

Abstract
This study has investigated a potential role of common Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) gene variants in the etiology of noncystic fibrosis bronchiectasis in Serbian children. The study has encompassed 48 patients (19 male and 29 female, aged between 5 and 18 years, median age 10.6±3.3), diagnosed with idiopathic bronchiectasis based on high-resolution computed tomography of thorax and pathologic examination of lobectomy materials. The CFTR gene analysis was performed on genomic DNA extracted from peripheral blood samples of patients by polymerase chain reaction (PCR)-Mediated Site-Directed Mutagenesis method, Denaturing Gradient Gel Electrophoresis method, and DNA sequencing. Mutation c.1521_1523delCTT (F508del) was detected with an allelic frequency of 1.0%, and c.224G>A (R75Q) variant. Carriers of c.1210-12T[5] (IVS8-5T) allele were significantly more common than in the general population (10.4% vs. 5.0%, P=0.0302). The frequency of homozygotes for Met 470 allele was higher in patients than in the general population (33% vs. 20%), while heterozygotes for p.Met470Val were less frequent (31% vs. 50%), and this difference was statistically significant (P=0.0222). The results obtained in this study indicate involvement of 2 common CFTR variants, c.1210-12T[5] and c.1408A, in idiopathic bronchiectasis in children, but this observation should be further confirmed by more extensive analysis of the CFTR gene in a larger group of patients.
Introduction
Bronchiectasis is a lung disease in which deregulated inflammatory response and recurrent bacterial infection result in a progressive lung damage and irreversible dilatation of bronchi. The estimates of disease incidence vary between 1/10,000 and 100/10,000.1 The true prevalence of bronchiectasis is most likely underestimated, as less severe forms of bronchiectasis have been documented with the increased use of high-resolution computed tomography (HRCT). The presentation of this condition has significantly changed since the introduction of antibiotics, while with the advent of HRCT technique diagnostics became much easier. Despite that, bronchiectasis continues to be an important cause of chronic respiratory disease and is increasingly recognized as a major cause of respiratory morbidity, especially in developing countries.
Among many known causes of bronchiectasis, the most common are mechanical bronchial obstruction, postinfectious bronchial damage (tuberculosis and allergic bronchopulmonary aspergillosis), abnormal host defense (ciliary dyskinesia and hypogammaglobulinemia), genetic defects (cystic fibrosis and alpha-1-antitrypsin deficiency), and autoimmune disease (systemic lupus erythematosus, rheumatoid arthritis, and bulcerative colitis).2 However, underlying cause of bronchiectasis is found in less than 50% of patients, while in others it remains unknown, in spite of extensive investigations.2 Idiopathic bronchiectasis is currently considered to be a multifactorial disorder, whose pathogenesis is influenced by environmental factors and probably several undetermined genes.
Genetic variations in the Cystic Fibrosis Transmembrane Conductance Regulator (CFTR) gene are studied as a potential cause of idiopathic bronchiectasis due to the importance of the role played by the CFTR protein in the lung.3,4 The CFTR protein is a cAMP-activated chloride channel, expressed in the apical membrane of ciliated surface cells and gland ducts. The permeability of CFTR to HCO3−, the relative acidity of the gland secretions pH in cystic fibrosis, and the apical localization of the protein support the notion that CFTR activity alkalinizes airway surface liquid (ASL) pH.5 Available evidence indicates that the defects in the CFTR protein affect physiological functions of both surface and submucosal gland epithelia.6 Defective CFTR function at the airway surface can lead to impaired regulation of ASL volume or composition and, therefore, to impaired mucociliary clearance. In submucosal glands, CFTR defects can lead to defects in water and salt secretion, compromising the clearance of mucins and a variety of defense substances onto the airway surface. Impaired mucociliary clearance, together with CFTR-related changes in the airway surface microenvironment, leads to a progressive cycle of infection, inflammation, and declining lung function. It is known that the CFTR protein may be defective in a variety of diseases affecting lung, but its role in the etiology of bronchiectasis remains unclear, considering controversial findings published so far.7–12
