

Abstract
We report a new synthesis of phenanthridines based on palladium-catalyzed picolinamide-directed sequential C–H functionalization reactions starting from readily available benzylamine and aryl iodide precursors. Under the catalysis of Pd(OAc)2, the ortho-C–H bond of benzylpicolinamides is first arylated with an aryl iodide. The resulting biaryl compound is then subjected to palladium-catalyzed picolinamide-directed intramolecular dehydrogenative C–H amination with PhI(OAc)2 oxidant to form the corresponding cyclized dihydrophenanthridines. The benzylic position of these dihydrophenanthridines could be further oxidized with Cu(OAc)2, removing the picolinamide group and providing phenathridine products. The cyclization and oxidation could be carried out in a single step and afford phenathridines in moderate to good yields.
Keywords: C–H functionalization, palladium, phenanthridine, picolinamide
Introduction
Phenanthridines and 5,6-dihydro-phenanthridines are important core structures found in a variety of natural products and functional molecules (Scheme 1A) [1–8]. Synthetic methods for their preparation include the classical Pictet–Hubert condensation [9], radical-mediated reactions [10–13], metal-catalyzed cross-couplings [14–18], cycloadditions [19], and others [20–22]. More recently, methods based on the metal-catalyzed functionalization of carbon–hydrogen (C–H) bonds have also emerged as viable strategies for synthesizing phenanthridines [23–25]. Despite these advances, construction of phenanthridines with complex substitution patterns remains difficult and often requires lengthy and inefficient synthetic sequences. Herein, we report a novel method for phenanthridine synthesis based on sequential palladium-catalyzed picolinamide (PA)-directed C–H functionalization reactions beginning from easily accessible PA-protected benzylamine and aryl iodide precursors.
Scheme 1.

Representative phenanthridine compounds and our synthetic strategy based on Pd-catalyzed sequential C–H functionalizations.
Results and Discussion
New synthetic strategy for phenanthridine compounds. The picolinamide (PA) group has been shown to be an excellent directing group for a range of Pd-catalyzed C–H functionalization reactions [26–35]. In 2005, the Daugulis laboratory first reported that the ortho-C(sp2)−H bond of benzylpicolinamides could be arylated with aryl iodides under Ag-promoted Pd-catalyzed conditions [26]. In 2012, our laboratory [28] as well as that of Daugulis [27] independently reported that picolinamide substrates can undergo intramolecular dehydrogenative C–H amination reactions to afford medium-sized N-heterocycles under the catalysis of Pd(OAc)2 with PhI(OAc)2 oxidant. These discoveries led us to explore whether we could develop a new strategy for synthesizing phenanthridines. As outlined in Scheme 1B, we envisioned that ortho-arylated benzylamine picolinamides could undergo an intramolecular amination at the ortho ε-C–H position of the newly installed arene group to form cyclized dihydrophenanthridines, which could be further converted to phenanthridine products under oxidative conditions. Ideally, we hoped to perform both the intramolecular C–H amination and subsequent oxidation in a single step [36].
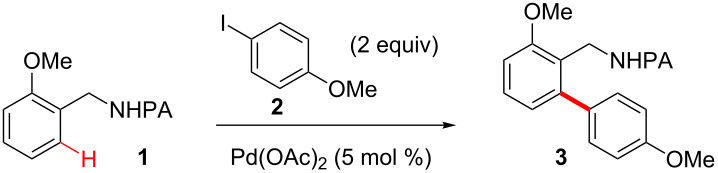
Arylation of 2-methoxybenzyl picolinamide 1 with 4-iodoanisole (2) under various conditions. We commenced the study by investigating the arylation of 2-methoxybenzyl picolinamide 1 with 4-iodoanisole (2) under various conditions (Table 1) to form our desired arylated product 3. Our initial attempt under the original Pd(OAc)2-catalyzed AgOAc-promoted solvent-free condition afforded the desired arylated product 3 in good yield (Table 1, entry 1). This method, however, required the use of expensive silver salt as an additive and high reaction temperature (150 °C). We next sought to replace the silver salts with cheaper reagents and lower the reaction temperature [12]. Not surprisingly, the arylation yield dropped significantly when the reaction was performed in toluene solvent at 120 °C (Table 1, entry 2). Addition of PivOH (0.3 equiv) gave little improvement (Table 1, entries 3 and 4). To our delight, the desired arylation reaction was largely restored with the application of 2 equiv of K2CO3 at 120 °C for 24 h (Table 1, entry 5). Furthermore, an excellent yield was obtained when K2CO3 was replaced with KHCO3 and 0.3 equiv of PivOH was applied (Table 1, entry 7). The most effective carboxylate ligand and solvent was found to be PivOH and toluene, respectively.
Table 1.
