

Abstract
Acute lung injury (ALI) results from loss of alveolar-capillary barrier integrity and the evolution of high-permeability pulmonary edema resulting in alveolar flooding and significant morbidity and mortality. HMGB1 is a late mediator of sepsis which uniquely participates in the evolution of sepsis and sepsis-induced ALI. The molecular events by which HMGB1 contributes to ALI remain poorly characterized. We characterized the role of HMGB1 in endothelial cell (EC) cytoskeletal rearrangement and vascular permeability, events essential to paracellular gap formation and barrier dysfunction characteristic of ALI. Initial experiments demonstrated HMGB1-mediated dose-dependent (5–20 μg/ml) decreases in transendothelial cell electrical resistance (TER) in human pulmonary artery EC, a reflection of loss of barrier integrity. Furthermore, HMGB1 produced dose-dependent increases in paracellular gap formation in concert with loss of peripheral organized actin fibers, dissociation of cell-cell junctional cadherins, and development of central stress fibers, a phenotypic change associated with increased contractile activity and increased EC permeability. Using siRNA strategies directed against known HMGB1 receptors (RAGE, TLR2, TLR4), we systematically determined that the receptor for advanced glycation end products (RAGE) is the primary receptor signaling HMGB1-induced TER decreases and paracellular gap formation via p38 MAP kinase activation and phosphorylation of the actin-binding protein, Hsp27. These studies add to understanding of HMGB1-induced inflammatory events and vascular barrier disruption and offer the potential for clinical intervention in sepsis-induced ALI.
Keywords: HMGB1, RAGE, acute lung injury, endothelium, MAP kinase, Hsp27
Introduction
The hallmark of acute lung injury (ALI), an inflammatory process involving leukocyte infiltration and generation of proinflammatory cytokines, is the loss of alveolar-capillary integrity with resultant high-permeability, non-hydrostatic pulmonary edema (Hyers et al., 1991; Millar et al., 1989; Suter et al., 1992). The pulmonary endothelium plays a critical role in maintaining a cellular barrier between the vascular compartment and the pulmonary interstitium, with barrier integrity regulated by competing EC contractile forces and adhesive cell-cell tethering forces, both of which are intimately linked to the endothelial cytoskeleton (Dudek and Garcia, 2001). Movement of fluid and solutes across the endothelium primarily occurs via this paracellular pathway (Dudek and Garcia, 2001) with barrier-disrupting mediators producing EC cytoskeletal rearrangement, increased paracellular gap formation, and alveolar flooding, the pathognomonic feature of ALI.
High-mobility group box 1 (HMGB1), a nuclear transcription factor, was first implicated as an important endogenous signaling molecule when it was identified that extracellular HMGB1 released by necrotic and inflammatory cells functions as a late-acting cytokine mediating endotoxin-related lethality in mice (Wang et al., 1999). Extracellular, acetylated endogenous HMGB1 released by macrophages (Park et al., 2003) and monocytes (Andersson et al., 2000), acts as an alarmin to signal danger to neighboring cells (Klune et al., 2008). Since mice challenged with LPS developed increased intestinal barrier dysfunction many hours after the injection of LPS, HMGB1 was hypothesized to be the late-acting mediator that is pathophysiologically responsible for LPS-induced toxicity in that model. It has been demonstrated that HMGB1 is released with a lag phase of 8–32 hr after endotoxin exposure, and that direct administration of purified recombinant HMGB1 induces lethality (Wang et al., 1999). Interestingly, delayed administration of an HMGB1 neutralizing antibody (up to 24 hr after induction of experimental sepsis) attenuated lethality which offers a clinically relevant therapeutic window that is significantly wider than for other known cytokines.
Subsequent studies have increased understanding of the mechanisms by which HMGB1 mediates delayed toxicity. HMGB1 contributes to the development of acute lung injury after hemorrhage (Kim et al., 2005) and intestinal barrier dysfunction after hemorrhagic shock (Raman et al., 2006), suggesting an impact on epithelial and endothelial cell function with HMGB1 increasing in vitro permeability of Caco-2 intestinal epithelial monolayers (Sappington et al., 2002). However, endothelial cells are the prime targets in the vasculature for circulating inflammatory cytokines and thus an effect of HMGB1 on endothelial cells would be logical for eliciting a systemic inflammatory response.
HMGB1 ligates three known receptors all expressed on the surface of endothelial cells—the receptor for advanced glycation end products (RAGE), toll-like receptor 2 (TLR2), and TLR4. RAGE functions as a pattern recognition receptor and binds a variety of ligands, including HMGB1 and AGEs, which are important in the vascular complications of diabetes (Bierhaus et al., 2005). RAGE ligation leads to sustained activation of NFκB and increased RAGE expression, which insure maintenance and amplification of an inflammatory signal (Bierhaus et al., 2005). Signal transduction through RAGE utilizes many mechanisms, including the MAP kinases ERK1/2, p38, and SAPK/JNK, as well as rho-GTPases, phophoinositol-3-kinase, and the JAK/STAT pathway, and via the direct generation of reactive oxygen species (Bierhaus et al., 2005). RAGE participates in murine sepsis, with RAGE−/− KO mice protected against the lethal effects of cecal ligation and puncture via alterations of the innate immune response. This protection was abolished by reconstitution of RAGE in endothelial and hematopoietic cells (Liliensiek et al., 2004). RAGE is also the primary receptor for HMGB1 in bone marrow-derived macrophages, with macrophages from RAGE−/− mice releasing lower amounts of proinflammatory cytokines in response to HMGB1 than macrophages from control or TLR2−/− mice (Kokkola et al., 2005). HMGB1 also interacts directly with TLR2 and TLR4 on macrophages (Park et al., 2006). Both TLR2 and TLR4 are HMGB1 receptors and potentially exert greater influence than RAGE in HMGB1-mediated activation of NFκB in cultured macrophages (Park et al., 2004). Macrophages from genetically engineered mice show the importance of TLR4 and MyD88 in HMGB1-mediated TNF release, while anti-TLR2 antibodies decrease HMGB1 cell surface binding on cultured murine macrophage (Yu et al., 2006).