This study has investigated a potential role of CFTR gene variants in the etiology of idiopathic bronchiectasis in Serbian children and is based on the hypothesis that CFTR gene mutations may play a role in the development of noncystic fibrosis bronchiectasis. Previous studies have indicated that a unique CFTR mutation may have pathogenic consequences in patients with bronchiectasis and it could therefore be expected that CFTR gene mutation heterozygotes will be more common among idiopathic bronchiectasis patients.13,14
Materials and Methods
Patients
The study has encompassed 48 children (19 male and 29 female) with idiopathic bronchiectasis who were referred to the Department of Pulmonology and Allergology of the University Children's Hospital during the period 2008–2010. Informed consent was obtained from all patients' parents and the study was approved by the Ethical Committee of the Medical Faculty, University of Belgrade. Children of all ages who were diagnosed with noncystic fibrosis bronchiectasis were prospectively recruited for this study. Bronchiectasis was diagnosed by HRCT of thorax in all patients and was confirmed on pathologic exemination of lobectomy materials in patients who underwent surgical removal of lung due to hemorrhage. Other known causes of bronchiectasis were ruled out in every patient. Immunodeficiency disorders were excluded based on total immunoglobulins and T lymphocyte phenotype. Ciliary dyskinesia was excluded based on electron microscopy of bronchial brushing cytology specimens collected during flexible bronchoscopy. Tuberculosis was excluded based on negative purified protein derivative (PPD) skin test and negative polymerase chain reaction (PCR) for Mycobacterium tuberculosis in bronchoalveolar lavage (BAL). Spirometry was performed in all patients. Lower respiratory tract infection was defined as increased cough and sputum production and/or any change in chest X-ray.
Controls
Considering that no age-matched control group was available for this study, the data previously published for general population were used instead. The following estimated frequencies based on previously published data for general population were used: 0.05 for c.1210-12T[5] carriers, 0.04 for CFTR mutation carriers, and 0.55 for c.1408G (470 Val allele).15–17 The suitability of the used data was confirmed by the comparison to the group of previously analyzed healthy individuals from Serbia, which did not significantly differ from the data published for the general population.18 This previously studied group has encompassed 102 healthy unrelated subjects from the same geographic region as the patients encompassed by this study. Their mean age was 50.5±13.8 years and 36.3% were male. All control subjects showed normal pulmonary function on spirometry (FEV1/FVC >70% predicted and FEV1 >80% predicted).
Genetic analysis
Genomic DNA was isolated from peripheral blood samples, taken with sodium citrate as anticoagulant, using commercially available GFX Genomic Blood DNA Purification Kit (GE Healthcare).
The detection of mutation c.1521_1523delCTT and the analysis of c.1210-12T[5–9] polymorphism were performed by previously described protocols based on PCR-Mediated Site-Directed Mutagenesis method.19,20
The screening of the exons 3, 10, and 11 of the CFTR gene for the presence of genetic variations was performed using Denaturing Gradient Gel Electrophoresis (DGGE) method. The fragments encompassing the exons and flanking sequences were amplified in PCR reactions and analyzed by DGGE according to the previously published protocol.21 Amplified fragments of CFTR exons 3, 10, and 11 were analyzed on the DGGE gels with concentration of denaturants 10%–60%. The electrophoresis was performed for 4 h at 230 V, followed by silver staining of the DGGE gels.22
The automated DNA sequencing was performed for all samples showing aberrant band patterns on DGGE gels. The fragments for sequencing were amplified using the primers CTTGGGTTAATCTCCTGGGA and ATTCACCAGATTTCGTAGTC for exon 3. The PCR was conducted in a 100μL reaction mixture containing: 15 mM Tris-HCl pH 8.0, 50 mM KCl, 2.5 mM MgCl2, 0.2 mM each dNTP, 20 pmol of each primer, 5 U of Taq polymerase (FIREPol; Solis BioDyne), and approximately 500 ng of DNA. The amplifications were performed as follows: initial denaturation for 5 min at 94°C; 30 cycles consisting of 1 min at 94°C, 1 min at 55°C, and 1 min at 72°C; and final extension for 10 min at 72°C. Before use in sequencing reactions, PCR products were purified by QIAquick PCR Purification Kit (QIAGEN). The sequencing reactions were performed in both forward and reverse direction, with the same primers as for the amplification, using the ABI PRISM Big Dye Terminator system (Applied Biosystems). Sequences were analyzed using the Sequencing Analysis software (Applied Biosystems).
