PubMed] [Google Scholar]
- 71.Cao Q, et al. Repression of E-cadherin by the polycomb group protein EZH2 in cancer. Oncogene. 2008;27(58):7274–84. doi: 10.1038/onc.2008.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fujii S, Ochiai A. Enhancer of zeste homolog 2 downregulates E-cadherin by mediating histone H3 methylation in gastric cancer cells. Cancer Sci. 2008;99(4):738–46. doi: 10.1111/j.1349-7006.2008.00743.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lu C, et al. Regulation of tumor angiogenesis by EZH2. Cancer Cell. 18(2):185–97. doi: 10.1016/j.ccr.2010.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zeidler M, Kleer CG. The Polycomb group protein Enhancer of Zeste 2: its links to DNA repair and breast cancer. J Mol Histol. 2006;37(5-7):219–23. doi: 10.1007/s10735-006-9042-9. [DOI] [PubMed] [Google Scholar]
- 75.Zeidler M, et al. The Polycomb group protein EZH2 impairs DNA repair in breast epithelial cells. Neoplasia. 2005;7(11):1011–9. doi: 10.1593/neo.05472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tonini T, et al. Importance of Ezh2 polycomb protein in tumorigenesis process interfering with the pathway of growth suppressive key elements. J Cell Physiol. 2008;214(2):295–300. doi: 10.1002/jcp.21241. [DOI] [PubMed] [Google Scholar]
- 77.Sauvageau M, Sauvageau G. Polycomb group proteins: multi-faceted regulators of somatic stem cells and cancer. Cell Stem Cell. 7(3):299–313. doi: 10.1016/j.stem.2010.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Jiang L, Li J, Song L. Bmi-1, stem cells and cancer. Acta Biochim Biophys Sin (Shanghai) 2009;41(7):527–34. doi: 10.1093/abbs/gmp040. [DOI] [PubMed] [Google Scholar]
- 79.Lee TI, et al. Control of developmental regulators by Polycomb in human embryonic stem cells. Cell. 2006;125(2):301–13. doi: 10.1016/j.cell.2006.02.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Molofsky AV, et al. Bmi-1 dependence distinguishes neural stem cell selfrenewal from progenitor proliferation. Nature. 2003;425(6961):962–7. doi: 10.1038/nature02060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ohm JE, et al. A stem cell-like chromatin pattern may predispose tumor suppressor genes to DNA hypermethylation and heritable silencing. Nat Genet. 2007;39(2):237–42. doi: 10.1038/ng1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Martinez-Romero C, et al. The epigenetic regulators Bmi1 and Ring1B are differentially regulated in pancreatitis and pancreatic ductal adenocarcinoma. J Pathol. 2009;219(2):205–13. doi: 10.1002/path.2585. [DOI] [PubMed] [Google Scholar]
- 83.Tschen SI, et al. Age-dependent decline in beta-cell proliferation restricts the capacity of beta-cell regeneration in mice. Diabetes. 2009;58(6):1312–20. doi: 10.2337/db08-1651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Dhawan S, Tschen SI, Bhushan A. Bmi-1 regulates the Ink4a/Arf locus to control pancreatic beta-cell proliferation. Genes Dev. 2009;23(8):906–11. doi: 10.1101/gad.1742609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sangiorgi E, Capecchi MR. Bmi1 lineage tracing identifies a self-renewing pancreatic acinar cell subpopulation capable of maintaining pancreatic organ homeostasis. Proc Natl Acad Sci U S A. 2009;106(17):7101–6. doi: 10.1073/pnas.0902508106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Guerra C, et al. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell. 2007;11(3):291–302. doi: 10.1016/j.ccr.2007.01.012. [DOI] [PubMed] [Google Scholar]