Optimization of the Pd-catalyzed ortho-C–H arylation of benzylpicolinamide. All screening reactions were carried out in a 10 mL glass vial on a 0.2 mmol scale.
 | ||||
| entry | additives (equiv) | temperature (°C) | solvent | yield of 3 (%)a |
| 1 | AgOAc (1.5) | 150 | no solvent | 76 |
| 2 | AgOAc (1.5) | 120 | toluene | 6 |
| 3 | AgOAc (1.5), PivOH (0.3) | 120 | toluene | 3 |
| 4 | PivOH (0.3) | 120 | toluene | 5 |
| 5 | K2CO3 (2) | 120 | toluene | 57 |
| 6 | PivOH (0.3), K2CO3 (2) | 120 | toluene | 90 |
| 7 | PivOH (0.3), KHCO3 (2) | 120 | toluene | 95 (91)b |
| 8 | AcOH (0.3), KHCO3 (2) | 120 | toluene | 78 |
| 9 | oPBAc (0.3), KHCO3 (2) | 120 | toluene | 84 |
| 10 | PivOH (0.3), KHCO3 (2) | 90 | toluene | 29 |
aYields are based on 1H NMR analysis of the reaction mixture after workup; bIsolated yield; coPBA: ortho-phenylbenzoic acid.
The determination of the scope of this reaction with benzylpicolinamide and aryl iodide substrates. With the optimized conditions in hand, we next explored the scope of benzylpicolinamide and aryl iodide substrates (Figure 1). The electronic properties of benzylpicolinamide and aryl iodides had little influence on the reactivity, as benzylpicolinamide and aryl iodide substrates bearing electron-donating and withdrawing substituents react in good yields (3, 8, and 12). Significantly decreased arylation yield was observed for ortho-substituted aryl iodides (e.g., 9). The sterics of the benzylpicolinamides is also important for the regioselectivity of the arylation reaction. For instance, the less hindered ortho position is preferentially arylated (e.g., 14) when a meta substitutent is present on the benzylpicolinamide. Aryl bromides are much less reactive compared with aryl iodide substrates 4. This is in accordance with results on the Pd-catalyzed PA-directed arylation of more inert C(sp3)−H bonds [29].
Figure 1.

Substrate scope of the Pd-catalyzed PA-directed C–H arylation reaction. All reactions were carried out in a 10 mL glass vial on a 0.2 mmol scale; yields are based on the isolated product.
Cyclization of biaryl compounds to form dihydrophenanthridines. Next, we investigated the cyclization of biaryl compounds to form dihydrophenanthridines via Pd-catalyzed intramolecular dehydrogenative amination of ε-C(sp2)–H bonds [37–45]. To our delight, treatment of 3 in the presence of 5 mol % of Pd(OAc)2 and 2 equiv of PhI(OAc)2 in toluene at 120 °C for 24 h gave the desired dihydrophenanthridine 16 in good yield (Table 2, entry 1). In addition, a further oxidized phenanthridine 17 was obtained as a side product. Compound 17 is presumably generated through the PhI(OAc)2-mediated oxidation of the benzylic C–H bond to form a phenanthridinium intermediate 18, which then undergoes a removal of the PA group. Encouraged by these observations, we proceeded to explore whether the cyclization and oxidation steps can be performed in one step to give the phenanthridines in a shorter procedure. A variety of oxidants, such as 1,4-benzoquinone (BQ), KMnO4, ceric ammonium nitrate (CAN), and copper salts were examined [46]. The combination of PhI(OAc)2 (2 equiv) and Cu(OAc)2 (2 equiv) afforded the phenanthridine product 17 in highest yield (Table 2, entry 7). The yield can be further improved using 10 mol % of Pd(OAc)2 catalyst (Table 2, entry 8). In a control experiment, dihydrophenanthridine 16 was oxidized with the application of Cu(OAc)2 (2 equiv) in toluene at 120 °C for 24 h, forming 17 in excellent yield. We believe that PhI(OAc)2 serves as the oxidant for the initial Pd-catalyzed intramolecular C–H amination step, in which a PdII/IV catalytic manifold might be operative. Cu(OAc)2 is responsible for the subsequent oxidation of the benzylic C–H bond of dihydrophenanthridine.
Table 2.
Formation of phenanthridine 17 in a single step by Pd-catalyzed intramolecular C–H amination followed by oxidation. All screening reactions were carried out in a 10 mL glass vial on a 0.2 mmol scale.
 | ||||
| entry | Pd(OAc)2 (mol %) | additives (equiv) | yield (%)a | |
| 16 | 17 | |||
| 1 | 5 | PhI(OAc)2 (2) | 40 | 5 |
| 2 | 5 | PhI(OAc)2 (2), AcOH (2) | 23 | 8 |
| 3 | 5 | PhI(OAc)2 (2), BQ (2) | 35 | 10 |
| 4 | 5 | PhI(OAc)2 (2), KMnO4 (2) | 56 | 3 |
| 5 | 5 | PhI(OAc)2 (2), CAN (2) | 37 | 25 |
| 6 | 5 | PhI(OAc)2 (2), CuCl2 (2) | 29 | 34 |
| 7 | 5 | PhI(OAc)2 (2), Cu(OAc)2 (2) | 17 | 51 |
| 8 | 10 | PhI(OAc)2 (2), Cu(OAc)2 (2) | 15 | 62 (58)b |
aYields are based on 1H NMR analysis of the reaction mixture after workup; bIsolated yield.
