

Abstract
Biliary atresia (BA) is the most common identifiable hepatobiliary disease affecting infants, in which there are defects in intra- and extrahepatic bile ducts and progressive fibrosis. Activation of interferon-gamma (IFNγ) appears to be critical in both patients with BA and in rodent models of BA. We have recently reported a zebrafish model of biliary disease that shares features with BA, in which inhibition of DNA methylation leads to intrahepatic biliary defects and activation of IFNγ target genes. Here we report that ifng genes are hypomethylated and upregulated in zebrafish larvae treated with azacytidine (azaC), an inhibitor of DNA methylation. Injection of IFNγ protein into developing zebrafish larvae leads to biliary defects, suggesting that activation of the IFNγ pathway is sufficient to cause developmental biliary defects. These defects are associated with decreased cholangiocyte proliferation and with a decrease in the expression of vhnf1 (hnf1b, tcf2), which encodes a homeodomain protein with previously reported roles in biliary development in multiple models. These results support an importance of IFNγ in mediating biliary defects, and also demonstrate the feasibility of direct injection of intact protein into developing zebrafish larvae.
Introduction
Defects in the developing biliary system can lead to severe liver disease in infants. The most common identifiable disease affecting bile ducts in infants is biliary atresia (BA), a progressive fibro-inflammatory disorder for which the etiology is unclear1. Studies in patients and in the mouse model of BA demonstrate a central importance of interferon-gamma (IFNγ) activation.2,3 Recently, we reported activation of IFNγ in a zebrafish model of liver disease resembling BA, in which DNA methylation is inhibited.4 In this previous study, we observed severe developmental intrahepatic biliary defects in several zebrafish models in which DNA methylation is inhibited. In addition, we observed decreased DNA methylation in cholangiocytes from patients with BA, suggesting that inhibition of DNA methylation may play an important role in the pathogenesis of BA. Others have reported similar results in lymphocytes of patients with BA, and have also shown that the IFNG gene is hypomethylated in lymphocytes from BA patients.5
In several studies, we and others have demonstrated the ability of zebrafish to model human hepatobiliary diseases, including BA.4,6 Other zebrafish models of hepatobiliary diseases support a relatively strong conservation of molecular pathways such as hnf6/hnf1b7, jagged/notch8, and intracellular trafficking pathways.9
Our zebrafish models of BA in which DNA methylation is inhibited include genetic models (dtp, dnmt1-morpholino injected larvae) and chemically-treated models (azacytidine, azaC).4 Here we report further analysis of the azaC model showing increased expression and decreased methylation of ifng genes. To determine the potential role of IFNγ in mediating biliary defects, we injected human recombinant IFNγ protein into developing zebrafish larvae.
Injection of protein into developing zebrafish larvae is a relatively novel technique. Stoll and colleagues10 previously injected IL-8 into the developing heart of 24 hpf larvae,10 while Shibata et al. injected recombinant Musashi1 into embryos.11 These studies demonstrate the feasibility of injecting protein into developing zebrafish, and as our phenotype appears late (3–5 dpf), we were concerned that earlier effects of increased ifng expression could have deleterious effects on earlier developmental processes. Thus, we injected recombinant human IFNγ protein into 2 dpf larvae to determine the effect on hepatobiliary development.
Here we report that injection of IFNγ into developing zebrafish larvae does in fact lead to defects in biliary development, similar to defects seen after injection of azaC. Also similar to azaC-injected larvae, we observe decreased expression of vhnf1 (hnf1b, tcf2), which encodes a homeodomain protein previously shown to be important in biliary development in mammals and zebrafish.7,12 These results support a central role of IFNγ in mediating biliary defects in developing vertebrates, and further support a role for IFNγ in the pathogenesis of disorders such as biliary atresia.
Materials and Methods
Fish lines and injections
Zebrafish were TL strain in all studies, and were housed in the laboratory animal facility at The Children's Hospital of Philadelphia Research Institute, in accordance with protocols approved by the institutional animal care and use committee (IACUC).
For the azaC studies, azaC (1 mg/mL) was injected into 2 dpf larvae, identical to previous studies.4 For the IFNγ injection studies, recombinant human IFNγ was kindly provided by Dr. Jordan Orange (Children's Hospital of Philadelphia Research Institute). The protein was diluted to 40 μg/mL in sterile water and injected into the yolk of 2 dpf larvae, similar to our previous studies injecting morpholino or drug at that stage. As a control, IFNγ protein was inactivated by adding dithiothreitol (0.1 M final concentration) and boiling for 5 min.
Morpholino injections were performed similar to previous studies. The sequence of the crfb17 MOs are shown in Supplementary Table S1 (Supplementary Data are available online at www.liebertpub.com/zeb). Confirmation of knockdown by the splice-blocking crfb17 MO is shown in Supplementary Figure 1. Rescue to demonstrate specificity is shown by the studies of IFNγ and crfb17 MO co-injection (see below).