Statistical analysis
Descriptive statistics are presented as mean values with standard deviations for continuous variables. Categorical data are presented by absolute numbers with percentages. The obtained allele and genotype frequencies were compared to the frequencies in general population and analyzed using the Chi-square test.15–18 In all tests, P value<0.05 was considered to be statistically significant. Considering that 4 consecutive statistical analyses are performed, a corrected level of significance (alpha adjustment) of 0.0125 (0.05/4) according to modified Bonferroni procedure was also discussed. Statistical analysis was performed using SPSS statistical software (SPSS for Windows, release 17.0, SPSS).
Results
This study has encompassed a group of 48 children (19 male and 29 female) between 5 and 18 years of age (median 10.6±3.3 years) diagnosed with idiopathic (noncystic fibrosis) bronchiectasis. All patients had normal sweat chloride values (<40 mEq/L), while only 3 patients had sweat test values near borderline (30–40 mmol/L). Baseline demographic and clinical characteristics of the patients are given in Table 1. Bronchiectasis was diagnosed by HRCT of thorax in all patients. Findings on HRCT were similar in all patients and a representative CT scan is shown in Fig. 1. Sputum cultures were obtained from all patients and 97.9% of children tested positive. The most common was colonization by Streptococcus pneumoniae, Pseudomonas aeruginosa, Haemophilus influenzae, Moraxella catarrhalis, and Staphylococcus aureus.
Table 1.
Baseline Demographic and Clinical Characteristics of Patients
| Gender, n (%) | |
| Male | 19 (39.6) |
| Female | 29 (60.4) |
| Age | |
| Age on diagnosis, mean±SD (years) | 10.6±3.3 |
| Age at disease onset, mean±SD (years) | 9.7±3.2 |
| Spirometry | |
| FEV1, mean±SD (%) | 85.0±14.5 |
| FVC, mean±SD (%) | 80.0±14.5 |
| Sweat test (mEq/L) | 12.5±8.2 |
| Microorganisms isolated from sputum, n (%) | |
| Normal flora | 1 (2.1) |
| Pseudomonas | 10 (20.8) |
| Streptococcus pneumoniae | 14 (29.2) |
| Moraxella catarrhalis | 5 (10.4) |
| Haemophilus influenzae | 10 (20.8) |
| Staphylococcus aureus | 4 (8.3) |
| Acinetobacter | 1 (2.1) |
| Burkholderia cepacia | 1 (2.1) |
| Acinetobacter, Burkholderia cepacia | 1 (2.1) |
| Staphylococcus (coagulase-negative) | 1 (2.1) |
SD, standard deviation; FEV1, forced expiratory volume in 1 s; FVC, forced vital capacity.
FIG. 1.
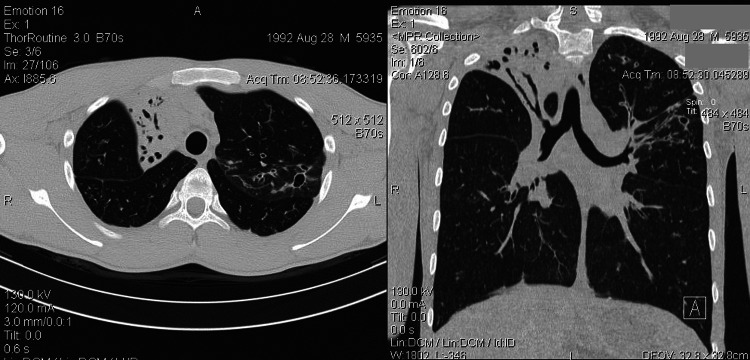
A representative computed tomography (CT) scan of a patient with idiopathic bronchiectasis. A, anterior; R, right; L, left; S, superior.
The presence of the mutation c.1521_1523delCTT, analysis of common c.1210-12T[5–9] polymorphism and screening of exons 3, 10, and 11 in the CFTR gene were performed in 48 children diagnosed with idiopathic disseminated bronchiectasis. The obtained allele frequencies are shown in Table 2, while the obtained genotype frequencies are shown in Table 3.
Table 2.
CFTR Alleles Detected in Children with Idiopathic Bronchiectasis
| Variant | Alleles | Number | % |
|---|---|---|---|
| F508del | Mutant | 1 | 1.0 |
| Wild type | 95 | 99.0 | |
| R75Q | Mutant | 1 | 1.0 |
| Wild type | 95 | 99.0 | |
| Tn polymorphism | IVS8-5T | 5 | 5.2 |
| IVS8-7T | 81 | 84.4 | |
| IVS8-9T | 10 | 10.4 | |
| M470V | M470 | 47 | 49.0 |
| 470V | 49 | 51.0 |
CFTR, cystic fibrosis transmembrane conductance regulator.