Extension of the cyclization–oxidation step to other arylated picolinamide substrates. The coupled cyclization–oxidation step detailed above was then used to synthesize phenanthridines from other arylated picolinamide substrates (Figure 2). In general, electron-rich arene motifs, installed by C–H arylation, gave a higher yield of phenanthridine products; electron-deficient substrates provide a lower yield. For instance, substrate 8 with a para-nitro group failed to give any cyclized product under the standard conditions. Substrates with moderately electron-withdrawing groups, such as 20 bearing a para-ester group, reacted in moderate yield. The electronic properties of the benzylpicolinamide scaffold had much less influence on the reaction. For example, product 22 bearing an ortho-CF3 substituent was obtained in 51% yield. Finally, it is noteworthy that all of the above phenanthridine products show intense blue fluorescence. We expect our synthetic strategy will afford access to phenanthridines bearing varied substitution patterns, enabling applications in biology and materials science.
Figure 2.

Substrate scope of this phenanthridine synthesis. All reactions were carried out in a 10 mL glass vial on a 0.2 mmol scale; yields are based on isolated product.
Conclusion
In summary, we have developed a readily applicable two-step method for the synthesis of phenanthridines from easily accessible benzylamine picolinamides and aryl iodides. In the first step, an improved protocol allows us to carry out the Pd-catalyzed PA-directed C–H arylation reaction without the use of expensive silver additives. In the second step, application of PhI(OAc)2 and Cu(OAc)2 oxidant under the catalysis of Pd(OAc)2 affords phenanthridines in moderate to good yields. Applications of this method to the synthesis of more complex phenanthridines with novel photophysical properties are currently underway.
Experimental
General conditions: All commercial materials were used as received unless otherwise noted. All solvents were obtained from a JC Meyer solvent dispensing system and used without further purification. Flash chromatography was performed using 230–400 mesh SiliaFlash 60® silica gel (Silicycle Inc.). PhI(OAc)2 (98%, Aldrich), Pd(OAc)2 (98%, Aldrich) were used in the Pd-catalyzed reactions. NMR spectra were recorded on Bruker CDPX-300, DPX-300, DPX-400 instruments and calibrated by using residual solvent peaks as the internal reference. Multiplicities are recorded as: s = singlet, d = doublet, t = triplet, dd = doublet of doublets, m = multiplet. High-resolution ESI mass experiments were operated on a Waters LCT Premier instrument.
Standard procedure for the Pd-catalyzed ortho C–H arylation reaction: A mixture of picolinamide 1 [30] (48 mg, 0.2 mmol, 1 equiv), aryl iodide 2 (94 mg, 0.4 mmol, 2 equiv), Pd(OAc)2 (2.2 mg, 0.01 mmol, 0.05 equiv), KHCO3 (40 mg, 0.4 mmol, 2.0 equiv), and PivOH (6 mg, 0.06 mmol, 0.3 equiv) in anhydrous toluene (4 mL) in a 10 mL glass vial (purged with N2, sealed with PTFE cap) was heated at 120 °C for 24 h. The reaction mixture was filtered through a short pad of celite and concentrated in vacuo. The resulting residue was purified by silica-gel flash chromatography (hexanes/EtOAc 3:1) to give the product 3 as a pale white solid (64 mg, 91%). Compounds 4–15 were prepared from the known precursors [30] by using the standard C–H arylation procedure.
Standard procedure for the Pd-catalyzed cyclization and oxidation reaction to form phenanthridines: A mixture of picolinamide 3 (70 mg, 0.2 mmol, 1 equiv), Pd(OAc)2 (4.4 mg, 0.02 mmol, 0.1 equiv), PhI(OAc)2 (129 mg, 0.4 mmol, 2.0 equiv), and Cu(OAc)2 (72 mg, 0.4 mmol, 2 equiv) in anhydrous toluene (4 mL) in a 10 mL glass vial (purged with N2, sealed with PTFE cap) was heated at 120 °C for 24 h. The reaction mixture was filtered through a short pad of celite and concentrated in vacuo. The resulting residue was purified by silica gel flash chromatography (hexanes/EtOAc 4:1) to give the product 17 as a pale white solid (28 mg, 58%). Compounds 19–24 were prepared by using the standard cyclization–oxidation procedure.
Compound 3. 1H NMR (CDCl3, 300 MHz) δ 8.53 (d, J = 4.2 Hz, 1H), 8.40 (s, 1H), 8.21 (d, J = 7.5 Hz, 1H), 7.85–7.80 (m, 1H), 7.42–7.30 (m, 3H), 6.99–6.93 (m, 4H), 4.65 (d, J = 5.4 Hz, 2H), 3.95 (s, 3H), 3.85 (s, 3H); 13C NMR (CDCl3, 75 MHz) δ 163.3, 158.8, 158.6, 150.2, 147.9, 143.6, 137.1, 132.7, 130.3, 128.3, 125.8, 123.4, 122.7, 122.1, 113.6, 109.1, 55.7, 55.2, 36.8; HRMS (m/z): [M + H]+ calcd for C21H21N2O3, 349.1552; found, 349.1546.