In situ hybridizations
The in situ hybridization studies were performed essentially as described previously13, in accordance to standard protocols. PCR primers to generate riboprobes for ifng1-1 and ifng1-2 are shown in Supplementary Table S1.
PED-6 and cytokeratin staining
These studies were performed identically to previous studies.4,14,15 Briefly, 5 dpf larvae were given the fluorescent phospholipid PED-6, and gallbladder uptake was assayed 3 h later. Intensity was classified as “normal,” “weak,” or “absent,” and the relative number in each category was scored and compared using chi square analysis. For cytokeratin staining, 5 dpf larvae were fixed in methanol/DMSO, stained with anti-keratin (Ks 18.04) and appropriate secondary. Livers were then examined by confocal microscopy.
Quantitative PCR
For the quantitative PCR (QPCR) studies shown here, livers were isolated from 4 or 5 dpf larvae. Ten livers per condition were pooled in RNAlater®, and RNA extraction and cDNA synthesis were then performed in accordance with standard protocols. Expression QPCR studies were essentially performed as per previous studies, with primers (vhnf1, hprt) also used in previous studies.13 For the methylation-specific QPCR (MSP), in silico analysis of sequence surrounding the transcription start site (−2.5 to +0.5 kb) was performed using MethPrimer (http://www.urogene.org/methprimer/), which identified putative CpG islands and methylated and unmethylated primers. MSP primers are also shown in Supplementary Table S1. In the MSP studies, genomic DNA was isolated from 5 dpf larvae using standard procedures, and then treated with sodium bisulfite using EpiTect (Qiagen). The bisulfite-treated DNA was then used as template for MSP, with detection by SYBR Green using a StepOne Plus. For MSP, primers are designed that target the same region as methylated or unmethylated sequence, taking advantage of bisulfite treatment converting unmethylated cytosine to uracil. Because of the sequence differences, amplification of both the unmethylated and methylated versions can be quantified differentially.
PCNA and apoptosis detection
For proliferating cell nuclear antigen (PCNA) detection, larvae at 5 dpf were fixed in 4% paraformaldehyde. The skin was removed, similar to the larvae prepared for cytokeratin staining, and the larvae were then stained with anti-PCNA antibody (FL-261, Santa Cruz Biotechnologies, Santa Cruz, CA) and appropriate secondary antibody. The larvae were also stained with 2F11 antibody16 and appropriate secondary. The larvae were counterstained with DAPI and examined by confocal microcsopy. Cells in the liver stained with 2F11 and 2F11 plus PCNA were quantified in six samples from both control and IFNγ-injected larvae. Statistical comparisons utilized Student's t-test.
For apoptosis detection, larvae at 5 dpf were again fixed in 4% paraformaldehyde. We used the Roche in situ cell death detection kit with TMR-dUTP as per the manufacturer's instructions. This method involves TUNEL staining to add the labeled dUTP to DNA strand breaks. Larvae were then examined under fluorescence.
Results
Elevation of ifng in azaC-treated larvae
We have reported an increase in IFNγ pathway activity in livers from zebrafish larvae treated with azacytidine (azaC),4 an inhibitor of DNA methylation.17 Because our models were based on inhibition of DNA methylation, we hypothesized that decreased methylation of specific genes within the IFNγ pathway would result in increased expression of these genes, thus leading to activation of the IFNγ pathway. The IFNγ signaling pathway is complex, as there are numerous mechanisms by which IFNγ signaling can be activated.18 While increased expression of numerous genes could thus lead to activation of IFNγ, we initially examined genes explicitly in the IFNγ pathway, such as the ifng, crfb17 (IFNγ receptor), and irf (interferon regulatory factor) genes. In zebrafish, there are two ifng genes and at least 13 irf genes (www.ensembl.org; Zv9); the irf genes are specific to the IFNγ pathway but are also target genes19 and thus would be expected to be elevated in our models regardless of methylation status.
In the azaC-treated larvae, we observed a modest increase in expression of both ifng1-1 and ifng1-2 by quantitative PCR and by in situ hybridization, with increased liver expression (Fig. 1). Expression of crfb17 (IFNγ receptor) did not appear to be increased in the azaC-treated larvae, while expression of the IFNγ target irf1 was increased, as expected (Supplementary Figure S2).
FIG. 1.

