

Abstract
Drug discovery and therapeutic development for disorders of the central nervous system (CNS) represents one of the largest unmet markets in modern medicine. We have increasingly recognized that the lack of stringent assessment of mitochondrial function during the discovery process has resulted in drug recalls, black box warnings, and an urgent need to understand the metabolic liability of small molecules in neural systems. Given that the brain is the most energetically demanding organ, even modest perturbations in neuronal energetic pathways have been shown to impact growth, signaling, connectivity, and the restorative capacity of the CNS. In this work, we describe several tools to assess metabolic activity of primary neuronal cultures and neural cell lines using an acute model of injury induced by oxygen glucose deprivation. Methods include the measurement of total ATP and NADH, enzymatic assessment of lactate production by anaerobic respiration, as well as viability assays. We also present a modified screening method for assessing aerobic respiration of immortalized cell lines using galactose challenge.
Keywords: Mitochondria, Oxidative phosphorylation, Neuron, ATP, Lactate, Energetics, Oxygen glucose deprivation, Neurotoxicity
1. Introduction
There is an increasing awareness that appropriate metabolic tone is a key feature of biological processes including neurotransmission, cellular differentiation, and development as well as adaptation to stress (1–4). The brain represents the most energy demanding organ in the body accounting for 20% of total oxygen consumption and 25% of total body glucose utilization (5–7). Failure to maintain energetic status has been linked to a host of neurological disorders (8–15). Moreover, the drug discovery market has revealed that failure to account for off-target drug actions which impact aerobic respiration or mitochondrial function is a major source of liability (16, 17). In each of the acute and chronic disorders of CNS function, fundamental shifts in aerobic respiration, metabolic coupling, ionic homeostasis, and reactive oxygen species generation, which compromise neuronal health, have been observed. In the most simplified models, it is postulated that the ability to maintain sufficient energetic reserves even under stress redirects cell death from necrotic loss to apoptotic death and these features dictate the targets that are appropriate for drug discovery (18, 19). In this work, we discuss the advantages of several methods and model systems to assess metabolic function, adaptation, and survival in neural platforms in the context of an acute injury model of stroke. Methodologies are presented in a manner to facilitate cross purposing to assess energetic tone in cells exposed acutely or chronically to agents with possible neurotoxic action.
1.1. Essential Salient Features of the Krebs Cycle and Oxidative Phosphorylation
To accurately interpret the data acquired using the methods presented within this chapter, a basic understanding of the underlying principles of energetic status is required. In neurons, pyruvate is created within the cytoplasm through glycolysis and serves as the dominant mitochondrial substrate for aerobic respiration (20). It is transported into mitochondria and catalyzed into acetyl-CoA for use in the Krebs cycle to produce the reducing equivalents NADH and FADH2.
The electron transport chain is located within the inner mitochondrial membrane and transfers electrons from NADH and to molecular oxygen forming a proton gradient that FADH2 drives the formation of high-energy phosphate bonds on ATP from ADP. This aerobic pathway of energy also known as oxidative phosphorylation results in a combined total of 36 ATP. A far less efficient means of producing high-energy phosphate in the form of ATP involves pyruvate conversion to lactate setting the stage for glycolysis to occur by the liberation of NAD+. Lactate is actively extruded from cells along with hydrogen and the accumulation of lactate is an excellent means to quickly assess dependence upon anaerobic metabolism. In the CNS, it is becoming increasingly clear that astrocytes, the predominate glia cell type in the brain, provide an essential metabolic support role for neurons by releasing lactate which can be converted back to pyruvate for aerobic respiration (Fig. 1).
Fig. 1.