Compound 4. 1H NMR (CDCl3, 300 MHz) δ 8.52 (d, J = 4.5 Hz, 1H), 8.36 (s, 1H), 8.19 (d, J = 7.8 Hz, 1H), 7.84 (td, J = 7.8, 1.4 Hz, 1H), 7.55 (d, J = 8.3 Hz, 2H), 7.41 (dd, J = 5.0, 6.8 Hz, 1H), 7.35 (t, J = 8.0 Hz, 1H), 7.25 (d, J = 8.3 Hz, 2H), 6.97 (d, J = 8.2 Hz, 1H), 6.89 (d, J = 7.7 Hz, 1H), 4.59 (d, J = 5.5 Hz, 2H), 3.95 (s, 3H); 13C NMR (CDCl3, 75.5 MHz) δ 163.3, 158.7, 150.1, 148.0, 142.5, 139.4, 137.1, 131.3, 130.9, 128.6, 125.9, 123.5, 122.4, 122.1, 121.6, 109.9, 55.8, 36.7; HRMS (m/z): [M + H]+ calcd for C20H18BrN2O2, 397.0552; found, 397.0561.
Compound 5. 1H NMR (CDCl3, 400 MHz) δ 8.51 (d, J = 4.0 Hz, 1H), 8.33 (s, 1H), 8.15 (d, J = 7.6 Hz, 1H), 8.08 (d, J = 8.0 Hz, 1H), 7.80 (t, J = 7.6 Hz, 1H), 7.44–7.34 (m, 4H), 6.97 (d, J = 8.0 Hz, 1H), 6.90 (d, J = 7.6 Hz, 1H), 4.58 (d, J = 5.2 Hz, 2H), 3.95 (s, 3H), 3.92 (s, 3H); 13C NMR (CDCl3, 75 MHz) δ 166.9, 163.3, 158.7, 150.2, 148.0, 145.2, 142.9, 137.2, 129.5, 129.3, 129.0, 128.6, 125.9, 123.5, 122.3, 122.2, 110.0, 55.8, 52.1, 36.6; HRMS (m/z): [M + H]+ calcd for C22H21N2O4, 377.1501; found, 377.1509.
Compound 6. 1H NMR (CDCl3, 300 MHz) δ 8.51 (d, J = 4.2 Hz, 1H), 8.34 (s, 1H), 8.19 (d, J = 7.8 Hz, 1H), 7.40–7.31 (m, 1H), 6.98–6.91 (m, 5H), 4.66 (d, J = 5.4 Hz, 2H), 3.94 (s, 3H), 3.79 (s, 3H); 13C NMR (CDCl3, 75 MHz) δ 163.8, 159.7, 159.0, 150.7, 148.4, 144.4, 142.3, 137.6, 129.7, 128.9, 126.3, 124.0, 123.0, 122.6, 122.1, 114.9, 113.8, 110.1, 55.3, 55.6, 37.2; HRMS (m/z): [M + H]+ calcd for C21H21N2O3, 349.1552; found, 349.1564.
Compound 7. 1H NMR (CDCl3, 300 MHz) δ 8.53 (d, J = 4.2 Hz, 1H), 8.34 (s, 1H), 8.19 (d, J = 7.8 Hz, 1H), 7.84–7.80 (m, 1H), 7.40–7.29 (m, 2H), 6.96–6.80 (m, 5H), 6.00 (s, 2H), 4.64 (d, J = 5.4 Hz, 1H), 3.96 (s, 3H); 13C NMR (CDCl3, 75 MHz) δ 163.4, 158.7, 150.3, 148.0, 147.4, 146.9, 143.7, 137.1, 134.4, 128.4, 125.8, 123.7, 122.8, 122.7, 122.2, 109.9, 109.5, 108.1, 101.0, 55.8, 36.8; HRMS (m/z): [M + H]+ calcd for C21H20N2O4, 363.1345; found, 363.1355.
Compound 8. 1H NMR (CDCl3, 300 MHz) δ 8.53 (d, J = 4.1 Hz, 1H), 8.41 (s, 1H), 8.29 (d, J = 8.8 Hz, 2H), 8.16 (d, J = 7.8 Hz, 1H), 7.85 (td, J = 7.7, 1.6 Hz, 1H), 7.55 (d, J = 8.7 Hz, 2H), 7.42–7.34 (m, 2H), 7.03 (d, J = 8.2 Hz, 1H), 6.89 (d, J = 7.7 Hz, 1H), 4.58 (d, J = 5.7 Hz, 2H), 3.98 (s, 3H); 13C NMR (CDCl3, 75.5 MHz) δ 163.4, 158.8, 150.0, 148.1, 147.4, 147.1, 141.4, 137.3, 130.4, 128.9, 126.1, 123.7, 123.5, 122.2, 122.2, 110.7, 56.0, 36.6; HRMS (m/z): [M + H]+ calcd for C20H18N3O4, 364.1297; found, 364.1292.
Compound 9. 1H NMR (CDCl3, 300 MHz) δ 8.53 (d, J = 4.2 Hz, 1H), 8.32 (s, 1H), 8.18–8.16 (m, 1H), 7.85–7.80 (m, 1H), 7.38–7.30 (m, 4H), 7.25–7.13 (m, 2H), 7.02–6.92 (m, 2H), 4.64–4.90 (m, 2H), 3.98 (s, 3H); 13C NMR (CDCl3, 75 MHz) δ 163.8, 161.2, 158.9, 150.7, 148.4, 137.8, 137.6, 132.0, 129.9, 128.9, 126.2, 125.2, 124.5, 123.3, 122.6, 115.9, 115.8, 110.7, 56.22, 37.2; HRMS (m/z): [M + H]+ calcd for C20H18FN2O2, 337.1352; found, 337.1364.