Table 3.
CFTR Genotypes Detected in Children with Idiopathic Bronchiectasis
| Genotype | Number of patients | % | ||
|---|---|---|---|---|
| F508del | 7T/9T | M/V | 1 | 2.1 |
| R75Q | 5T/7T | M/V | 1 | 2.1 |
| — | 5T/7T | M/V | 2 | 4.2 |
| — | 5T/9T | V/V | 1 | 2.1 |
| — | 5T/9T | M/M | 1 | 2.1 |
| — | 7T/9T | M/M | 4 | 8.3 |
| — | 7T/9T | M/V | 2 | 4.2 |
| — | 7T/9T | V/V | 1 | 2.1 |
| — | 7T/7T | M/M | 11 | 22.9 |
| — | 7T/7T | M/V | 11 | 22.9 |
| — | 7T/7T | V/V | 13 | 27.0 |
The c.1521_1523delCTT mutation was detected in one patient, with allelic frequency of 1.0%. The screening of CFTR exon 3 showed the presence of the change in one patient, which was characterized by sequencing as c.224G>A. The CFTR mutation frequency in the group of patients did not significantly differ from the general population.
The c.1210-12T[5] variant was detected with relatively high genotype frequency in comparison to general population (10.4% vs. 5.0%).15,18 The frequency of genotypes c.1210-12T[5]+[7] and c.1210-12T[5]+[9] was statistically significantly higher in patients than in the general population (P=0.0302). However, after the correction for multiple comparisons statistical significance is lost.
The screening of CFTR exon 10 showed only the presence of the common polymorphism c.1408A>G (p.Met470Val). The c.1408A>G genotype was determined based on comparison of the DGGE exon 10 band pattern with samples previously characterized by sequencing. The alleles c.1408A and c.1408G of the c.1408A>G polymorphism were detected with the frequencies of 49.0% and 51.0%, respectively, which did not significantly differ from the general population frequencies. However, there was statistically significant difference in the distribution of Met470Val genotypes between the patients group and the general population (P=0.0222). The genotype Met/Met was more frequent in patients than in the general population (33% vs. 20%), while genotype Val/Val was less frequent (31% vs. 50%), but after the alpha adjustment for multiple comparisons the above-mentioned statistical significance is lost.16–18
Discussion
Several studies in the past decade have been aimed at revealing the genetic basis of bronchiectasis, but the existing data remain controversial. This study was conducted on a group of pediatric patients with idiopathic bronchiectasis and was aimed to correlate the presence of some common CFTR gene variants with the development of the disease. It was presumed that CFTR genetic variations should be most strongly associated with the disease development in a group of children, in which all other known causes and risk factors for bronchiectasis were excluded. The study was conducted on a well-defined group of children in whom diagnosis of bronchiectasis was confirmed by HRCT of thorax, considered the gold standard technique for diagnosis of this disease. Bronchiectasis was confirmed to be idiopathic by ruling out all known causes in each child. None of the children included in the study were suspected with cystic fibrosis, since no clinical evidence of CF was observed. Sweat test, considered the gold standard for the diagnosis of CF, was negative in all subjects (<40 mM), while only 3 patients had sweat test values near borderline (30–40 mmol/L). Previous studies indicated that atypical forms of the disease, with 2 CF disease-causing mutations, are associated with sweat test results above 30 mM.23,24 Previous studies have also shown that heterozygosity for CFTR mutations is more common in non-CF bronchiectasis than in healthy individuals, indicating that CFTR function may be defective even in those individuals with negative sweat test and one normal copy of the CFTR gene.13,25–27
All selected patients were subjected to genetic analysis to detect the presence of CFTR gene variants. In addition to the detection of the most common mutation, c.1521_1523delCTT, and analysis alleles of c.1210-12T[5–9] polymorphic locus, exons 3, 10, and 11 of the CFTR gene were screened for the presence of any genetic variations, considering that mutations in these exons were previously found to be commonly associated with pulmonary disorders in Serbia.12 Considering that no control group was available for this study, which is its main limitation, the data previously published for general population were used instead. The rationale for using such data relies on the fact that they are based on studies that have encompassed the screening of the entire CFTR gene in a relatively large sample mainly from Mediterranean area populations known to have similar genetic background and similar distribution of CFTR gene mutations as Serbia.12,16,18 The distribution of the CFTR genetic variations analyzed in this study in the general population is not significantly different from their distribution in Serbian general population, confirmed by a previous study that has encompassed a relatively small number of healthy individuals.16,18