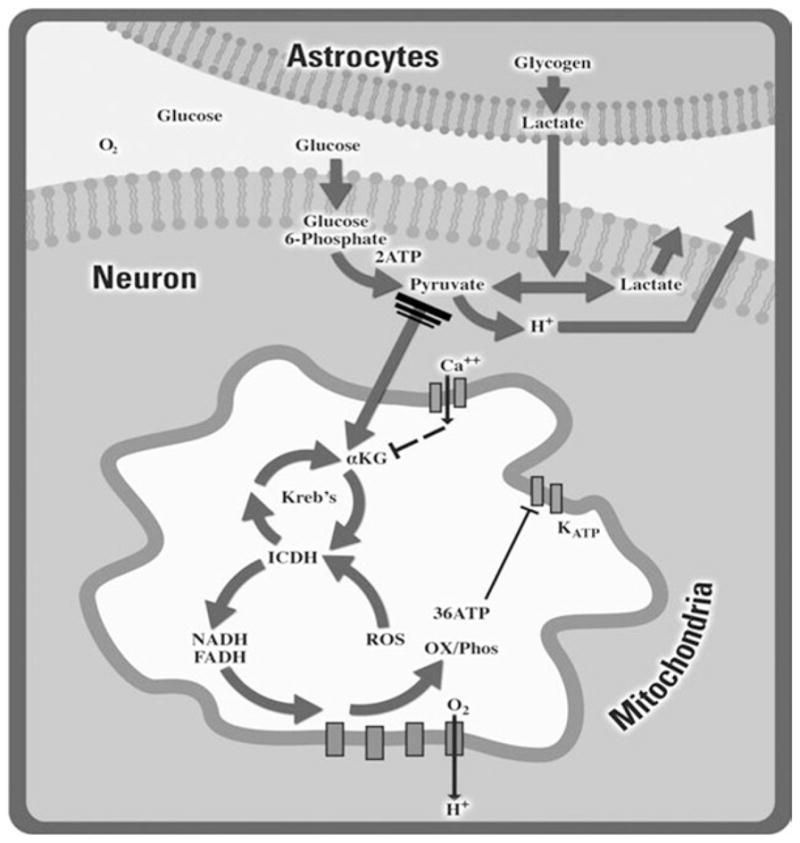
Many factors play a role in determining a neuron’s bioenergetic capacity. Through glycolysis, glucose is converted into pyruvate within the cytoplasm of the cell. In the presence of oxygen, this pyruvate is transported into the mitochondria and converted into acetyl-CoA to enter the Krebs cycle for further generation of NADH and FADH. The gradient formed by electron transfer from these reduced donors to oxygen leads to the production of ATP in the presence of ADP and phosphate via ATP synthase. When oxygen becomes limiting, anaerobic respiration can augment energetic capacity by converting pyruvate into lactate resulting in ATP production, albeit to a lesser extent than via aerobic respiration.
1.2. Cell Line Usage and Energetics
Prior to attempting to manipulate the cellular energetic status following various stimuli, knowledge of the cell’s baseline energetic profile is required. The use of tumor-derived, immortalized neuronal cell lines including PC-12, HT-22, and SY5Y cells has become increasingly popular due to their ease of passage and transfection, relative homogeneity, and low cost. As the tumor cells from which these lines are derived, these cell lines also maintain the ability to adapt to low oxygen conditions and effectively use glycolysis to produce sufficient ATP (21, 22). Despite abundant oxygen present under normal culture growth conditions and fully functional mitochondria, these highly proliferative cells continue to depend on anaerobic pathways to generate ATP. Similarly, under conditions of high extracellular glucose, these cells often rely solely on anaerobic respiration to fuel ATP production, a condition commonly described as the Crabtree effect (21–23). Today’s culture practices exacerbate these kinds of adaptations which normal neurons and glia could not endure in vivo as cell lines are often maintained in high-glucose medium. We believe that these kinds of adaptations at the energetic and molecular level are strongly linked to the poor predictive value of these cells in the context of understanding mitochondria in vitro. This fact is starkly evidenced by the fact that many cell lines can survive hours in conditions of complete oxygen and glucose deprivation which would cause rapid death in both primary neuronal cultures and in vitro (21, 24–26). High throughput screens using primary cultures is impractical as the cells are extremely expensive to maintain, are time intensive, and exhibit low yield. In our experience, the best use of neural and other cell lines is to force cultures to rely more heavily upon aerobic respiration using a galac-tose challenge than expose them to secondary stressors.