Taken together, HMGB1 receptors appear to exert cell type-dependent effects. The receptor most actively involved in vascular barrier regulation is unknown. HMGB1 increases expression of adhesion molecules in endothelium including ICAM-1 and VCAM-1 (Fiuza et al., 2003; Treutiger et al., 2003), and induces upregulation of inflammatory mediators such as TNFα, IL-8, monocyte chemotactic protein-1, and plasminogen activator inhibitor 1 (Fiuza et al., 2003). More recently, HMGB1 was recognized as a putative pro-angiogenic factor that stimulates endothelial cell proliferation, chemotaxis, and monolayer wound repair (Mitola et al., 2006; Schlueter et al., 2005). Furthermore, HMGB1 has been demonstrated to promote mesoangioblasts, vessel-associated stem cells that migrate to damaged tissues, to transmigrate across an endothelial monolayer (Palumbo et al., 2004). Such evidence points to endothelial cell participation in a pro-inflammatory cascade in response to HMGB1, but the question remains as to whether HMGB1 directly affects endothelial barrier regulation and if so, by which receptor pathway do these effects become transduced. HMGB1 produces transient phosphorylation of MAP kinases ERK, JNK, and p38 in endothelial cells (Fiuza et al., 2003), signaling pathways involved in EC activation and barrier function. Activation of the p38 MAP kinase is associated with EC barrier dysfunction via actin-binding protein Hsp27 (Garcia et al., 2002), a known downstream target of p38 MAP kinase whose phosphorylation status determines its ability to prevent actin polymerization (Landry and Huot, 1999; Pichon et al., 2004). Despite the circumstantial evidences, there are no published data on the direct effects of HMGB1 on endothelial barrier dysfunction.
In the present study, we characterized the effect of HMGB1 on pulmonary endothelial barrier function and investigated the contribution of known HMGB1 receptors to these processes. Using a variety of physiologic, molecular and cell biologic strategies to delineate the signaling pathway, we challenged human lung ECs with recombinant human HMGB1 and assessed the role of MAP kinases in HMGB1–mediated paracellular gap formation, endothelial barrier function, and HSP27 phosphorylation.
Materials and Methods
Reagents
Recombinant human HMGB1 was purchased from Sigma-Aldrich (St. Louis, MO). Protease inhibitor cocktail set III, phosphatase inhibitor cocktail set II, and pharmacologic inhibitors of p38 MAP kinase and MAPKAP kinase-2 were from EMD Chemicals (San Diego, CA). Silencing RNA transfection reagent, siPORT™ Amine, was purchased from Ambion (Austin, TX). Primary antibodies were purchased from Santa Cruz Biotechnology (Santa Cruz, CA) and Cell Signaling Technology (Danvers, MA). Anti-mouse and anti-rabbit secondary antibodies conjugated to horse radish peroxidase were from GE Health Sciences (Chalfont St. Giles, UK). Enhanced chemiluminescence (ECL), and Supersignal West Dura were from Pierce Biotechnology (Rockford, IL). Texas Red-phalloidin, anti-mouse and rabbit Alexa 488 secondary antibodies, and Prolong mounting solution were from Molecular Probes (Eugene, OR).
Cell Culture
Human pulmonary artery endothelial cells (HPAEC) were from Lonza Group, Ltd (Switzerland) and grown in manufacture’s recommended Endothelial Growth Medium-2 (EGM-2) consisting of defined growth factors and supplemented up to 10% FBS. Cells were grown at 37°C in 5% CO2 incubator and used from passage 6–9. For experiments, ECs were plated at appropriate density (875,000 cells/D60; 300,000 cells/D35; 100,000 cells/CultureSlide; 75,000 cells/ECIS well) and used 3 days after plating unless otherwise noted. Furthermore, medium was changed 1 day prior to all experiments.
RNA interference
On-targetPlus small interfering RNA (siRNA) against RAGE, TLR2, TLR4, p38α, p38β, and MAPKAPK-2 were obtained as pools of four siRNA duplexes from Dharmacon (Lafayette, CO). On-targetPlus siRNA (siCONTROL#2) targeting non-human protein, luciferase, was used as negative control siRNA with minimal off-target silencing. The silencing protocol was optimized to allow transfection of cells shortly after plating and on non-conventional substrates such as gold electrodes. 3–5 hours after cell plating, siRNA (calculated for final concentration of 100 nM siRNA) was premixed with transfection reagent (4 μl/ml siPORT™ Amine) for 5 min and then diluted with basal media based on using half volume typical for a dish/well. After 16–24 hr, equal volume of serum media was added to the media containing siRNA. 48hr post-transfection, the diluted siRNA media was replaced with serum media. Silenced cells were used 3–6 days post-transfection, and the media replaced one day prior to all experiments.
Immunofluorescence
ECs were grown to confluency on 12-chamber, collagen-coated Culture Slides (BD Biosciences, Lexington, KY). After agonist stimulation, cells were washed with phosphate buffered saline twice and fixed with 3.7% formaldehyde for 10 min, permeabilized with 0.25% Triton X-100 in PBS for 5 min, washed and probed with primary antibodies at 1:100–1:200 dilution for 45 min. F-actin was probed with Texas Red-phalloidin at 1:300 dilution. Incubation with secondary antibodies at 1:300 dilution was 30 min. Slides were mounted with Prolong™ anti-fade reagent (Invitrogen, Carlsbad, CA). Stained cells were visualized using a Nikon Eclipse TE2000 inverted microscope (Nikon Inc., Melville, NY) and images acquired using SPOT software (Diagnostic Instruments, Sterling Heights, MI). The integrated densities of paracellular gap area in the endothelium was quantified using Adobe Photoshop CS3 based on a modified protocol as described online by Luke Miller http://www.lukemiller.org/journal/2007/08/quantifying-western-blots-without.html. Briefly, gaps were selected by adjusting the level such that the images of the cells were overexposed with the cell-cell junctional gaps remaining black. Overexposed cells were selected, and using the inverse function under the select tab, the gaps were highlighted. Then, integrated density for the total gaps in the field of view was measured by using the record measurement feature under the analysis tab. At least 3 images of the same experimental conditions were averaged and standard error calculated.
Electrical Resistance Measurements
An electrical cell-substrate impedance sensing (ECIS) system (Applied Biophysics, Troy, NY) was used to measure transendothelial electrical resistance (TER) with EC grown on gold microelectrodes (Garcia et al., 2001). EC were plated directly onto gold microelectrodes of ECIS arrays (8W10E) and cultured for a minimum of 2 days to establish confluency. Data pooling and analysis were performed using in-house-created Epool software which has integrated graphing linked to Microsoft Excel (Schaphorst et al., 2003).