Elevation of ifng1-1 and ifng1-2 in azaC-treated larvae. (A, B) Real-time quantitative PCR of ifng1-1 and ifng1-2 in whole 5 dpf azaC-treated larvae, showing a trend towards increase in ifng1-1 expression (A; p=N.S.) and a significant increase in ifng1-2 expression (B; *p≤0.05). (C, D) In situ hybridization of 5 dpf control (C, cont) and azaC-treated (D, azaC) larvae with ifng1-1 showing modestly increased liver (black outline) expression in the azaC-treated larva (D). (E, F) In situ hybridization of 5 dpf control (E, cont) and azaC-treated (F, azaC) larvae with ifng1-2 showing more clearly increased liver (black outline) expression in the azaC-treated larva (F). (G, H) Methylation-specific quantitative PCR showing decreased ratio of methylated to unmethylated ifng1-1 (G) and ifng1-2 (H) target in the azaC-treated larvae (*p≤0.05 for both). Color images available online at www.liebertpub.com/zeb
Interestingly, ifng1-1 and ifng1-2 are only 8.5 kb apart on chromosome 4 and thus may be regulated jointly. In addition to recent studies implicating methylation at the IFNG locus as potentially important in lymphocytes from patients with BA,5 several other studies have demonstrated an importance of promoter methylation in IFNG expression.20–22 To determine whether the upregulation of ifng1-1 and ifng1-2 was due to decreased DNA methylation at the respective loci, we performed methylation-specific qPCR (MSP). We initially performed in silico studies on the region immediately around both transcription start sites (from −2.5 kb to +0.5 kb) to look for CpG islands and to identify methylation-specific primers. As depicted in Figure 1, there was statistically significant relative hypomethylation in the CpG islands neighboring both the ifng1-1 and ifng1-2 genes (p≤0.05 for both), suggesting that decreased DNA methylation may in fact be leading to the increased expression of these genes. These results are consistent with azaC treatment leading to increased expression of ifng genes, and thus we examined whether increased expression of IFNγ was sufficient to cause biliary defects.
Injection of IFNγ leads to biliary defects
To determine whether increased IFNγ activity was sufficient to mediate biliary defects in zebrafish larvae, we injected recombinant IFNγ protein into 2 dpf larvae. Human recombinant IFNγ was injected into the yolk of 2 dpf larvae, similar to our previous studies injecting morpholino antisense oligonucleotide (MO) or drug into the yolk at that stage. We examined biliary function and anatomy of the IFNγ -injected larvae, similar to previous studies. To examine biliary function, we administered the fluorescent phospholipid PED-6 to 5 dpf larvae and assayed gallbladder uptake. PED-6 is swallowed and absorbed through the intestine into the liver, and excreted into the bile, concentrating in the gallbladder. There was diminished gallbladder PED-6 uptake in larvae injected with IFNγ when compared to control larvae (Fig. 2; p≤0.0001). This decreased gallbladder function was attenuated if the IFNγ protein was treated by boiling and denaturing, and was also attenuated by co-injection with a MO directed against the IFNγ receptor-1 gene (crfb17) (Fig. 2; p=N.S. between control vs. inactive IFNγ and for control vs. IFNγ+crfb17 MO), suggesting that the injected IFNγ is biologically functional in the larvae.
FIG. 2.
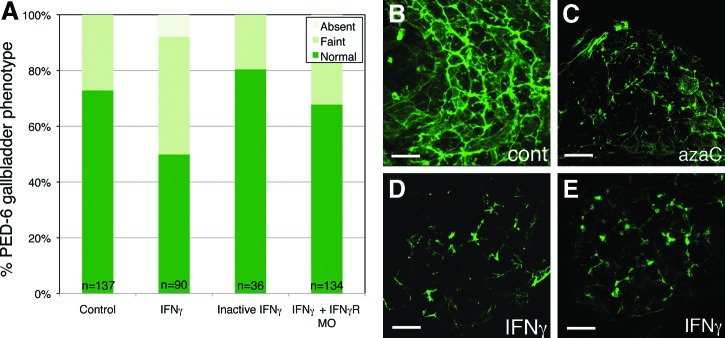
Biliary defects in IFNγ-injected larvae. (A) Gallbladder PED-6 uptake, showing decreased uptake in the IFNγ-injected larvae (p≤0.0001 relative to control by chi square). Uptake in larvae injected with inactivated IFNγ is also shown, as is uptake in larvae injected with IFNγ and morpholino antisense oligonucleotides directed against crfb17 (IFNγ receptor). There is no significant difference in uptake between these conditions and control (p=N.S.). (B–E) Confocal projections of cytokeratin immunostaining in whole-mount 5 dpf control (B), azaC-treated (C), and IFNγ -injected larvae (D, E). Note that the IFNγ -injected liver demonstrates only short, isolated ducts, similar to bile ducts noted in larvae injected in azaC reported previously4 and in (C). Scale bar, 25 μm. Color images available online at www.liebertpub.com/zeb
Similar to previous studies, we examined intrahepatic biliary anatomy by cytokeratin immunostaining. Confocal projections of livers from 5 dpf IFNγ-injected larvae demonstrated severe defects in the intrahepatic bile ducts (Fig. 2), similar to treatment with azaC reported previously. These results demonstrate that IFNγ is sufficient to cause disruption of biliary development in zebrafish, and support an importance of IFNγ in mediating the biliary defects noted after azaC treatment.