The CFTR mutation frequency in the group of patients did not significantly differ from the general population. The c.1521_1523delCTT mutation, detected in this study in one patient, is the most common mutation causing cystic fibrosis and is also found to be a risk factor in heterozygous state for the development of lung disease.28 The patient in whom c.224G>A variant was detected was also shown to be a carrier of c.1210-12T[5] variant. The c.224G>A variant is polymorphic at amino acid level, but appears to have channel activity not different from the wild-type CFTR.29 The c.224A allele was found to be significantly more frequent in patients with mild pulmonary manifestations, such as atypical cystic fibrosis.30 The increased frequency of this variant was also observed in Serbian patients with chronic obstructive pulmonary disease.31
The alleles of Tn polymorphic locus are not considered to be disease-causing mutations, but they affect CFTR mRNA splicing and can be associated with some mild clinical presentations. The c.1210-12T[7] and c.1210-12T[9] alleles are considered normal, since they generate sufficient amount of functional CFTR mRNA to prevent the development of the disease. The c.1210-12T[5] allele generates low levels of functional CFTR mRNA, due to which it has been considered a mild mutation rather than a polymorphism. In this study, the c.1210-12T[5] variant was detected with the allelic frequency of 10.4%, and carriers of this variant were statistically significantly overrepresented in patients than in the general population (χ2=4.696, P=0.0302), suggesting possible involvement of this variant in the etiology of idiopathic bronchiectasis.32 Our results are in concordance with the hypothesis that c.1210-12T[5] variant may be associated with obstructive pulmonary disorders, but this finding should be confirmed on a larger study sample.8
The c.1408A>G variant is also polymorphic at amino acid level and there is a difference in maturation and intrinsic chloride channel activity between the 2 alleles. The p.470Met variant of the CFTR protein matures more slowly and has 1.7-fold increased channel activity compared with p.470Val protein.33 Although p.470Met and p.470Val alleles differ functionally, there have been no reports that they alone could be associated with certain clinical conditions. However, it is possible that they may be of importance for mild clinical presentations if present in combination with some other sequence variants.34 The frequency of MM genotype was statistically significantly higher in the patients group than in the general population, while heterozygotes for c.1408A>G variant were less frequent (χ2=7.618, P=0.0222). Since in adult patients with bronchiectasis who carry CFTR mutations a very high frequency of Met/Met genotype was observed (90%), the increased frequency of Met/Met genotype in this study may rather indicate the presence of CFTR mutations that were not detected due to study limitations, than the association of c.1408A>G variant with the disease.25 Considering variability of CFTR mutations detected in patients with bronchiectasis, future studies should encompass the screening of the entire CFTR gene.
The analysis of polymorphic TG dinucleotide repeat adjacent to the c.1210-12T[5] variant would also be of importance for interpretation of the obtained results, since the clinical presentations of the 5T variant are associated with the 5T-12TG-470M haplotype.35,36 Such analysis was not possible in this study, considering that most children's parents were not available for analysis and no haplotype data could have been obtained. A possibility remains that study subjects are carriers of undetected mutations in the CFTR gene, which could significantly influence CFTR function and could also significantly contribute to airways functional abnormalities in these patients.
The findings of this study correlate with data published for adult bronchiectasis patients, which report generally high incidence of c.1210-12T[5] and c.1408A CFTR variants.13,25–27 These results can only be considered preliminary, considering lack of control group, relatively small group of patients, and loss of statistical significance after the correction for multiple comparisons. Also, the data on CFTR mutations in pediatric population with idiopathic bronchiectasis are virtually nonexistent and it is difficult to estimate the significance of the obtained results. Studies conducted in adults with bronchiectasis suggested that heterozygosity for CFTR mutations most likely has pathogenic consequences, contributing to the development of bronchiectasis, probably with other genetic and epigenetic associated factors.13,25–27
The results obtained in this study indicate involvement of 2 common CFTR variants, c.1210-12T[5] and c.1408A, in idiopathic bronchiectasis in children. However, analysis of larger sample size, more extensive analysis of the CFTR gene, and replication of findings in different populations are necessary to further investigate the role of these variants and confirm the observations of this study. More detailed and extensive studies should be conducted in children with idiopathic bronchiectasis in to evaluate the importance of CFTR mutations heterozygosity and investigate the role of CFTR in this disease.