Western Blots
Cells were washed with cold Endothelial Basal Medium (EBM) once and extracted with 0.3% SDS in 10mM Tris lysis buffer (300 μl/D60) containing protease and phosphatase cocktail inhibitors. DNA was sheared with a 26-gauge syringe. Each sample was boiled for 5 min, and diluted with 5X sample buffer (0.56 M Tris pH7.0, 10% SDS, 25% β-ME, 25% sucrose, 0.025% bromophenol blue). Sample proteins were separated with a 4–20% gradient SDS-PAGE gel using the Mini-Protean III (Bio-Rad, Hercules, CA). Proteins were transferred onto Immobilion-P PVDF membrane (Millipore, Bedford, MA), immunoblotted with primary antibodies (1:200–1:1000, 4°C, overnight) followed by secondary antibodies conjugated to HRP (1:5000, room temperature, 30 min), and detected with enhanced chemiluminescence (Pierce ECL or SuperSignal West Dura, Pierce Biotechnology, Rockford, IL) on Biomax MR film (Kodak, Rochester, NY). Densitometry was performed using Adobe Photoshop based on a modified protocol as described online by Luke Miller as referenced above for immunofluorescence.
Statistical analysis
Values are shown as the mean + SE. Data were analyzed using a standard Student’s t-test, and significance in all cases was defined at p < 0.05.
Results
HMGB1 induces dose-dependent decrease in transendothelial electrical resistance (TER), paracellular gap formation and actin cytoskeletal rearrangement
Initial studies measured continuous TER of cultured human lung microvascular EC grown on gold microelectrodes. Challenge with recombinant HMGB1 produced dose-dependent decreases in TER measurements suggesting increased EC barrier dysfunction (Figure 1A and 1B) compared to control buffer (20 mM HEPES, pH 7.8, 150 mM NaCl, 0.2 mM EDTA, 0.1 % Triton X-100, 2 mg/ml leupeptin, and 0.1 mM AEBSF). As HMGB1-induced barrier disruption was maximal ~1hr after challenge (Figure 1A), this time point was used to quantify HMGB1–mediated changes in paracellular gap formation and actin cytoskeletal rearrangement (Figure 2A & 2B). EC grown on glass coverslips were treated with varying concentrations (5–15μg/ml) of HMGB1 and assessed for alterations in polymerized actin (probed with Texas-Red phalloidin) and the adherens junctional protein, VE-cadherin (Figure 2A & 2B). HMGB1 produced prominent paracellular gap formation at 15μg/ml HMGB1. HMGB1 also induced actin cytoskeleton rearrangement, with loss of peripheral organized actin fibers and the development of central stress fibers, a phenotypic change associated with increased contractile activity and increased endothelial permeability as we have previously described (Birukova et al., 2009; Birukova et al., 2004; Dudek and Garcia, 2001). Therefore, the functional assessment of HMGB1-induced EC barrier disruption, TER, is mirrored by morphologic changes in paracellular gap formation and in the EC actomyosin contractile apparatus.
Figure 1. HMGB1 causes rapid and dose-dependent decrease in transendothelial electrical resistance (TER) in human lung endothelial cells.
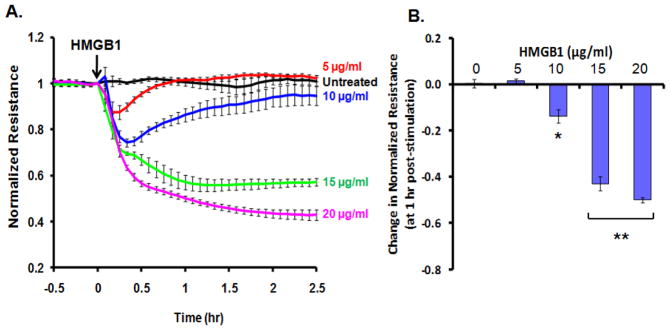
(Panel A) Cultured EC grown to confluency on gold electrodes were treated with recombinant HMGB1 (5, 10, 15, 20 μg/ml) and changes in TER was measured over time. Decreased TER represents endothelial barrier dysfunction or increased vascular permeability. (Panel B) Bar graph depicting dose-dependent changes in resistance at 1 hr post-HMGB1 stimulation. Asterisk (*) denotes p=0.0004, double asterisk (**) denotes p<0.0001 compared with untreated control.
Figure 2. HMGB1 induces paracellular gap formation in human lung EC.

(Panel A) EC were treated with varying concentrations of HMGB1 (1 hr), fixed and probed with antibody to VE-cadherin and Texas Red-phalloidin (to highlight F-actin). HMGB1 induced thinning of VE-cadherin borders and dissociation of junctional adhesion leading to increased paracellular gap formation (yellow arrows) and loss of peripheral F-actin at 15μg/ml HMGB1. (Panel B) Paracellular gaps are quantified using a densitometry software measuring pixels of the gap area. HMGB1 (15μg/ml) led to significant increases in paracellular gap area. Asterisk (*) denotes p=0.008 compared to unstimulated control).
RAGE is the primary receptor in HMGB1-induced TER decrease and paracellular gap formation in EC
Each of the three known HMGB1 receptors (RAGE, TLR2, and TLR4) has been suggested to be the primary receptor for untoward effects of HMGB1. We next sought to specifically determine which receptor was primarily involved in HMGB1-mediated EC barrier disruption and adopted a siRNA strategy targeting each receptor (RAGE, TLR2, or TLR4). Silenced cells were subsequently plated on gold microelectrodes allowing measurement of TER after HMGB1 challenge. Reduction in RAGE expression (siRNA), but not TLR4 or TLR2 silencing, resulted in attenuation of HMGB1-induced TER declines (Figure 3A). We next challenged EC with HMGB1 and fixed after 2 hours of exposure, the time point when HMGB1-induced TER barrier disruption was maximal. EC were probed with VE-cadherin and examined using immunofluorescence (Figure 3B). EC treated with siRNA targeting RAGE were relatively protected from HMGB1-induced paracellular gap formation whereas EC treated with siRNA targeting TLR2 or TLR4 exhibited prominent HMGB1-induced paracellular gap formation similar to controls. Decreased expression of both RAGE and TLR4 were confirmed by western blotting of EC lysates (Figure 3C). Densitometric assessment confirmed an ~50% decrease in RAGE expression in siRNA-treated cells which reflects the degree of attenuation in the TER response seen in these cells.