- 87.Chen H, et al. Polycomb protein Ezh2 regulates pancreatic beta-cell Ink4a/Arf expression and regeneration in diabetes mellitus. Genes Dev. 2009;23(8):975–85. doi: 10.1101/gad.1742509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.van Arensbergen J, et al. Derepression of Polycomb targets during pancreatic organogenesis allows insulin-producing beta-cells to adopt a neural gene activity program. Genome Res. 20(6):722–32. doi: 10.1101/gr.101709.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Gunster MJ, et al. Differential expression of human Polycomb group proteins in various tissues and cell types. J Cell Biochem Suppl. 2001;Suppl 36:129–43. doi: 10.1002/jcb.1093. [DOI] [PubMed] [Google Scholar]
- 90.Sanchez-Beato M, et al. Variability in the expression of polycomb proteins in different normal and tumoral tissues. A pilot study using tissue microarrays. Mod Pathol. 2006;19(5):684–94. doi: 10.1038/modpathol.3800577. [DOI] [PubMed] [Google Scholar]
- 91.Martínez-Romero C, et al. The epigenetic regulators Bmi1 and Ring1B are differentially regulated in pancreatitis and pancreatic ductal adenocarcinoma. J Pathol. 2009:205–13. doi: 10.1002/path.2585. [DOI] [PubMed] [Google Scholar]
- 92.Song W, et al. Bmi-1 is related to proliferation, survival and poor prognosis in pancreatic cancer. Cancer Sci. 101(7):1754–60. doi: 10.1111/j.1349-7006.2010.01577.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Wellner U, et al. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat Cell Biol. 2009;11(12):1487–95. doi: 10.1038/ncb1998. [DOI] [PubMed] [Google Scholar]
- 94.Li Y, et al. Up-regulation of miR-200 and let-7 by natural agents leads to the reversal of epithelial-to-mesenchymal transition in gemcitabine-resistant pancreatic cancer cells. Cancer Res. 2009;69(16):6704–12. doi: 10.1158/0008-5472.CAN-09-1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Karamitopoulou E, et al. Loss of the CBX7 protein expression correlates with a more aggressive phenotype in pancreatic cancer. Eur J Cancer. 46(8):1438–44. doi: 10.1016/j.ejca.2010.01.033. [DOI] [PubMed] [Google Scholar]
- 96.Bezsonova I, et al. Ring1B contains a ubiquitin-like docking module for interaction with Cbx proteins. Biochemistry. 2009;48(44):10542–8. doi: 10.1021/bi901131u. [DOI] [PubMed] [Google Scholar]
- 97.Maertens GN, et al. Several distinct polycomb complexes regulate and colocalize on the INK4a tumor suppressor locus. PLoS One. 2009;4(7):e6380. doi: 10.1371/journal.pone.0006380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ougolkov AV, Bilim VN, Billadeau DD. Regulation of pancreatic tumor cell proliferation and chemoresistance by the histone methyltransferase enhancer of zeste homologue 2. Clin Cancer Res. 2008;14(21):6790–6. doi: 10.1158/1078-0432.CCR-08-1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Chen Y, et al. RNAi targeting EZH2 inhibits tumor growth and liver metastasis of pancreatic cancer in vivo. Cancer Lett. 297(1):109–16. doi: 10.1016/j.canlet.2010.05.003. [DOI] [PubMed] [Google Scholar]
- 100.Sato N, et al. Epigenetic down-regulation of CDKN1C/p57KIP2 in pancreatic ductal neoplasms identified by gene expression profiling. Clin Cancer Res. 2005;11(13):4681–8. doi: 10.1158/1078-0432.CCR-04-2471. [DOI] [PubMed] [Google Scholar]
- 101.Fujii S, et al. Enhancer of zeste homologue 2 (EZH2) down-regulates RUNX3 by increasing histone H3 methylation. J Biol Chem. 2008;283(25):17324–32. doi: 10.1074/jbc.M800224200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Wada M, et al. Frequent loss of RUNX3 gene expression in human bile duct and pancreatic cancer cell lines. Oncogene. 2004;23(13):2401–7. doi: 10.1038/sj.onc.1207395. [DOI] [PubMed] [Google Scholar]
- 103.Toll AD, et al. Implications of enhancer of zeste homologue 2 expression in pancreatic ductal adenocarcinoma. Hum Pathol. 41(9):1205–9. doi: 10.1016/j.humpath.2010.03.004. [DOI] [PubMed] [Google Scholar]