Acknowledgment
This work was supported by the grant 173008 of the Ministry of Education and Science of Serbia.
Author Disclosure Statement
No competing financial interests exist.
References
- 1.Callahan CW. Redding GJ. Bronchiectasis in children: orphan disease or persistent problem? Pediatr Pulmonol. 2002;33:492–496. doi: 10.1002/ppul.10104. [DOI] [PubMed] [Google Scholar]
- 2.Cohen M. Sahn SA. Bronchiectasis in systemic disease. Chest. 1999;116:1063–1074. doi: 10.1378/chest.116.4.1063. [DOI] [PubMed] [Google Scholar]
- 3.Zielenski J. Genotype and phenotype in cystic fibrosis. Respiration. 2000;67:117–133. doi: 10.1159/000029497. [DOI] [PubMed] [Google Scholar]
- 4.Rowntree RK. Harris A. The phenotypic consequences of CFTR mutations. Ann Hum Genet. 2003;67(Pt 5):471–485. doi: 10.1046/j.1469-1809.2003.00028.x. [DOI] [PubMed] [Google Scholar]
- 5.Fischer H. Widdicombe JH. Mechanisms of acid and base secretion by the airway epithelium. J Membr Biol. 2006;211:139–150. doi: 10.1007/s00232-006-0861-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pilewski JM. Frizzell RA. Role of CFTR in airway disease. Physiol Rev. 1999;79(1 Suppl):S215–S255. doi: 10.1152/physrev.1999.79.1.S215. [DOI] [PubMed] [Google Scholar]
- 7.Luisetti M. Pignatti PF. Genetics of idiopathic disseminated bronchiectasis. Semin Respir Crit Care Med. 2002;24:179–184. doi: 10.1055/s-2003-39028. [DOI] [PubMed] [Google Scholar]
- 8.Pignatti PF. Bombieri C. Benetazzo M. Casartelli A. Trabetti E. Gile LS. Martinati LC. Boner AL. Luisetti M. CFTR gene variant IVS8-5T in disseminated bronchiectasis. Am J Hum Genet. 1996;58:889–892. [PMC free article] [PubMed] [Google Scholar]
- 9.Girodon E. Cazeneuve C. Lebargy F. Chinet T. Costes B. Ghanem N. Martin J. Lemay S. Scheid P. Housset B. Bignon J. Goossens M. CFTR gene mutations in adults with disseminated bronchiectasis. Eur J Hum Genet. 1997;5:149–155. [PubMed] [Google Scholar]
- 10.Bombieri C. Benetazzo M. Saccomani A. Belpinati F. Gilè LS. Luisetti M. Pignatti PF. Complete mutational screening of the CFTR gene in 120 patients with pulmonary disease. Hum Genet. 1998;103:718–722. doi: 10.1007/s004390050897. [DOI] [PubMed] [Google Scholar]
- 11.King PT. Freezer NJ. Holmes PW. Holdsworth SR. Forshaw K. Sart DD. Role of CFTR mutations in adult bronchiectasis. Thorax. 2004;59:357–358. doi: 10.1136/thx.2003.020263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Divac A. Nikolic A. Mitic-Milikic M. Nagorni-Obradovic L. Petrovic-Stanojevic N. Dopudja-Pantic V. Nadaskic R. Savic A. Radojkovic D. CFTR mutations and polymorphisms in adults with disseminated bronchiectasis: a controversial issue. Thorax. 2005;60:85. [PMC free article] [PubMed] [Google Scholar]
- 13.Bienvenu T. Sermet-Gaudelus I. Burgel PR. Hubert D. Crestani B. Bassinet L. Dusser D. Fajac I. Cystic fibrosis transmembrane conductance regulator channel dysfunction in non-cystic fibrosis bronchiectasis. Am J Respir Crit Care Med. 2010;181:1078–1084. doi: 10.1164/rccm.200909-1434OC. [DOI] [PubMed] [Google Scholar]
- 14.Sermet-Gaudelus I. Dechaux M. Vallee B. Fajac A. Girodon E. Nguyen-Khoa T. Marianovski R. Hurbain I. Bresson JL. Lenoir G, et al. Chloride transport in nasal ciliated cells of cystic fibrosis heterozygotes. Am J Respir Crit Care Med. 2005;171:1026–1031. doi: 10.1164/rccm.200406-740OC. [DOI] [PubMed] [Google Scholar]