Figure 3. Reduced RAGE expression (siRNA) protects against HMGB1-induced EC barrier dysfunction and gap formation.

(Panel A) EC were transfected with siRNA targeting RAGE (siRAGE), TLR2 (siTLR2), TLR4 (siTLR4), or a non-human protein used as control (siCONT) and grown to confluency on gold electrodes, treated with 15 μg/ml HMGB1, and TER measured. Silencing of RAGE attenuated HMGB1-induced decrease in TER while silencing of TLR4 augmented HMGB1-induced barrier dysfunction. (Panel B) EC were treated with siRNA and then replated on glass coverslips. EC were treated with 15 μg/ml HMGB1 for 2 hr (corresponding to the time of maximal TER disruption in Panel A), then fixed and probed with antibody to VE-cadherin to observe paracellular gap formation. This morphologic effect parallels the functional TER response of EC observed with the challenge of HMGB1 after treatment with siRNA for each HMGB1 receptor. Receptor silencing has been confirmed for RAGE and TLR4. Silencing of TLR2 could not be confirmed. Asterisks (*) denote significant reduction of receptor expression (p < 0.05) (Panel C).
MAP kinase signaling pathways mediate HMGB1-induced EC barrier disruption
Activation of p38 MAP kinase is central to specific models of agonist –induced EC barrier dysfunction and is associated with lipopolysaccharide-induced EC permeability (Dudek and Garcia, 2001). p38 MAPK activity is critically important to the lung EC barrier disruption induced by pertussis toxin (PTX), and PTX-mediated decreases in TER were related temporally to the phosphorylation of downstream targets of p38 MAPK, including Hsp27 and caldesmon (Garcia et al., 2002). In addition, p38 MAPK participates in EC permeability augmentation by microtubule disassembly (Birukova et al., 2005). Thus, we examined the role of p38 MAP kinase signal transduction pathways in HMGB1-induced EC barrier disruption using the pharmacologic inhibitor, SB203580, which suppresses activity of p38α and p38β isoforms. EC grown to confluency on gold electrodes were treated with 10μM SB203580 for 30 min and then challenged with HMGB1, with continuous TER measurement. Inhibition of the p38 MAP kinase signal transduction pathway resulted in significant attenuation of HMGB1-induced decreases in TER (Figure 4A). Similarly, pharmacologic inhibition of p38 MAP kinase also protected EC against HMGB1-induced paracellular gap formation and cytoskeletal changes with decreased central stress fiber formation in response to HMGB1 challenge (Figure 4B).
Figure 4. Pretreatment with p38 MAP kinase inhibitor attenuates HMGB1-induced decrease in TER, paracellular gap formation, and central stress fiber formation.
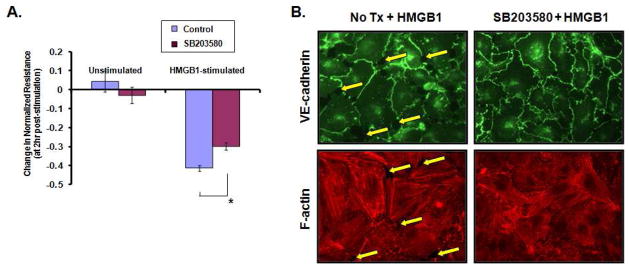
(Panel A) Confluent monolayer of EC pretreated with the p38 MAP kinase pharmacologic inhibitor SB203580 (10 μM, 30 min) were stimulated with 15 μg/ml HMGB1, and TER was measure over a period of time. Pretreatment with the inhibitor protected against HMGB1-mediated decrease in TER, shown 2 hr after HMGB1 stimulation. Asterisk (*) denotes statistical significance p<0.0075 of mean ± standard error (n=3 for SB203580/HMGB1). (Panel B) To visualize paracellular gap formation or stress fiber formation, EC were pretreated with SB203580 (10 μM, 30 min) or control basal media then stimulated with HMGB1 (15 μM/ml, 1hr), then fixed and probed with antibody to VE-cadherin, or Texas Red-phalloidin to highlight F-actin fibers. Pretreatment with p38 MAP kinase inhibitor protected EC from the prominent HMGB1-induced paracellular gap and central stress fiber formation. Yellow arrows denote interendothelial junctional gaps.
HMGB1 induces p38- and MK2-dependent Hsp27 phosphorylation
Hsp27 is a known downstream target of p38 whose phosphorylation status determines its capacity to function as a regulator of actin rearrangement. Therefore, we collected EC whole cell lysates from 0–120 minutes after treatment with 15μg/ml HMGB1 and probed for Hsp27 phosphorylated at ser15, ser78, and ser82. Western blot analysis demonstrated that HMGB1 significantly increases Hsp27 phosphorylation at each of these three known serine residues (Figure 5). The kinases known to catalyze Hsp27 phosphorylation include MAPKAPK-2 (MK2), MAPKAPK-3, MAPKAPK-5 (or PRAK), and p38 MAPK via MAPKAPK-2 activation (Kostenko and Moens, 2009). We investigated MAPKAPK-2 phosphorylation over the same time course and found that HMGB1 caused phosphorylation of MK2 (Figure 6A). HMGB1 also caused phosphorylation of p38 MAPK, although this was more rapid than MAPKAPK-2 or Hsp27 phosphorylation. We next sought to verify that HMGB1-induced Hsp27 phosphorylation depended upon activation of both p38 and MAPKAPK-2. We used an siRNA strategy targeting p38α/β, the predominant p38 isoforms in endothelial cells (Hale et al. 1999) and MAPKAPK-2 and found that decreased expression of both p38α/β and MAPKAPK-2 attenuated Hsp27 phosphorylation in response to HMGB1 (15μg/ml for 60 min) (Figure 6B). Densitometric analysis of Western blots confirmed decreased expression of p38α/β and MAPKAPK-2 (Figure 6C).
Figure 5. HMGB1 induces Hsp27 phosphorylation.

(Panel A) EC were unstimulated (UN) or treated with HMGB1 (15 μg/ml) for 5–120 minutes and whole cell lysates collected. Western blot shows significant phosphorylation of Hsp27 at serine residues 15, 78, and 82 in response to HMGB1. (Panel B) Aggregate densitometry from three separate experiments validates significant Hsp27 phosphorylation after EC treatment with HMGB1. Asterisks (*) denote p<0.05 compared to unstimulated EC.
Figure 6. HMGB1-induced Hsp27 phosphorylation is p38- and MAPKAPK-2-dependent.