Compound 10. 1H NMR (CDCl3, 300 MHz) δ 8.51 (d, J = 4.0 Hz, 1H), 8.25 (s, 1H), 8.17 (d, J = 7.8 Hz, 1H), 7.84 (td, J = 7.7, 1.7 Hz, 1H), 7.73 (d, J = 8.3 Hz, 2H), 7.66 (d, J = 8.3 Hz, 1H), 7.41–7.36 (m, 1H), 7.32–7.24 (m, 3H), 7.17 (d, J = 8.3 Hz, 1H), 7.06 (s, 1H), 6.94 (d, J = 8.2 Hz, 1H), 6.88 (d, J = 7.4 Hz, 1H), 4.57 (d, J = 5.4 Hz, 2H), 3.95 (t, J = 8.8 Hz, 2H), 3.92 (s, 3H), 2.93 (t, J = 8.4 Hz, 2H), 2.37 (s, 3H); 13C NMR (CDCl3, 75.5 MHz) δ 163.3, 158.7, 150.2, 148.1, 144.1, 143.4, 141.3, 137.2, 136.1, 133.9, 131.8, 129.8, 128.7, 128.5, 127.3, 126.2, 125.9, 123.5, 122.6, 122.1, 114.5, 109.5, 55.8, 50.1, 36.7, 27.8, 21.5; HRMS (m/z): [M + H]+ calcd for C29H18N3O4S, 514.1801; found: 514.1813.
Compound 11. 1H NMR (CDCl3, 300 MHz) δ 8.54 (d, J = 3.9 Hz, 1H), 8.21 (d, J = 7.5 Hz, 2H), 7.88–7.83 (m, 1H), 7.46–7.41 (m, 2 H), 7.34–7.23 (m, 4 H), 6.96 (d, J = 8.7 Hz, 1H), 4.72 (d, J = 5.4 Hz, 1H), 8.86 (s, 3H); 13C NMR (CDCl3, 75 MHz) δ 163.5, 159.1, 149.8, 148.0, 144.9, 137.2, 135.8, 132.9, 132.4, 130.1, 129.2, 128.7, 128.6, 126.0, 122.2, 113.8, 55.2, 39.6; HRMS (m/z): [M + H]+ calcd for C20H18ClN2O2, 353.1057; found, 353.1067.
Compound 12. 1H NMR (CDCl3, 300 MHz) δ 8.50 (d, J = 4.8 Hz, 1H), 8.14 (d, J = 7.8 Hz, 1H), 7.93 (s, 1H), 7.83–7.74 (m, 2H), 7.50–7.39 (m, 3H), 7.28–7.22 (m, 2H), 6.89 (d, J = 8.7 Hz, 2H), 4.71 (d, J = 4.5 Hz, 1H), 3.82 (s, 3H); 13C NMR (CDCl3, 75 MHz) δ 163.0, 159.1, 149.6, 148.0, 145.6, 137.1, 134.6, 133.5, 131.9, 130.1, 129.9, 127.8, 126.0, 125.4, 122.1, 113.8, 55.2, 38.2; HRMS (m/z): [M + H]+ calcd for C21H18F3N2O2, 387.1320; found, 387.1328.
Compound 13. 1H NMR (CDCl3, 300 MHz) δ 8.51 (d, J = 4.2 Hz, 1H), 8.18 (d, J = 7.8 Hz, 2H), 7.83–7.79 (m, 1H), 7.42–7.38 (m, 1H), 7.33–7.30 (m, 3H), 7.11 (d, J = 7.5 Hz, 2H), 6.97 (d, J = 8.7 Hz, 2H), 4.68 (d, J = 5.4 Hz, 2H), 3.84 (s, 3H); 13C NMR (CDCl3, 75 MHz) δ 163.7, 160.4, 159.1, 149.7, 147.9, 144.4, 137.1, 131.6, 130.1, 128.9, 128.7, 126.0, 122.6, 122.1, 114.3, 113.8, 55.2, 35.6; HRMS (m/z): [M + H]+ calcd for C20H18FN2O2, 337.1352; found, 337.1357.
Compound 14. 1H NMR (CDCl3, 300 MHz) δ 8.50 (d, J = 4.5 Hz, 1H), 8.27–8.21 (m, 2H), 7.82 (t, J = 7.5 Hz, 1H), 7.41–7.29 (m, 4H), 7.19 (dd, J = 14.1 and 7.8 Hz, 2H), 6.98 (d, J = 8.4 Hz, 2H), 4.64 (d, J = 6.0 Hz, 2H), 3.84 (s, 3H), 2.38 (s, 3H); 13C NMR (CDCl3, 75 MHz) δ 163.8, 158.5, 149.6, 147.8, 138.4, 137.0, 136.9, 135.2, 132.8, 130.0, 129.1, 127.9, 125.9, 122.0, 113.5, 55.0, 41.1, 20.9; HRMS (m/z): [M + H]+ calcd for C21H21N2O2, 333.1603; found, 333.1609.