- 104.Winter JM, et al. Absence of E-cadherin expression distinguishes noncohesive from cohesive pancreatic cancer. Clin Cancer Res. 2008;14(2):412–8. doi: 10.1158/1078-0432.CCR-07-0487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wei Y, et al. Loss of trimethylation at lysine 27 of histone H3 is a predictor of poor outcome in breast, ovarian, and pancreatic cancers. Mol Carcinog. 2008;47(9):701–6. doi: 10.1002/mc.20413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Cha TL, et al. Akt-mediated phosphorylation of EZH2 suppresses methylation of lysine 27 in histone H3. Science. 2005;310(5746):306–10. doi: 10.1126/science.1118947. [DOI] [PubMed] [Google Scholar]
- 107.Li C, et al. Identification of pancreatic cancer stem cells. Cancer Res. 2007;67(3):1030–7. doi: 10.1158/0008-5472.CAN-06-2030. [DOI] [PubMed] [Google Scholar]
- 108.Hermann PC, et al. Distinct populations of cancer stem cells determine tumor growth and metastatic activity in human pancreatic cancer. Cell Stem Cell. 2007;1(3):313–23. doi: 10.1016/j.stem.2007.06.002. [DOI] [PubMed] [Google Scholar]
- 109.Marechal R, et al. High expression of CXCR4 may predict poor survival in resected pancreatic adenocarcinoma. Br J Cancer. 2009;100(9):1444–51. doi: 10.1038/sj.bjc.6605020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Singh S, et al. CXCL12-CXCR4 signalling axis confers gemcitabine resistance to pancreatic cancer cells: a novel target for therapy. Br J Cancer. 103(11):1671–9. doi: 10.1038/sj.bjc.6605968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Bayraktar S, Bayraktar UD, Rocha-Lima CM. Recent developments in palliative chemotherapy for locally advanced and metastatic pancreas cancer. World J Gastroenterol. 16(6):673–82. doi: 10.3748/wjg.v16.i6.673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Shah AP, Strauss JB, Abrams RA. Review and commentary on the role of radiation therapy in the adjuvant management of pancreatic cancer. Am J Clin Oncol. 33(1):101–6. doi: 10.1097/COC.0b013e31819171b9. [DOI] [PubMed] [Google Scholar]
- 113.Tan J, et al. Pharmacologic disruption of Polycomb-repressive complex 2-mediated gene repression selectively induces apoptosis in cancer cells. Genes Dev. 2007:1050–63. doi: 10.1101/gad.1524107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Miranda TB, et al. DZNep is a global histone methylation inhibitor that reactivates developmental genes not silenced by DNA methylation. Mol Cancer Ther. 2009;8(6):1579–88. doi: 10.1158/1535-7163.MCT-09-0013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Chen DH, et al. Effects of adenosine dialdehyde treatment on in vitro and in vivo stable protein methylation in HeLa cells. J Biochem. 2004;136(3):371–6. doi: 10.1093/jb/mvh131. [DOI] [PubMed] [Google Scholar]
- 116.Kirmizis A, Bartley SM, Farnham PJ. Identification of the polycomb group protein SU(Z)12 as a potential molecular target for human cancer therapy. Mol Cancer Ther. 2003:113–21. [PubMed] [Google Scholar]
- 117.Pizzatti L, et al. SUZ12 is a candidate target of the non-canonical WNT pathway in the progression of chronic myeloid leukemia. Genes Chromosomes Cancer. 49(2):107–18. doi: 10.1002/gcc.20722. [DOI] [PubMed] [Google Scholar]
- 118.Liu S, et al. Hedgehog signaling and Bmi-1 regulate self-renewal of normal and malignant human mammary stem cells. Cancer Res. 2006;66(12):6063–71. doi: 10.1158/0008-5472.CAN-06-0054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Rao PS, et al. RNF2 is the target for phosphorylation by the p38 MAPK and ERK signaling pathways. Proteomics. 2009;9(10):2776–87. doi: 10.1002/pmic.200800847. [DOI] [PMC free article] [PubMed] [Google Scholar]