(Panel A). EC were unstimulated (UN) or treated with HMGB1 (15 μg/ml) for 5–120 minutes and whole cell lysates collected. Western blot analysis shows phosphorylation of both p38 and MAPKAPK-2 (referred to in this Figure as MK-2). (Panel B) EC were transfected with siRNA targeting p38α and p38β. (sip38α/β), MAPKAPK-2 (siMK-2), or a non-human protein used as control (siCONT) and grown to confluence. EC then remained unstimulated (UN) or were treated with HMGB1 (15 μg/ml) for 60 minutes and whole cell lysates collected. Western blot analysis shows attenuation of Hsp27 phosphorylation at serine residues 15, 78, and 82 in EC with decreased expression of p38α/β or MK2. (Panel C). Aggregate densitometry of Western blots confirms decreased expression of the target proteins with representative blots shown (p < 0.03 for each).
Discussion
A mechanistic understanding of the events which increase endothelial permeability, the hallmark of ALI, is critical to the ultimate development of therapeutic strategies targeting this cellular pathogenetic process. Our data show that HMGB1, a late mediator of sepsis, plays a critical role in EC barrier disruption, and that this process is mediated by rearrangement of the actin cytoskeleton into a contractile phenotype. Our initial TER data show that HMGB1 causes EC barrier disruption in a dose-dependent manner (Figure 1), results that are consistent with immunofluorescence studies of EC after treatment with HMGB1 (Figure 2). We recognize that the concentration of HMGB1 required to elicit barrier disruption (5–20μg/ml) was significantly greater than levels of HMGB1 noted in the serum of septic animals (Wang et al., 1999) and in human patients with sepsis (Sunden-Cullberg et al., 2005). However, Ueno and colleague reported levels of HMGB1 in pulmonary epithelial lining fluid (ELF) (obtained by bronchoscopic microsampling) from patients with ALI that are comparable to the concentrations utilized in our experiments (Ueno et al., 2004). ALI patients were reported to have HMGB1 concentrations in ELF approximately 1000-fold higher than in plasma, with a median peak ELF concentration of 9.2μg/ml. Interestingly, control patients had a median peak ELF HMGB1 level of 4.9μg/ml, suggesting a physiologic role for HMGB1 in the lung at lower levels and offering an explanation for the delayed increase in TER above baseline seen when EC are treated with 5μg/ml HMGB1.
Injury to the pulmonary endothelium is an essential characteristic of ALI and is critical to the pathophysiology of high-permeability pulmonary edema (Ware, 2006). The EC lining of the pulmonary vasculature forms a semi-permeable barrier between the blood and the interstitial space. Disruption of this barrier results in fluid flux into the interstitium and ultimately into the airspaces, leading to the clinical and physiological derangements observed in ALI. The primary route of this fluid flux is via paracellular gaps that form in proinflammatory states. Formation of paracellular gaps can be understood using a model of competing forces, in which contractile forces within the cell generate centripetal tension and compete against the tethering generated by cell-cell adhesive properties (Dudek and Garcia, 2001). These opposing forces are functionally linked to the actin cytoskeleton of EC. Actin filaments are central to EC barrier regulation-- disruption of actin increases permeability (Dudek and Garcia, 2001) while actin stabilization prevents mediator-induced barrier disruption (Phillips et al., 1989). The actin cytoskeleton also interacts with various adhesion molecules, including zona occludens, zona adherens, and focal adhesion complex proteins, cytoskeletal targets in EC barrier function. In this study, we demonstrate that HMGB1 causes reorganization of the actin cytoskeleton and disruption of the key junctional protein, VE-cadherin (Figure 2). Furthermore, HMGB1 induces paracellular gap formation, which correlates with increased barrier dysfunction (Figure 1), highlighting the importance of the actin cytoskeleton in mediating endothelial barrier function.
A clear objective of this work was to identify the receptor or receptors involved in HMGB1–mediated vascular permeability. To target the three putative HMGB1 receptors, we utilized siRNA technology to individually downregulate the protein expression of RAGE, TLR2, or TLR4, followed by either measurements of endothelial barrier function or staining for interjunctional gap formation. In comparison to control siRNA, silencing of RAGE attenuated HMGB1-induced endothelial barrier dysfunction to approximately the same degree as the level of decreased RAGE expression, whereas silencing of TLR2 failed to significantly affect the HMGB1 response with the caveat that significant knockdown of TLR2 expression could not be confirmed (Figure 3A,C). The degree of decreased RAGE expression directly correlates with the degree of attenuation of HMGB1-induced barrier disruption, suggesting that RAGE is the primary receptor mediating HMGB1 effects. Similar results were observed with gap formation in which silencing of RAGE (but not TLR2 or TLR4), attenuated HMGB1-induced gap formation (Figure 3B), suggesting RAGE is the primary receptor by which HMGB1 induces increased permeability via cytoskeletal rearrangement leading an increase in gap formation. Significantly decreased expression of both RAGE and TLR4 by siRNA treatment were confirmed (Figure 3C). Interestingly, decreased TLR4 expression actually augmented the loss of TER seen after HMGB1 stimulation. This was an unexpected finding and suggests that TLR4 may participate in a complex barrier-protective role via an as yet unknown mechanism. Preliminary investigation did not find increased expression of RAGE in response to siRNA targeting TLR4, although other signaling proteins may have been affected. These effects clearly merit further investigation.
Our data indicate that RAGE is the main barrier-regulatory HMGB1 receptor in ECs, a finding consistent with the demonstration that macrophages from RAGE−/− mice release lower amounts of proinflammatory cytokines in response to HMGB1 than macrophages from control or TLR2−/− mice (Kokkola et al., 2005). Furthermore, in a murine model of sepsis, RAGE−/− mice were protected against the lethal effects of cecal ligation and puncture, and this protection was abolished by reconstitution of RAGE in endothelial and hematopoietic cells (Liliensiek et al., 2004). Our findings are particularly interesting in light of the significant literature linking endothelial RAGE to the vascular hyperpermeability that is the hallmark of diabetic vasculopathy. AGEs are associated with diabetic complications via RAGE, with increased vascular permeability being a hallmark of diabetic vasculopathy (Wautier et al., 1996). Red blood cells bearing AGEs increase vascular permeability in normal rats, and hyperpermeability in diabetic rats was prevented by treatment with soluble RAGE, which competes with cell surface RAGE for AGE binding (Wautier et al., 1996). In cultured HUVEC, AGE-modified albumin disrupts the vascular endothelial (VE)-cadherin complex, which functions at the EC adherens junction, by decreasing levels of VE-cadherin, β-catenin, and γ-catenin (Otero et al., 2001). Furthermore, EC treated with AGE-modified albumin showed increased permeability and actin cytoskeleton rearrangement with stress fiber formation, effects which were attenuated by treatment with anti-RAGE antibody, specific MAP kinase inhibitors, or dominant negative forms of ERK and p38 MAP kinase (Guo et al., 2006). RAGE is a known receptor of HMGB1, and plays a role in AGE-mediated changes in EC structure and function.