Gene expression changes in IFNγ-injected larvae
We have previously reported a downregulation of Hnf6 target genes in azacytidine-treated larvae, which is consistent with the known role of HNF6 in biliary development in zebrafish and mammals.7,23 We examined expression of vhnf1, an Hnf6 target gene, in the IFNγ-injected larvae. There is decreased expression of the Hnf6 target gene in livers isolated from 5 dpf IFNγ-injected larvae when compared to control (Fig. 3). A similar decrease in vhnf1 expression was also seen in livers isolated from 4 dpf IFNγ-injected larvae (data not shown). These results suggest a possible downstream effect of IFNγ on Hnf6 and consequently vhnf1. We were not able to rescue the IFNγ-mediated defects by co-injection with vhnf1 mRNA, however (not shown), suggesting that IFNγ may also affect other critical pathways.
FIG. 3.

Decreased expression of vhnf1 in IFNγ-injected larvae. Real-time quantitative PCR of vhnf1 in control (inactive IFNγ) and IFNγ-injected larvae showing a 40% decrease in vhnf1 expression (*p≤0.05).
Cell proliferation/death in IFNγ-injected larvae
In order to more fully understand the effects of IFNγ injection in mediating biliary defects, we examined cell proliferation and cell death in the IFNγ-injected larvae. We examined proliferation of cholangiocytes by immunostaining using the cholangiocyte marker 2F11 and the proliferation marker PCNA. At 3 dpf and 4 dpf, during early biliary growth, there were fewer cholangiocytes as well as a lower number of cholangiocytes undergoing cell division in the IFNγ-injected larvae (Table 1 and Figure 4). Of note, this effect seemed to be relatively specific to cholangiocytes, as at 3 dpf there was no difference in hepatocyte number or proliferation between control and IFNγ-injected larvae (Table 2). Moreover, at 4 dpf the relative number of cholangiocytes in the IFNγ-injected larvae is lower, which also supports a more specific effect on cholangiocytes (Table 2). By 5 dpf, there continued to be fewer cholangiocytes, but the number of dividing cholangiocytes was indistinguishable between control and the IFNγ-injected larvae (Fig. 4 and Table 1). This is consistent with an effect of IFNγ on cholangiocytes in the developing liver, possibly affecting the initial differentiation, but more clearly affecting proliferation.
Table 1.
Decreased Number of Cholangiocytes in IFNγ-Injected Larvae
| 2F11+ | PCNA+ and 2F11+ | Fraction PCNA/2F11 | |
|---|---|---|---|
| 3 dpf control | 10.3±3.1 | 8.0±2.1 | 0.78±0.09 |
| 3 dpf IFNγ-injected | 5.8±1.5* | 4.5±1.4* | 0.75±0.09 |
| 4 dpf control | 28.7±6.2 | 22.3±3.2 | 0.79±0.09 |
| 4 dpf IFNγ-injected | 14.3±6.2** | 10.5±4.5*** | 0.74±0.07 |
| 5 dpf control | 35.3±10.0 | 9.8±2.8 | 0.28±0.06 |
| 5 dpf IFNγ-injected | 20.3±8.9* | 9.2±3.8 | 0.47±0.12* |
Larvae were stained with PCNA and 2F11, and positive cells were counted from 6 samples in each condition. At 3 and 4 dpf, there are significantly fewer cholangiocytes in the IFNγ-injected larvae, and significantly fewer proliferating cholangiocytes. At 5 dpf, there continue to be fewer cholangiocytes in the IFNγ-injected larvae, but the number of PCNA-positive cells is similar in both conditions. Note that the relative increase in cholangiocytes is similar in both conditions (ie, the number of cholangiocytes at 4 dpf is approximately 3× the number at 3 dpf in both). The fraction of PCNA-positive cells is similar at both 3 and 4 dpf, but there is a higher percentage of PCNA positive cells at 5 dpf. (*p≤0.05; **p≤0.005; ***p≤0.0005, all relative to control for the same age; other comparisons not significant; comparisons by Student's t-test).
FIG. 4.