1.2.1. Principles of the Galactose Challenge Assay
Galactose is a hexose that differs from glucose only by the con-figuration of its carbon four hydroxyl group. The replacement of glucose with galactose in the culture medium to force cells to rely more heavily on oxidative phosphorylation for ATP formation was initially described by H.G. Crabtree in 1935 (27) and has been used to determine underlying metabolic defects in a host of other cell lines (21–23). Cells are placed in medium containing 25–100% galactose and mitochondrial activity is assessed by a 3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyl tetrazolium bromide (MTT) assay. Galactose metabolism promotes oxidative phosphorylation by promoting the use of glutamine and glutamate to fuel the Krebs cycle as opposed to pyruvate. In galactose-rich medium, cells which have increased proliferation and decreased production of lactate are the most aerobically active or adept at switching to an aerobic phenotype most readily. Establishing titering and proliferation curves for growth is essential to determine the set points at which proliferation is impeded, or in cells which have impaired oxidative phosphorylation, concentrations of galactose that cause cell death. Excellent reviews of establishing proliferation curves have been presented elsewhere (28, 29).
1.2.2. Principles of the MTT Assay
The MTT assay is used most often to assess cellular viability, but in the case of the galactose challenge, where mitochondrial activity is compromised, this technique can be used as an indirect measurement of cellular respiration. The method is based on the cleavage of the yellow tetrazolium salt MTT by active mitochondrial dehydrogenases to generate purple formazan crystals, a process that requires NADH and NADPH. The crystals are dissolved in acidified isopropanol and the purple solution is then measured spectrophotometrically. Other processes in addition to oxidative phosphorylation can consume the NADH as well as NADPH and combining this technique with other biochemical assays of total ATP reserves, lactate generation or pyruvate stores presents a fuller picture of the cell’s metabolic profile and are typically considered next step strategies once proliferation curves for various galactose levels are established (30). ATP assays as described in Subheading 1.4.1 of cells grown under galactose challenge vs. full glucose provides an excellent means to determine at what point metabolic compensations are brought online.
1.3. Assessing Primary Neuronal Survival Following Oxygen Glucose Deprivation
1.3.1. Principles of Primary Neuronal Cultures
Primary neuronal cultures are the gold standard for assessing neurotoxicity in vitro. They represent the most physiologically relevant metabolic model available for evaluating bioenergetic effects of a host of stressors including oxygen glucose deprivation (OGD) to model the loss of blood experienced during ischemic stroke. The composition of primary cultures can be altered by adding mitotic inhibitors at various times following isolation and plating. The default cellular program for isolated cells from CNS at embryonic day 16 or beyond in rat will promote the proliferation of glial components as they continue to proliferate following dissociation whereas the neuronal cell population is postmitotic. Glial inhibition in the 48 h following plating promotes formation of neuron-enriched cultures resulting in cultures which contain in the excess of 95% neurons and 5% astrocytes. Neurobasal medium provides a low serum base that can be supplemented with growth factors, neurotransmitters, antioxidants, vitamins, and fatty acid precursors to promote long-term survival and cell to cell communication.
1.3.2. Principles of OGD
Cell cultures exposed to OGD capture many of the essential features of the CNS when blood flow is halted in ischemic stroke. These cells undergo rapid energetic decline, release of glutamate, failure of the Na+–K+ ATPases, generation of oxygen-derived free radicals, glutamate transporter reversal, NMDA receptor hyper-stimulation, ionic dysfunction, mitochondrial swelling, abnormal protein trafficking, and signaling cascades associated with both apoptotic and necrotic cell death. (31, 32).
The OGD method consists of placing neurons into glucose-free medium bubbled with an anaerobic mix to remove oxygen. A sealed hypoxic chamber containing the anaerobic gas mixture is used to maintain neurons in the oxygen deprived state at physiological temperatures for various durations. By conducting experiments 3 weeks following dissection (21–25 days in vitro), excitotoxicity is fully expressed as a consequence of neuronal expression of a full compliment of NMDA receptors (33, 34). Using this methodology, we have found that in culture neurons can survive 5 and 15 min of OGD, but longer exposures become increasingly toxic with maximal cell death observed 24 h after incubations longer than a single hour. Similar experiments in neural cell lines often require 4–6 h of OGD to elicit death (35–37), which highlights the glycolytic nature of these cells and their ability to survive anaerobic conditions for periods that are not physiologically relevant to humans.