Although TLR2 and TLR4 have also been described as HMGB1 receptors, neither TLR2 nor TLR4 appeared to be directly involved HMGB1-induced gap formation (Figure 3B) and endothelial barrier dysfunction (Figure 3A), although decreased expression of TLR2 could not be confirmed. However, we suspect that these receptors may indirectly contribute to endothelial barrier dysfunction by activation of independent signaling pathways that release additional cytokines such as TNF-α and interleukins that do not signal through RAGE. For example, Yu and colleague have used macrophages from knockout mice to demonstrate the importance of TLR4 and MyD88 in HMGB1-mediated TNF release (Yu et al., 2006). Since HMGB1-induced activation of NFκB is known to involve TLR2 and TLR4 via downstream targets such as MyD88, TIRAP, IRAK-1 and IRAK-2 (Park et al., 2004), we hypothesize that HMGB1 may activate the NFκB signaling pathway to cause cytokine production and thereby indirectly promote endothelial barrier dysfunction. This will be thoroughly investigated in the future.
Ligation of each of the three HMGB1 receptors leads to activation of MAP kinases as well as the NFκB pathway. Downstream targets of activated p38 MAP kinase are MAPK-activated protein kinase 2 (MAPKAPK-2 or MK-2), MK-3, and PRAK (MK-5), as well as the transcription factors ATF-2, Mac, and MEF2. MAPKAPK-2 is believed to be among the most important p38 substrates in mediating proinflammatory effects since MAPKAPK-2 knockout mice have increased stress resistance and survive LPS-induced endotoxic shock (Kotlyarov et al., 1999). A key downstream target of p38 MAP kinase and MAPKAPK-2/3 is heat shock protein, Hsp27, which binds and modulates the actin cytoskeleton. Hsps are induced when cells are exposed to environmental stresses and act to protect the cell from apoptosis via actin microfilament stability (Lavoie et al., 1995). In unstressed cells, Hsp27 forms large aggregates of non-phosphorylated monomers, which may cap the plus ends of the actin filament to regulate microfilament assembly. Phosphorylation of Hsp27 disrupts the large aggregates into smaller oligomers, which interact along the length of F-actin to protect the filament against breakage by actin-severing proteins (Mounier and Arrigo, 2002). For example, Hsp27 is found localized in the leading edge of lamellipodia bound to barbed end of F-actin. Upon phosphorylation of Hsp27, the barbed ends are uncapped allowing for elongation of F-actin while the phosphorylated Hsp27 binds along the F-actin to stabilize the microfilament. Thus, the phosphorylation status of Hsp27 is critical to its ability to stabilize the actin cytoskeleton and therefore regulate endothelial barrier function. Phosphorylation of Hsp27 has been shown to be a common effector target in response to agonists that cause ALI-like pulmonary edema, including TNF-α (Arrigo, 1990), thrombin (Mendelsohn et al., 1991), and hydrogen peroxide (Huot et al., 1997), confirming that Hsp27 phosphorylation may be important in the endothelial disruption. More recently, Hirano and colleague reported that increased Hsp27 phosphorylation was associated with pathological lung injury in an animal model of sepsis (Hirano et al., 2004). We interrogated the HMGB1 signaling pathway and found direct involvement of specific isoforms of p38 MAP kinase, as SB203580 blocks only p38α and p38β and not p38γ or p38δ isoforms (Figure 4). Furthermore, our siRNA approach revealed the involvement of the p38/MAPKAPK-2 signaling pathway in HMGB1-induced Hsp27 phosphorylation (Figure 6). Two interesting points must be addressed with regard to our results. First, the attenuation of HMGB1-induced TER disruption resulting from pretreatment with SB203580 was incomplete, highlighting the likelihood that other signaling pathways are involved. We have begun preliminary work to investigate the involvement of NFkB in this effect, and TER data suggests that NFkB is also important in HMGB1-induced TER disruption (data not shown). Second, there is an apparent discrepancy between MK-2 silencing (~20%) and the pronounced effect of this knockdown on HSP27 phosphorylation at serine 15 and serine 78. One possible explanation for this effect derives from the presence of other kinases in the signaling pathway linking p38 activation to HSP27 phosphorylation, namely MK-3 and MK-5, which phosphorylate HSP27 in vitro (Gaestel 2006). While we did not specifically investigate these kinases, it is possible that they contribute to HMGB1-induced phosphorylation of HSP27 in our system, and that the siRNA targeting MK-2 may be non-specific, allowing knockdown of these kinases as well. This may explain the relatively robust attenuation of HSP27 phosphorylation in the context of only moderate MK-2 knockdown.
In summary, we characterized HMGB1 effects on endothelial barrier function and explored the role of MAP kinases in this response. Using pharmacologic and molecular strategies, we demonstrate that HMGB1 produces dose-dependent paracellular gap formation and decreases in TER, a reflection of loss of barrier integrity. These events were in concert with loss of peripheral organized actin fibers and development of actin stress fibers, a phenotypic change associated with increased contractile activity and increased EC permeability. HMGB1 induces these events via ligation of the RAGE receptor, resulting in downstream activation of p38 MAP kinase and MAPKAPK-2, and phosphorylation of the actin-binding protein Hsp27. While RAGE is clearly involved in the functional effect of HMGB1 on TER and paracellular gap formation, TLR2 and TLR4 may also be involved in mediating Hsp27 phosphorylation induced by HMGB1 via mechanisms not yet investigated (Figure 7). Additional studies should address the role of TLR2 and TLR4 and potential interaction with the NFκB pathway, as well as the efficacy of HMGB1 neutralization on mouse models of sepsis and HMGB1-induced ALI.
Figure 7. Cartoon schematic of proposed HMGB1 pathway leading to paracellular gap formation and endothelial barrier disruption.