Compound 15. 1H NMR (CDCl3, 300 MHz) δ 8.45 (d, J = 4.7 Hz, 1H), 8.01 (d, J = 7.8 Hz, 1H), 7.78 (m, 2H), 7.42 (s, 2H), 7.37 (m, 1H), 7.30 (d, J = 8.7 Hz, 4H), 6.90 (d, J = 8.7 Hz, 4H), 4.47 (d, J = 5.1 Hz, 2H), 3.79 (s, 6H); 13C NMR (CDCl3, 75.5 MHz) δ 162.9, 159.1, 149.5, 147.8, 145.2, 137.0, 132.3, 132.2, 132.1, 129.9, 125.9, 121.8, 121.0, 113.8, 55.2, 39.0; HRMS (m/z): [M + H]+ calcd for C27H24Br3N2O3, 503.0970; found, 503.0975.
Compound 17. 1H NMR (CDCl3, 300 MHz) δ 9.72 (s, 1H), 8.45 (d, J = 9.0 Hz, 1H), 8.08 (d, J = 8.2 Hz, 1H), 7.75 (t, J = 8.1 Hz, 1H), 7.61 (d, J = 2.6 Hz, 1H), 7.33–7.29 (m, 1H), 6.99 (d, J = 7.8 Hz, 1H), 4.09 (s, 3H), 4.00 (s, 3H); 13C NMR (CDCl3, 75.5 MHz) δ 160.5, 157.5, 149.1, 147.2, 134.6, 132.1, 124.3, 118.5, 117.1, 113.8, 110.1, 105.9, 56.2, 56.0; HRMS (m/z): [M + H]+ calcd for C15H14NO2, 240.1025; found, 240.1030.
Compound 19. 1H NMR (CDCl3, 400 MHz) δ 9.68 (s, 1H), 8.33 (m, 2H), 8.04 (d, J = 8.3 Hz, 1H), 7.75 (m, 2H), 7.04 (d, J = 7.9 Hz, 1H), 4.05 (s, 3H); 13C NMR (CDCl3, 75.5 MHz) δ 157.6, 149.5, 145.7, 133.8, 132.5, 132.2, 130.1, 124.3, 122.5, 117.4, 113.7, 107.2, 55.9; HRMS (m/z): [M + H]+ calcd for C14H11BrNO, 228.0024; found, 228.0032.
Compound 20. 1H NMR (CDCl3, 300 MHz) δ 9.75 (s, 1H), 8.84 (s, 1H), 8.54 (d, J = 8.7 Hz, 1H), 8.24 (d, J = 7.5 Hz, 1H), 8.14 (d, J = 8.4 Hz, 1H), 7.79 (d, J = 8.1 Hz, 1H), 7.09 (d, J = 8.1 Hz, 1H), 4.06 (s, 3H), 4.01 (s, 3H); 13C NMR (CDCl3, 75 MHz) δ 166.8, 157.4, 149.2, 144.0, 133.2, 132.1, 132.0, 130.1, 127.0, 126.7, 122.9, 117.9, 114.2, 107.8, 55.8, 52.4; HRMS (m/z): [M + H]+ calcd for C16H13NO3, 268.0974, found, 268.0970.
Compound 21. 1H NMR (CDCl3, 300 MHz) δ 9.59 (s, 1H), 8.46 (d, J = 9.0 Hz, 1H), 8.29 (d, J = 8.4 Hz, 1H), 7.82–7.74 (m, 1H), 7.64 (s, 1H), 7.37–7.26 (m, 2H), 4.01 (s, 3H); 13C NMR (CDCl3, 75 MHz) δ 160.6, 147.4, 146.6, 134.6, 131.7, 131.6, 123.8, 118.7, 117.4, 117.3, 111.1, 110.8, 110.1, 55.7; HRMS (m/z): [M + H]+ calcd for C14H10FNO, 228.0825; found, 228.0830.
Compound 22. 1H NMR (CDCl3, 300 MHz) δ 9.62 (s, 1H), 8.72 (d, J = 8.4 Hz, 1H), 8.47 (d, J = 9.0 Hz, 1H), 7.95 (d, J = 7.2 Hz, 1H), 7.85 (t, J = 8.1 Hz, 1H), 7.62 (d, J = 2.4 Hz, 1H), 7.36 (dd, J = 9.0 Hz and 2.7 Hz, 1H), 4.01 (s, 3H); 13C NMR (CDCl3, 75 MHz) δ 160.7, 149.3, 145.8, 133.8, 129.5, 125.8, 124.4, 124.3, 123.4, 122.2, 119.1, 117.6, 109.8, 55.6; HRMS (m/z): [M + H]+ calcd for C15H12F3NO, 278.0793; found, 278.0797.
Compound 23. 1H NMR (CDCl3, 300 MHz) δ 9.72 (s, 1H), 8.46–8.42 (m, 2H), 7.72–7.62 (m, 3H), 7.35 (dd, J = 9.0 and 2.4 Hz, 1H), 4.02 (s, 3H); 13C NMR (CDCl3, 75 MHz, ppm) δ 160.5, 150.1, 146.1, 134.5, 133.9, 131.0, 126.9, 123.6, 122.3, 120.4, 118.8, 117.1, 109.9, 55.6; HRMS (m/z): [M + H]+ calcd for C14H11ClNO, 244.0529; found, 244.0534.