- 15.Groman JD. Hefferon TW. Casals T. Bassas L. Estivill X. Des Georges M, et al. Variation in a repeat sequence determines whether a common variant of the cystic fibrosis transmembrane conductance regulator gene is pathogenic or benign. Am J Hum Genet. 2004;74:176–179. doi: 10.1086/381001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bombieri C. Giorgi S. Carles S, et al. A new approach for identifying non-pathogenic mutations: an analysis of the cystic fibrosis transmembrane regulator gene in normal individuals. Hum Genet. 2000;106:172–178. doi: 10.1007/s004390051025. [DOI] [PubMed] [Google Scholar]
- 17.Cuppens H. Marynen P. de Boeck C. Cassiman JJ. Detection of 98.5% of the mutations in 200 Belgian cystic fibrosis alleles by reverse dot-blot and sequencing of the complete coding region and exon=intron junctions of the CFTR gene. Genomics. 1993;18:693–697. doi: 10.1016/s0888-7543(05)80376-3. [DOI] [PubMed] [Google Scholar]
- 18.Stankovic M. Nikolic A. Divac A. Tomovic A. Petrovic-Stanojevic N. Andjelic M. Dopudja-Pantic V. Surlan M. Vujicic I. Ponomarev D. Mitic-Milikic M. Kusic J. Radojkovic D. The CFTR M470V gene variant as a potential modifier of COPD severity: study of Serbian population. Genet Test. 2008;12:357–362. doi: 10.1089/gte.2007.0069. [DOI] [PubMed] [Google Scholar]
- 19.Friedman KJ. Highsmith E. Silverman LM. Detecting multiple cystic fibrosis mutations by polymerase chain reaction-mediated site-directed mutagenesis. Clin Chem. 1991;37:753. [PubMed] [Google Scholar]
- 20.Shrimpton A. R117H and IVS8-5T cystic fibrosis mutation detection by restriction enzyme digestion. Mol Diagn. 2000;5:235. doi: 10.1054/modi.2000.9730. [DOI] [PubMed] [Google Scholar]
- 21.Fanen P. Ghanem N. Vidaud M. Besmond C. Martin J. Costes B. Plassa F. Goossens M. Molecular characterization of cystic fibrosis: 16 novel mutations identified by analysis of the whole cystic fibrosis conductance transmembrane regulator (CFTR) coding regions and splice site junctions. Genomics. 1992;13:770–776. doi: 10.1016/0888-7543(92)90152-i. [DOI] [PubMed] [Google Scholar]
- 22.Radojkovic D. Kusic J. Silver staining of denaturing gradient gel electrophoresis gels. Clin Chem. 2000;46(6 Pt 1):883–884. [PubMed] [Google Scholar]
- 23.Padoan R. Bassotti A. Seia M. Corbetta C. Negative sweat test in hypertrypsinaemic infants with cystic fibrosis carrying rare CFTR mutations. Eur J Pediatr. 2002;161:212–215. doi: 10.1007/s00431-001-0910-8. [DOI] [PubMed] [Google Scholar]
- 24.Castellani C. Benetazzo MG. Tamanini A. Begnini A. Mastella G. Pignatti P. Analysis of the entire coding region of the cystic fibrosis transmembrane regulator gene in neonatal hypertrypsinaemia with normal sweat test. J Med Genet. 2001;38:202–205. doi: 10.1136/jmg.38.3.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Casals T. De-Gracia J. Gallego M. Dorca J. Rodríguez-Sanchón B. Ramos MD. Giménez J. Cisteró-Bahima A. Olveira C. Estivill X. Bronchiectasis in adult patients: an expression of heterozygosity for CFTR gene mutations? Clin Genet. 2004;65:490–495. doi: 10.1111/j.0009-9163.2004.00265.x. [DOI] [PubMed] [Google Scholar]