HMGB1 induces EC barrier disruption via ligation of the RAGE receptor, resulting in downstream activation of p38 MAP kinase and MAPKAPK-2, and phosphorylation of the actin-binding protein Hsp27. While RAGE is clearly involved in the functional effect of HMGB1 on TER and paracellular gap formation, TLR2 and TLR4 may also be involved in mediating Hsp27 phosphorylation induced by HMGB1 via mechanisms not yet investigated.
Acknowledgments
The authors gratefully acknowledge the expert technical assistance and contributions of Sara Camp and thank Lakshmi Natarajan for continued maintenance and supply of high quality endothelial cell culture stocks. This work was supported by grants from the National Institute of Child Health and Human Development (K12 HD043387) and the National Heart, Lung, and Blood Institute (K08 HL093359, HL 58064).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Andersson U, et al. High mobility group 1 protein (HMG-1) stimulates proinflammatory cytokine synthesis in human monocytes. J Exp Med. 2000;192:565–70. doi: 10.1084/jem.192.4.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrigo AP. Tumor necrosis factor induces the rapid phosphorylation of the mammalian heat shock protein hsp28. Mol Cell Biol. 1990;10:1276–80. doi: 10.1128/mcb.10.3.1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bierhaus A, et al. Understanding RAGE, the receptor for advanced glycation end products. J Mol Med. 2005;83:876–86. doi: 10.1007/s00109-005-0688-7. [DOI] [PubMed] [Google Scholar]
- Birukova AA, et al. Endothelial permeability is controlled by spatially defined cytoskeletal mechanics: atomic force microscopy force mapping of pulmonary endothelial monolayer. Nanomedicine. 2009;5:30–41. doi: 10.1016/j.nano.2008.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birukova AA, et al. MAP kinases in lung endothelial permeability induced by microtubule disassembly. Am J Physiol Lung Cell Mol Physiol. 2005;289:L75–84. doi: 10.1152/ajplung.00447.2004. [DOI] [PubMed] [Google Scholar]
- Birukova AA, et al. Role of Rho GTPases in thrombin-induced lung vascular endothelial cells barrier dysfunction. Microvasc Res. 2004;67:64–77. doi: 10.1016/j.mvr.2003.09.007. [DOI] [PubMed] [Google Scholar]
- Dudek SM, Garcia JG. Cytoskeletal regulation of pulmonary vascular permeability. J Appl Physiol. 2001;91:1487–500. doi: 10.1152/jappl.2001.91.4.1487. [DOI] [PubMed] [Google Scholar]
- Fiuza C, et al. Inflammation-promoting activity of HMGB1 on human microvascular endothelial cells. Blood. 2003;101:2652–60. doi: 10.1182/blood-2002-05-1300. [DOI] [PubMed] [Google Scholar]
- Gaestel M. MAPKAP kinases--MKs--two’s company, three’s a crowd. Nature Reviews Molecular Cell Biology. 2006;7:120–30. doi: 10.1038/nrm1834. [DOI] [PubMed] [Google Scholar]
- Garcia JG, et al. Sphingosine 1-phosphate promotes endothelial cell barrier integrity by Edg-dependent cytoskeletal rearrangement. J Clin Invest. 2001;108:689–701. doi: 10.1172/JCI12450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia JG, et al. Critical involvement of p38 MAP kinase in pertussis toxin-induced cytoskeletal reorganization and lung permeability. Faseb J. 2002;16:1064–76. doi: 10.1096/fj.01-0895com. [DOI] [PubMed] [Google Scholar]
- Guo XH, et al. Advanced glycation end products induce actin rearrangement and subsequent hyperpermeability of endothelial cells. Apmis. 2006;114:874–83. doi: 10.1111/j.1600-0463.2006.apm_372.x. [DOI] [PubMed] [Google Scholar]
- Hale KK, et al. Differential expression and activation of p38 mitogen-activated protein kinase alpha, beta, gamma, and delta in inflammatory cell lineages. J Immunol. 1999;162:4246–52. [PubMed] [Google Scholar]
- Hirano S, et al. Endothelial barrier dysfunction caused by LPS correlates with phosphorylation of HSP27 in vivo. Cell Biol Toxicol. 2004;20:1–14. doi: 10.1023/b:cbto.0000021019.50889.aa. [DOI] [PubMed] [Google Scholar]
- Huot J, et al. Oxidative stress-induced actin reorganization mediated by the p38 mitogen-activated protein kinase/heat shock protein 27 pathway in vascular endothelial cells. Circ Res. 1997;80:383–92. doi: 10.1161/01.res.80.3.383. [DOI] [PubMed] [Google Scholar]
- Hyers TM, et al. Tumor necrosis factor levels in serum and bronchoalveolar lavage fluid of patients with the adult respiratory distress syndrome. Am Rev Respir Dis. 1991;144:268–71. doi: 10.1164/ajrccm/144.2.268. [DOI] [PubMed] [Google Scholar]
- Kim JY, et al. HMGB1 contributes to the development of acute lung injury after hemorrhage. Am J Physiol Lung Cell Mol Physiol. 2005;288:L958–65. doi: 10.1152/ajplung.00359.2004. [DOI] [PubMed] [Google Scholar]
- Klune JR, et al. HMGB1: endogenous danger signaling. Mol Med. 2008;14:476–84. doi: 10.2119/2008-00034.Klune. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kokkola R, et al. RAGE is the major receptor for the proinflammatory activity of HMGB1 in rodent macrophages. Scand J Immunol. 2005;61:1–9. doi: 10.1111/j.0300-9475.2005.01534.x. [DOI] [PubMed] [Google Scholar]
- Kostenko S, Moens U. Heat shock protein 27 phosphorylation: kinases, phosphatases, functions and pathology. Cell Mol Life Sci. 2009;66:3289–3307. doi: 10.1007/s00018-009-0086-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotlyarov A, et al. MAPKAP kinase 2 is essential for LPS-induced TNF-alpha biosynthesis. Nat Cell Biol. 1999;1:94–7. doi: 10.1038/10061. [DOI] [PubMed] [Google Scholar]
- Landry J, Huot J. Regulation of actin dynamics by stress-activated protein kinase 2 (SAPK2)-dependent phosphorylation of heat-shock protein of 27 kDa (Hsp27) Biochem Soc Symp. 1999;64:79–89. [PubMed] [Google Scholar]