Compound 24. 1H NMR (CDCl3, 300 MHz) δ 9.23 (s, 1H), 8.48–8.42 (m, 2H), 7.82 (s, 1H), 7.69 (d, J = 8.1 Hz, 1H), 7.62 (s, 1H), 7.34–7.30 (m, 1H), 4.02 (s, 3H), 2.62 (s, 3H); 13C NMR (CDCl3, 75 MHz) δ 160.2, 154.2, 146.3, 137.4, 136.7, 133.4, 131.1, 128.5, 123.6, 121.7, 118.2, 118.4, 110.3, 56.0, 21.9; HRMS (m/z): [M + H]+ calcd for C15H13NO, 244.1075; found, 244.1079.
Acknowledgments
We gratefully thank The Pennsylvania State University, NSF (CAREER CHE-1055795), and ACS-PRF (51705-DNI1) for financial support of this work.
This article is part of the Thematic Series "Transition-metal and organocatalysis in natural product synthesis".
References
- 1.Theobald R S, Schofield K. Chem Rev. 1950;46:170–189. doi: 10.1021/cr60143a004. [DOI] [PubMed] [Google Scholar]
- 2.Kock I, Heber D, Weide M, Wolschendorf U, Clement B. J Med Chem. 2005;48:2772–2777. doi: 10.1021/jm0490888. [DOI] [PubMed] [Google Scholar]
- 3.Zhang J, Lakowicz J R. J Phys Chem B. 2005;109:8701–8706. doi: 10.1021/jp046016j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stevens N, O’Connor N, Vishwasrao H, Samaroo D, Kandel E R, Akins D L, Drain C M, Turro N J. J Am Chem Soc. 2008;130:7182–7183. doi: 10.1021/ja8008924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chen J-J, Li K-T, Yang D-Y. Org Lett. 2011;13:1658–1661. doi: 10.1021/ol200117b. [DOI] [PubMed] [Google Scholar]
- 6.Barthelmes H U, Niederberger E, Roth T, Schulte K, Tang W C, Boege F, Fiebig H H, Eisenbrand G, Marko D. Br J Cancer. 2001;85:1585–1591. doi: 10.1054/bjoc.2001.2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bailly C, Arafa R K, Tanious F A, Laine W, Tardy C, Lansiaux A, Colson P, Boykin D W, Wilson W D. Biochemistry. 2005;44:1941–1952. doi: 10.1021/bi047983n. [DOI] [PubMed] [Google Scholar]
- 8.Park G Y, Wilson J J, Song Y, Lippard S J. Proc Natl Acad Sci U S A. 2012;109:11987–11992. doi: 10.1073/pnas.1207670109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pictet A, Hubert A. Ber Dtsch Chem Ges. 1896;29:1182–1189. doi: 10.1002/cber.18960290206. [DOI] [Google Scholar]
- 10.Alonso R, Campos P J, García B, Rodríguez M A. Org Lett. 2006;8:3521–3523. doi: 10.1021/ol061258i. [DOI] [PubMed] [Google Scholar]
- 11.McBurney R T, Slawin A M Z, Smart L A, Yu Y, Walton J C. Chem Commun. 2011;47:7974–7976. doi: 10.1039/c1cc12720a. [DOI] [PubMed] [Google Scholar]
- 12.Linsenmeier A M, William C M, Bräse S. J Org Chem. 2011;76:9127–9132. doi: 10.1021/jo201542x. [DOI] [PubMed] [Google Scholar]
- 13.Tobisu M, Koh K, Furukawa T, Chatani N. Angew Chem, Int Ed. 2012;51:11363–11366. doi: 10.1002/anie.201206115. [DOI] [PubMed] [Google Scholar]
- 14.Yanada R, Hashimoto K, Tokizane R, Miwa Y, Minami H, Yanada K, Ishikura M, Takemoto Y. J Org Chem. 2008;73:5135–5138. doi: 10.1021/jo800474c. [DOI] [PubMed] [Google Scholar]
- 15.Donaldson L R, Haigh D, Hulme A N. Tetrahedron. 2008;64:4468–4477. doi: 10.1016/j.tet.2008.02.044. [DOI] [Google Scholar]
- 16.Candito D A, Lautens M. Angew Chem, Int Ed. 2009;48:6713–6716. doi: 10.1002/anie.200902400. [DOI] [PubMed] [Google Scholar]
- 17.Gerfaud T, Neuville L, Zhu J. Angew Chem, Int Ed. 2009;48:572–577. doi: 10.1002/anie.200804683. [DOI] [PubMed] [Google Scholar]
- 18.Zhang L, Ang G Y, Chiba S. Org Lett. 2010;12:3682–3685. doi: 10.1021/ol101490n. [DOI] [PubMed] [Google Scholar]
- 19.Sripada L, Teske J A, Deiters A. Org Biomol Chem. 2008;6:263–265. doi: 10.1039/b716519f. [DOI] [PubMed] [Google Scholar]