- 26.Ziedalski TM. Kao PN. Henig NR. Jacobs SS. Ruoss SJ. Prospective analysis of cystic fibrosis transmembrane regulator mutations in adults with bronchiectasis or pulmonary nontuberculous mycobacterial infection. Chest. 2006;130:995–1002. doi: 10.1378/chest.130.4.995. [DOI] [PubMed] [Google Scholar]
- 27.Bombieri C. Claustres M. De Boeck K. Derichs N. Dodge J. Girodon E, et al. Recommendations for the classification of diseases as CFTR-related disorders. J Cyst Fibros. 2011;10(Suppl 2):S86–S102. doi: 10.1016/S1569-1993(11)60014-3. [DOI] [PubMed] [Google Scholar]
- 28.Dahl M. Nordestgaard BG. Lange P. Tybjaerg-Hansen A. Fifteen-year follow-up of pulmonary function in individuals heterozygous for the cystic fibrosis phenylalanine-508 deletion. J Allergy Clin Immunol. 2001;107:818–823. doi: 10.1067/mai.2001.114117. [DOI] [PubMed] [Google Scholar]
- 29.Gene GG. Llobet A. Larriba S. de Semir D. Martínez I. Escalada A. Solsona C. Casals T. Aran JM. N-terminal CFTR missense variants severely affect the behavior of the CFTR chloride channel. Hum Mutat. 2008;29:738–749. doi: 10.1002/humu.20721. [DOI] [PubMed] [Google Scholar]
- 30.Hughes D. Dork T. Stuhrmann M. Graham C. Mutation and haplotype analysis of the CFTR gene in atypically mild cystic fibrosis patients from Northern Ireland. J Med Genet. 2001;38:136–139. doi: 10.1136/jmg.38.2.136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Divac A. Nikolic A. Mitic-Milikic M. Nagorni-Obradovic L. Petrovic-Stanojevic N. Dopudja-Pantic V. Nadaskic R. Savic A. Radojkovic D. High frequency of the R75Q CFTR variation in patients with chronic obstructive pulmonary disease. J Cyst Fibros. 2004;3:189–191. doi: 10.1016/j.jcf.2004.05.049. [DOI] [PubMed] [Google Scholar]
- 32.Chillon M. Casals T. Mercier B. Bassas L. Lissens W. Silber S, et al. Mutations in the cystic fibrosis gene in patients with congenital absence of the vas deferens. N Engl J Med. 1995;332:1475–1480. doi: 10.1056/NEJM199506013322204. [DOI] [PubMed] [Google Scholar]
- 33.Cuppens H. Lin W. Jaspers M. Costes B. Teng H. Vankeerberghen A. Jorissen M. Droogmans G. Reynaert I. Goossens M. Nilius B. Cassiman JJ. Polyvariant mutant cystic fibrosis transmembrane conductance regulator genes. J Clin Invest. 1998;101:487–496. doi: 10.1172/JCI639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee JH. Choi JH. Namkung W. Hanrahan JW. Chang J. Song SY. Park SW. Kim DS. Yoon JH. Suh Y. Jang IJ. Nam JH. Kim SJ. Cho MO. Lee JE. Kim KH. Lee MG. A haplotype-based molecular analysis of CFTR mutations associated with respiratory and pancreatic diseases: a haplotype-based molecular analysis of CFTR mutations associated with respiratory and pancreatic diseases. Hum Mol Genet. 2003;12:2321–2332. doi: 10.1093/hmg/ddg243. [DOI] [PubMed] [Google Scholar]
- 35.Sun W. Anderson B. Redman J. Milunsky A. Buller A. McGinniss MJ. Quan F. Anguiano A. Huang S. Hantash F. Strom C. CFTR 5T variant has a low penetrance in females that is partially attributable to its haplotype. Genet Med. 2006;8:339–345. doi: 10.1097/01.gim.0000223549.57443.16. [DOI] [PubMed] [Google Scholar]
- 36.Noone PG. Pue CA. Zhou Z. Friedman KJ. Wakeling EL. Ganeshananthan M. Simon RH. Silverman LM. Knowles MR. Lung disease associated with the IVS8 5T allele of the CFTR gene. Am J Respir Crit Care Med. 2000;162:1919–1924. doi: 10.1164/ajrccm.162.5.2003160. [DOI] [PubMed] [Google Scholar]