- Lavoie JN, et al. Modulation of cellular thermoresistance and actin filament stability accompanies phosphorylation-induced changes in the oligomeric structure of heat shock protein 27. Mol Cell Biol. 1995;15:505–16. doi: 10.1128/mcb.15.1.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liliensiek B, et al. Receptor for advanced glycation end products (RAGE) regulates sepsis but not the adaptive immune response. J Clin Invest. 2004;113:1641–50. doi: 10.1172/JCI18704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendelsohn ME, et al. The 29-kDa proteins phosphorylated in thrombin-activated human platelets are forms of the estrogen receptor-related 27-kDa heat shock protein. Proc Natl Acad Sci U S A. 1991;88:11212–6. doi: 10.1073/pnas.88.24.11212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millar AB, et al. Tumour necrosis factor in bronchopulmonary secretions of patients with adult respiratory distress syndrome. Lancet. 1989;2:712–4. doi: 10.1016/s0140-6736(89)90772-1. [DOI] [PubMed] [Google Scholar]
- Mitola S, et al. Cutting edge: extracellular high mobility group box-1 protein is a proangiogenic cytokine. J Immunol. 2006;176:12–5. doi: 10.4049/jimmunol.176.1.12. [DOI] [PubMed] [Google Scholar]
- Mounier N, Arrigo AP. Actin cytoskeleton and small heat shock proteins: how do they interact? Cell Stress Chaperones. 2002;7:167–76. doi: 10.1379/1466-1268(2002)007<0167:acashs>2.0.co;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otero K, et al. Albumin-derived advanced glycation end-products trigger the disruption of the vascular endothelial cadherin complex in cultured human and murine endothelial cells. Biochem J. 2001;359:567–74. doi: 10.1042/0264-6021:3590567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palumbo R, et al. Extracellular HMGB1, a signal of tissue damage, induces mesoangioblast migration and proliferation. J Cell Biol. 2004;164:441–9. doi: 10.1083/jcb.200304135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JS, et al. Activation of gene expression in human neutrophils by high mobility group box 1 protein. Am J Physiol Cell Physiol. 2003;284:C870–9. doi: 10.1152/ajpcell.00322.2002. [DOI] [PubMed] [Google Scholar]
- Park JS, et al. High mobility group box. 1 protein interacts with multiple Toll-like receptors. Am J Physiol Cell Physiol. 2006;290:C917–24. doi: 10.1152/ajpcell.00401.2005. [DOI] [PubMed] [Google Scholar]
- Park JS, et al. Involvement of toll-like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. J Biol Chem. 2004;279:7370–7. doi: 10.1074/jbc.M306793200. [DOI] [PubMed] [Google Scholar]
- Phillips PG, et al. Phallacidin prevents thrombin-induced increases in endothelial permeability to albumin. Am J Physiol. 1989;257:C562–7. doi: 10.1152/ajpcell.1989.257.3.C562. [DOI] [PubMed] [Google Scholar]
- Pichon S, et al. Control of actin dynamics by p38 MAP kinase - Hsp27 distribution in the lamellipodium of smooth muscle cells. J Cell Sci. 2004;117:2569–77. doi: 10.1242/jcs.01110. [DOI] [PubMed] [Google Scholar]
- Raman KG, et al. The role of RAGE in the pathogenesis of intestinal barrier dysfunction after hemorrhagic shock. Am J Physiol Gastrointest Liver Physiol. 2006;291:G556–65. doi: 10.1152/ajpgi.00055.2006. [DOI] [PubMed] [Google Scholar]
- Sappington PL, et al. HMGB1 B box increases the permeability of Caco-2 enterocytic monolayers and impairs intestinal barrier function in mice. Gastroenterology. 2002;123:790–802. doi: 10.1053/gast.2002.35391. [DOI] [PubMed] [Google Scholar]
- Schaphorst KL, et al. Role of sphingosine-1 phosphate in the enhancement of endothelial barrier integrity by platelet-released products. Am J Physiol Lung Cell Mol Physiol. 2003;285:L258–67. doi: 10.1152/ajplung.00311.2002. [DOI] [PubMed] [Google Scholar]
- Schlueter C, et al. Angiogenetic signaling through hypoxia: HMGB1: an angiogenetic switch molecule. Am J Pathol. 2005;166:1259–63. doi: 10.1016/S0002-9440(10)62344-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunden-Cullberg J, et al. Persistent elevation of high mobility group box-1 protein (HMGB1) in patients with severe sepsis and septic shock. Crit Care Med. 2005;33:564–73. doi: 10.1097/01.ccm.0000155991.88802.4d. [DOI] [PubMed] [Google Scholar]
- Suter PM, et al. High bronchoalveolar levels of tumor necrosis factor and its inhibitors, interleukin-1, interferon, and elastase, in patients with adult respiratory distress syndrome after trauma, shock, or sepsis. Am Rev Respir Dis. 1992;145:1016–22. doi: 10.1164/ajrccm/145.5.1016. [DOI] [PubMed] [Google Scholar]
- Treutiger CJ, et al. High mobility group 1 B-box mediates activation of human endothelium. J Intern Med. 2003;254:375–85. doi: 10.1046/j.1365-2796.2003.01204.x. [DOI] [PubMed] [Google Scholar]
- Ueno H, et al. Contributions of high mobility group box protein in experimental and clinical acute lung injury. Am J Respir Crit Care Med. 2004;170:1310–6. doi: 10.1164/rccm.200402-188OC. [DOI] [PubMed] [Google Scholar]
- Wang H, et al. HMG-1 as a late mediator of endotoxin lethality in mice. Science. 1999;285:248–51. doi: 10.1126/science.285.5425.248. [DOI] [PubMed] [Google Scholar]
- Ware LB. Pathophysiology of acute lung injury and the acute respiratory distress syndrome. Semin Respir Crit Care Med. 2006;27:337–49. doi: 10.1055/s-2006-948288. [DOI] [PubMed] [Google Scholar]
- Wautier JL, et al. Receptor-mediated endothelial cell dysfunction in diabetic vasculopathy. Soluble receptor for advanced glycation end products blocks hyperpermeability in diabetic rats. J Clin Invest. 1996;97:238–43. doi: 10.1172/JCI118397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu M, et al. HMGB1 signals through toll-like receptor (TLR) 4 and TLR2. Shock. 2006;26:174–9. doi: 10.1097/01.shk.0000225404.51320.82. [DOI] [PubMed] [Google Scholar]