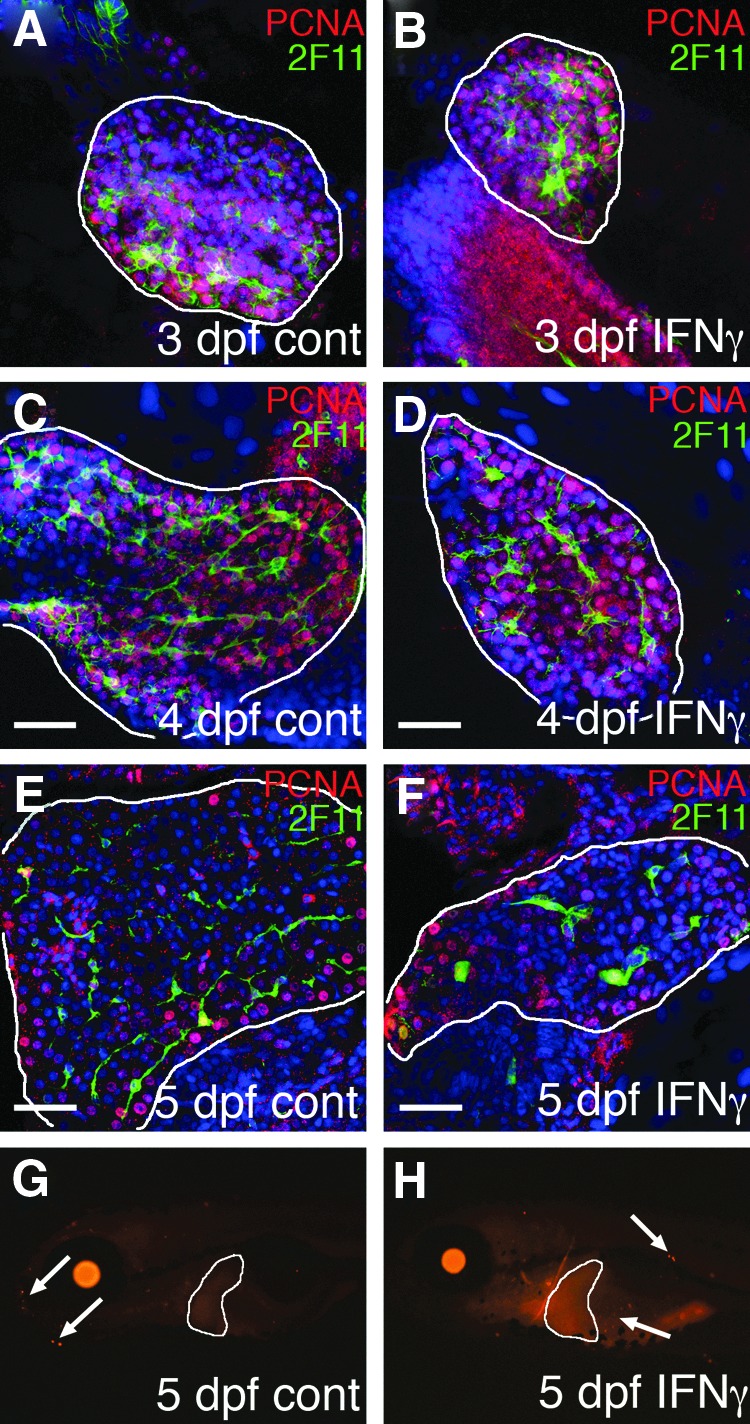
Cell proliferation and cell death in IFNγ-injected larvae. (A–F) Cell proliferation in control (A, C, E) and IFNγ-injected (B, D, F) larvae. Livers (white outline) were stained with anti-PCNA (red) to label proliferating cells and 2F11 (green) to label cholangiocytes, and were counterstained with DAPI. Representative livers from 3 dpf (A, B), 4 dpf (C, D), and 5 dpf (E, F) are shown. Note that there are fewer cholangiocytes in the IFNγ-injected larvae and fewer proliferating cholangiocytes in the IFNγ-injected at 3 dpf and 4 dpf. This is quantified in Table 1. (G, H) TUNEL staining of whole-mount 5 dpf larvae show no positive cells (white arrows) in the liver (white outline) in either control (G) and IFNγ-injected (H) larvae. Scale bar 25 μm for (A–F).
Table 2.
Modest Decrease in Number of Hepatocytes in IFNγ-Injected Larvae
| Non-2F11+ | PCNA+ non-2F11+ | Fraction PCNA/2F11 | Ratio 2F11+/non-2F11+ | |
|---|---|---|---|---|
| 3 dpf control | 202.5±37.4 | 189.8±35.9 | 0.94±0.02 | 0.05±0.02 |
| 3 dpf IFNγ-injected | 172.2±62.0 | 158.5±56.1 | 0.92±0.04 | 0.04±0.01 |
| 4 dpf control | 356.8±36.1 | 276.8±19.6 | 0.78±0.07 | 0.08±0.01 |
| 4 dpf IFNγ-injected | 265.8±48.5** | 199.5±37.3** | 0.75±0.07 | 0.05±0.02* |
In the same 6 larvae shown in Table 1, non-2F11-expressing cells±PCNA were counted. Of note, the total number of cells counted in the livers is the sum of the 2F11+ and non-2F11+ cells. At 3 dpf, there is not a statistically significant difference between non-2F11+ cells (mostly hepatocytes) in control and in IFNγ-injected larvae. At 4 dpf, there is a difference between the number of hepatocytes and proliferating hepatocytes in control and in the IFNγ-injected larvae. The last column shows the ratio of cholangiocytes to noncholangiocytes in the livers, showing that at 4 dpf there are proportionally fewer cholangiocytes in the IFNγ-injected larvae. (*p≤0.05; **p≤0.005, all relative to control for the same age; other comparisons not significant; comparisons by Student's t-test).