- 20.Shou W-G, Yang Y-Y, Wang Y-G. J Org Chem. 2006;71:9241–9243. doi: 10.1021/jo061648i. [DOI] [PubMed] [Google Scholar]
- 21.Budén M E, Dorn V B, Gamba M, Pierini A B, Rossi R A. J Org Chem. 2010;75:2206–2218. doi: 10.1021/jo9025918. [DOI] [PubMed] [Google Scholar]
- 22.Wu Y, Wong S M, Mao F, Chan T L, Kwong F Y. Org Lett. 2012;14:5306–5309. doi: 10.1021/ol302489n. [DOI] [PubMed] [Google Scholar]
- 23.Shabashov D, Daugulis O. J Org Chem. 2007;72:7720–7725. doi: 10.1021/jo701387m. [DOI] [PubMed] [Google Scholar]
- 24.Maestri G, Larraufie M-H, Derat É, Ollivier C, Fensterbank L, Lacôte E, Malacria M. Org Lett. 2010;12:5692–5695. doi: 10.1021/ol102509n. [DOI] [PubMed] [Google Scholar]
- 25.Peng J, Chen T, Chen C, Li B. J Org Chem. 2011;76:9507–9513. doi: 10.1021/jo2017108. [DOI] [PubMed] [Google Scholar]
- 26.Zaitsev V G, Shabashov D, Daugulis O. J Am Chem Soc. 2005;127:13154–13155. doi: 10.1021/ja054549f. [DOI] [PubMed] [Google Scholar]
- 27.Nadres E T, Daugulis O. J Am Chem Soc. 2012;134:7–9. doi: 10.1021/ja210959p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tran L D, Popov I, Daugulis O. J Am Chem Soc. 2012;134:18237–18240. doi: 10.1021/ja3092278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.He G, Chen G. Angew Chem, Int Ed. 2011;50:5192–5196. doi: 10.1002/anie.201100984. [DOI] [PubMed] [Google Scholar]
- 30.Zhao Y, Chen G. Org Lett. 2011;13:4850–4853. doi: 10.1021/ol201930e. [DOI] [PubMed] [Google Scholar]
- 31.He G, Zhao Y, Zhang S, Lu C, Chen G. J Am Chem Soc. 2012;134:3–6. doi: 10.1021/ja210660g. [DOI] [PubMed] [Google Scholar]
- 32.Zhang S-Y, He G, Zhao Y, Wright K, Nack W A, Chen G. J Am Chem Soc. 2012;134:7313–7316. doi: 10.1021/ja3023972. [DOI] [PubMed] [Google Scholar]
- 33.He G, Lu C, Zhao Y, Nack W A, Chen G. Org Lett. 2012;14:2944–2947. doi: 10.1021/ol301352v. [DOI] [PubMed] [Google Scholar]
- 34.Zhao Y, He G, Nack W A, Chen G. Org Lett. 2012;14:2948–2951. doi: 10.1021/ol301214u. [DOI] [PubMed] [Google Scholar]
- 35.Zhang S-Y, He G, Nack W A, Zhao Y, Li Q, Chen G. J Am Chem Soc. 2013;135:2124–2127. doi: 10.1021/ja312277g. [DOI] [PubMed] [Google Scholar]
- 36.Thansandote P, Lautens M. Chem–Eur J. 2009;15:5874–5883. doi: 10.1002/chem.200900281. [DOI] [PubMed] [Google Scholar]
- 37.Zaitsev V G, Daugulis O. J Am Chem Soc. 2005;127:4156–4157. doi: 10.1021/ja050366h. [DOI] [PubMed] [Google Scholar]
- 38.Shabashov D, Daugulis O. Org Lett. 2005;7:3657–3659. doi: 10.1021/ol051255q. [DOI] [PubMed] [Google Scholar]
- 39.Tsang W C P, Zheng N, Buchwald S L. J Am Chem Soc. 2005;127:14560–14561. doi: 10.1021/ja055353i. [DOI] [PubMed] [Google Scholar]
- 40.Inamoto K, Saito T, Katsuno M, Sakamoto T, Hiroya K. Org Lett. 2007;9:2931–2934. doi: 10.1021/ol0711117. [DOI] [PubMed] [Google Scholar]
- 41.Wasa M, Yu J-Q. J Am Chem Soc. 2008;130:14058–14059. doi: 10.1021/ja807129e. [DOI] [PubMed] [Google Scholar]
- 42.Jordan-Hore J A, Johansson C C C, Gulias M, Beck E M, Gaunt M J. J Am Chem Soc. 2008;130:16184–16186. doi: 10.1021/ja806543s. [DOI] [PubMed] [Google Scholar]
- 43.Mei T-S, Wang X, Yu J-Q. J Am Chem Soc. 2009;131:10806–10807. doi: 10.1021/ja904709b. [DOI] [PubMed] [Google Scholar]
- 44.Tan Y, Hartwig J F. J Am Chem Soc. 2010;132:3676–3677. doi: 10.1021/ja100676r. [DOI] [PubMed] [Google Scholar]
- 45.Cho S H, Yoon J, Chang S. J Am Chem Soc. 2011;133:5996–6005. doi: 10.1021/ja111652v. [DOI] [PubMed] [Google Scholar]
- 46.Li W-R, Hsu N-M, Chou H-H, Lin S T, Lin Y-S. Chem Commun. 2000:401–402. doi: 10.1039/a909236f. [DOI] [Google Scholar]