We also examined the IFNγ-injected larvae for apoptotic cell death of cholangiocytes in the developing liver. Examination of apoptotic cell death by TUNEL staining did not demonstrate an increase in apoptosis in the IFNγ-injected larvae (Fig. 4). This contrasts with BA and with the mouse model of BA, in which apoptosis appears to be playing a larger role.24
Discussion
Here we have shown that IFNγ is sufficient to elicit biliary defects in developing zebrafish larvae. In addition, in azaC-injected larvae, there is hypomethylation of ifng1-1 and increased liver expression of this gene as well. These findings suggest that biliary defects caused by inhibition of DNA methylation in developing zebrafish larvae are at least partially due to activation of IFNγ signaling. This has implications for diseases such as biliary atresia, in which there are biliary defects and activation of IFNγ, as it suggests that the IFNγ may be directly leading to the defects, at least in part.
Possible mechanisms by which IFNγ may lead to biliary defects
Our results above demonstrate that injection of IFNγ leads to fewer cholangiocytes in the developing zebrafish larvae, seemingly as a result of decreased proliferation, and possibly as a result of decreased differentiation of progenitor cells into cholangiocytes. We also observed decreased expression of vhnf1, a downstream target of HNF6, in a genetic pathway known to be important in biliary development.7,23 While this could be secondary to the decreased number of cholangiocytes, our previous results have demonstrated diffuse expression of vhnf1 in the developing liver,7 suggesting that the decrease in vhnf1 expression is due to globally decreased liver expression and not simply secondary to fewer cholangiocytes in the IFNγ-injected larvae.
Any effect of activation of IFNγ signaling on biliary development would most likely be mediated via the IFNγ receptor, as our results demonstrate that MO-mediated knockdown of the receptor at least partially reverses the defects. Activation of this pathway leads to increased expression of interferon regulatory factors (IRFs), which in turn activate additional downstream genes.19 These secondary effects of IFNγ activation govern several cellular processes, including inhibition of cell growth and proliferation, and increased cell death. Of note, the liver appears to be particularly sensitive to excess IFNγ activation, as mice deficient in the IFNγ inhibitor Socs1 demonstrate liver degeneration,25 and mice overexpressing Ifng in their liver demonstrate chronic hepatitis.26 Thus, our results are consistent with previous studies showing an effect on liver cells. We do see inhibition of cell proliferation in our IFNγ-injected larvae, although our effect does appear more pronounced in cholangiocytes than in hepatocytes. We cannot exclude an additional effect of IFNγ on earlier differentiation of progenitor cells into cholangiocytes, but as there is limited evidence for IFNγ directly inhibiting epithelial cell development in other systems, inhibition of proliferation appears the more likely mechanism.
There may be indirect effects of IFNγ on biliary development, or effects of IFNγ not mediated through IRFs that could lead to an inhibition of cholangiocyte differentiation. As noted, the homeodomain gene vhnf1 has a clear role in biliary development in zebrafish and in mice. The inability of vhnf1 mRNA injection to rescue the defects caused by IFNγ injection suggests that the downregulation of vhnf1 by IFNγ is not sufficient to lead to biliary defects, however. Alternative downstream targets of IFNγ that may have effects on biliary development include members of the GTPase family.19 Although this family of proteins has a wide variety of functions, GTPases play an important role in intracellular trafficking and the establishment of cell polarity—processes clearly important in biliary development and disease pathogenesis.6,9,13,27
IFNγ in biliary atresia
As noted above, IFNγ appears to play a critical role in both the animal model of BA and in patients with BA.2,3 Our studies here support the utility of zebrafish as a model to study BA, as we have demonstrated an importance of IFNγ in mediating biliary defects similar to other zebrafish models resembling BA. Moreover, our results support the critical role for IFNγ in mediating biliary defects, and suggest that the defects in patients may be mediated by IFNγ.
In addition, we have demonstrated that in our zebrafish model with features of BA in which DNA methylation is inhibited, there is upregulation and hypomethylation of ifng genes. Regulation of IFNG and IRF genes by DNA methylation has been reported in several diseases, including BA,5 and other conditions ranging from ulcerative colitis to various cancers.22,28,29 The previous report of IFNG hypomethylation in BA focused on lymphocytes, but our studies suggest the possibility that these genes could be hypomethylated in the liver as well, consistent with our previous report showing decreased DNA methylation in cholangiocytes from BA patients.4 Thus, our results here and elsewhere are consistent with a model in which decreased DNA methylation in cholangiocytes leads to aberrant activation of IFNG and other genes, resulting in biliary defects.
Successful injection of intact protein into larval zebrafish
We have shown that injection of protein into the yolk of 2 dpf larvae leads to an effect on biliary development. This suggests that similar approaches could provide important methods to examine the effects of proteins on development. As noted above, others have recently injected IL-8 into the developing heart of 24 hpf larvae,10 and Musashi1 into early embryos.11 In our studies, we believe that the effect is relatively limited to the liver, as yolk is initially metabolized in the liver and thus the injected IFNγ likely enters the liver readily. Injection of protein then represents a technique useful in modulating developmental processes locally, and could thus be applied to multiple developing tissues in zebrafish embryos and larvae.
Supplementary Material
Acknowledgments
We thank Danielle Belardo for excellent technical assistance, and Megan Ross for preliminary work on this project. We also thank Dr. Jordan Orange and members of his laboratory for the donation of IFNγ protein. This work was supported by funding from the Fred and Suzanne Biesecker Pediatric Liver Center at The Children's Hospital of Philadelphia, and from the NIDDK (R01 DK90260 to R.P.M.).
Disclosure Statement
No competing financial interests exist.
References
- 1.Haber BA. Russo P. Biliary atresia. Gastroenterol Clin North Am. 2003;32:891–911. doi: 10.1016/s0889-8553(03)00049-9. [DOI] [PubMed] [Google Scholar]
- 2.Bezerra JA. Tiao G. Ryckman FC, et al. Genetic induction of proinflammatory immunity in children with biliary atresia. Lancet. 2002;360:1653–1659. doi: 10.1016/S0140-6736(02)11603-5. [DOI] [PubMed] [Google Scholar]
- 3.Shivakumar P. Campbell KM. Sabla GE, et al. Obstruction of extrahepatic bile ducts by lymphocytes is regulated by IFN-gamma in experimental biliary atresia. J Clin Invest. 2004;114:322–329. doi: 10.1172/JCI21153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Matthews RP. Eauclaire SF. Mugnier M, et al. DNA hypomethylation causes bile duct defects in zebrafish and is a distinguishing feature of infantile biliary atresia. Hepatology. 2011;53:905–914. doi: 10.1002/hep.24106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dong R. Zhao R. Zheng S, et al. Abnormal DNA methylation of ITGAL (CD11a) in CD4+ T cells from infants with biliary atresia. Biochem Biophys Res Commun. 2012;417:986–990. doi: 10.1016/j.bbrc.2011.12.054. [DOI] [PubMed] [Google Scholar]
- 6.Sadler KC. Amsterdam A. Soroka C, et al. A genetic screen in zebrafish identifies the mutants vps18, nf2 and foie gras as models of liver disease. Development. 2005;132:3561–3572. doi: 10.1242/dev.01918. [DOI] [PubMed] [Google Scholar]
- 7.Matthews RP. Lorent K. Russo P, et al. The zebrafish onecut gene hnf-6 functions in an evolutionarily conserved genetic pathway that regulates vertebrate biliary development. Dev Biol. 2004;274:245–259. doi: 10.1016/j.ydbio.2004.06.016. [DOI] [PubMed] [Google Scholar]
- 8.Lorent K. Yeo SY. Oda T, et al. Inhibition of Jagged-mediated Notch signaling disrupts zebrafish biliary development and generates multi-organ defects compatible with an Alagille syndrome phenocopy. Development. 2004;131:5753–57. doi: 10.1242/dev.01411. -66. [DOI] [PubMed] [Google Scholar]
- 9.Matthews RP. Plumb-Rudewiez N. Lorent K, et al. Zebrafish vps33b, an ortholog of the gene responsible for human arthrogryposis-renal dysfunction-cholestasis syndrome, regulates biliary development downstream of the onecut transcription factor hnf6. Development. 2005;132:5295–5306. doi: 10.1242/dev.02140. [DOI] [PubMed] [Google Scholar]
- 10.Stoll SJ. Bartsch S. Augustin HG, et al. The transcription factor HOXC9 regulates endothelial cell quiescence and vascular morphogenesis in zebrafish via inhibition of interleukin 8. Circ Res. 2011;108:1367–1377. doi: 10.1161/CIRCRESAHA.111.244095. [DOI] [PubMed] [Google Scholar]
- 11.Shibata S. Umei M. Kawahara H, et al. Characterization of the RNA-binding protein Musashi1 in zebrafish. Brain Res. 2012;1462:162–173. doi: 10.1016/j.brainres.2012.01.068. [DOI] [PubMed] [Google Scholar]
- 12.Coffinier C. Gresh L. Fiette L, et al. Bile system morphogenesis defects and liver dysfunction upon targeted deletion of HNF1beta. Development. 2002;129:1829–1838. doi: 10.1242/dev.129.8.1829. [DOI] [PubMed] [Google Scholar]
- 13.Eauclaire SF. Cui S. Ma L, et al. Mutations in vacuolar H(+)-ATPase subunits lead to biliary developmental defects in zebrafish. Dev Biol. 2012;365:434–444. doi: 10.1016/j.ydbio.2012.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cui S. Capecci LM. Matthews RP. Disruption of planar cell polarity activity leads to developmental biliary defects. Dev Biol. 2011;351:229–241. doi: 10.1016/j.ydbio.2010.12.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cui S. Erlichman J. Russo P, et al. Intrahepatic biliary anomalies in a patient with Mowat-Wilson syndrome uncover a role for the zinc finger homeobox gene zfhx1b in vertebrate biliary development. J Pediatr Gastroenterol Nutr. 2011;52:339–344. doi: 10.1097/MPG.0b013e3181ff2e5b. [DOI] [PubMed] [Google Scholar]
- 16.Crosnier C. Vargesson N. Gschmeissner S, et al. Delta-Notch signalling controls commitment to a secretory fate in the zebrafish intestine. Development. 2005;132:1093–1104. doi: 10.1242/dev.01644. [DOI] [PubMed] [Google Scholar]
- 17.Jones PA. Taylor SM. Cellular differentiation, cytidine analogs and DNA methylation. Cell. 1980;20:85–93. doi: 10.1016/0092-8674(80)90237-8. [DOI] [PubMed] [Google Scholar]
- 18.Schoenborn JR. Wilson CB. Regulation of interferon-gamma during innate and adaptive immune responses. Adv Immunol. 2007;96:41–101. doi: 10.1016/S0065-2776(07)96002-2. [DOI] [PubMed] [Google Scholar]
- 19.Saha B. Jyothi Prasanna S. Chandrasekar B, et al. Gene modulation and immunoregulatory roles of interferon gamma. Cytokine. 2010;50:1–14. doi: 10.1016/j.cyto.2009.11.021. [DOI] [PubMed] [Google Scholar]
- 20.Zhang S. Crivello A. Offenbacher S, et al. Interferon-gamma promoter hypomethylation and increased expression in chronic periodontitis. J Clin Periodontol. 2010;37:953–961. doi: 10.1111/j.1600-051X.2010.01616.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gonsky R. Deem RL. Targan SR. Distinct methylation of IFNG in the gut. J Interferon Cytokine Res. 2009;29:407–414. doi: 10.1089/jir.2008.0109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gonsky R. Deem RL. Landers CJ, et al. Distinct IFNG methylation in a subset of ulcerative colitis patients based on reactivity to microbial antigens. Inflamm Bowel Dis. 2011;17:171–178. doi: 10.1002/ibd.21352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Clotman F. Lannoy VJ. Reber M, et al. The onecut transcription factor HNF6 is required for normal development of the biliary tract. Development. 2002;129:1819–1828. doi: 10.1242/dev.129.8.1819. [DOI] [PubMed] [Google Scholar]
- 24.Erickson N. Mohanty SK. Shivakumar P, et al. Temporal-spatial activation of apoptosis and epithelial injury in murine experimental biliary atresia. Hepatology. 2008;47:1567–1577. doi: 10.1002/hep.22229. [DOI] [PubMed] [Google Scholar]
- 25.Alexander WS. Starr R. Fenner JE, et al. SOCS1 is a critical inhibitor of interferon gamma signaling and prevents the potentially fatal neonatal actions of this cytokine. Cell. 1999;98:597–608. doi: 10.1016/s0092-8674(00)80047-1. [DOI] [PubMed] [Google Scholar]
- 26.Toyonaga T. Hino O. Sugai S, et al. Chronic active hepatitis in transgenic mice expressing interferon-gamma in the liver. Proc Natl Acad Sci USA. 1994;91:614–618. doi: 10.1073/pnas.91.2.614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gissen P. Johnson CA. Morgan NV, et al. Mutations in VPS33B, encoding a regulator of SNARE-dependent membrane fusion, cause arthrogryposis-renal dysfunction-cholestasis (ARC) syndrome. Nat Genet. 2004;36:400–404. doi: 10.1038/ng1325. [DOI] [PubMed] [Google Scholar]
- 28.Yamashita M. Toyota M. Suzuki H, et al. DNA methylation of interferon regulatory factors in gastric cancer and noncancerous gastric mucosae. Cancer Sci. 2010;101:1708–1716. doi: 10.1111/j.1349-7006.2010.01581.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tshuikina M. Jernberg-Wiklund H. Nilsson K, et al. Epigenetic silencing of the interferon regulatory factor ICSBP/IRF8 in human multiple myeloma. Exp Hematol. 2008;36:1673–1681. doi: 10.1016/j.exphem.2008.08.001. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.