1.3.3. Principles of the Lactate Dehydrogenase Assay
To assess neuronal survival following OGD exposure, we utilize a toxicity assay based on lactate dehydrogenase (LDH), an enzyme that catalyzes the formation of pyruvate to lactate. Upon cell damage or lysis, membrane integrity is compromised and LDH is released into the medium. Medium containing this enzyme is then used to catalyze the reduction of NAD+ to NADH and H+ by the oxidation of lactate to pyruvate. The resulting NADH reacts with a tetrazolium dye to form a pink color that can be measured spectrophotometrically. To account for variation in total LDH content between experiments, raw LDH values can be normalized to the toxicity caused by 100 μM NMDA plus 10 μM glycine, which is known to cause 100% cell death in this system (33, 34). To ensure predictability across culture preparations, all experiments should be performed using cells derived from multiple independent dissections.
1.4. Evaluating Neuronal Energetic Status (ATP, Lactate, and Pyruvate Assays)
1.4.1. Principles of the ATP Assay
The ATP assay is based on the bioluminescence formed from the interaction of the enzyme luciferase with luciferin and ATP present in the cell. Following OGD treatment, cells are lysed and mixed with the enzyme luciferase mixture to generate light in an ATP-dependent manner that can be measured with a luminometer. Care must be given to the appropriate handling of cells to ensure that external ATP and adenosine are removed and that hydrolysis of ATP is minimized by rapid sample preparation. While this common bench assay is widely available and amenable to equipment in nonspecialized labs, higher fidelity results are obtained by the use of high performance liquid chromatography to measure adenosine, ADP, and ATP to assess the total energetic reserves. This platform also allows for the assessment of other high-energy phosphate molecules including GTP. The bench assay does, however, provide a quick and inexpensive measure of total ATP which can be used in combination with a protein assay to account for variability in plating density that can arise in cell culture. This method alone does not discriminate between anaerobic or aerobic ATP content, but is rather a measure of total ATP.
1.4.2. Principles of the Lactate and Pyruvate Assays
As previously mentioned, pyruvate metabolism is a key determinate of metabolic efficiency. It can be converted into acetyl-CoA for use in the Krebs cycle or into lactate for anaerobic respiration. Measurements of pyruvate and lactate concentrations can be used to determine a cell’s aerobic versus anaerobic respiration rates. An increase in lactate production over time with a decrease in pyruvate following a cellular stressor would suggest an increased anaerobic pathway of ATP generation. For each assay, cells are first lysed and lactate or pyruvate is enzymatically oxidized by the corresponding substrate to produce a color that can be measured spectrophotometrically at the 570 nm wavelength or alternatively through fluorescence using an excitation of 535 nm and emission at 587 nm.
1.5. Future Methodologies for Assessing Cellular Energetic Status
Though beyond the scope of this chapter, new methodologies are emerging for noninvasively measuring cellular bioenergetic profiles. These technologies utilize live mammalian cells in a convenient, microplate format for fast and sensitive measurement of respiration and ATP utilization determined simultaneously and noninvasively by quantitative assessment of the oxygen consumption rate, extracellular acidification rate, lactate production, and carbon dioxide production (38, 39). This instrumentation has been adapted with high reliability using electrochemical sensors (21) and in a more mainstream platform using fluorophores and a microplate reader. Cost of the commercially available platforms range in the hundreds of thousands, while lab built devices require an extensive knowledge of mechanical and electrical engineering. However, this technology can be combined with measures of cell fate, protein expression, and traditional biochemistry to provide a comprehensive and powerful platform for basic research and drug discovery.
2. Materials
2.1. Galactose Challenge
Control medium: This medium should be the normal medium used for proper cell-line maintenance with its appropriate glucose concentration.
Galactose/glucose medium: This medium will consist of a percentage of galactose and glucose for a total concentration equal to the glucose concentration of the control medium. For example, HT-22 cell control medium contains 10 mM glucose. Using a glucose-free version of this control medium, a 25% galactose/75% glucose challenge medium will consist of 2.5 mM galactose and 7.5 mM glucose.
Sterile tissue culture plates and tissue culture hood.
Incubator appropriate for neuronal cell line.
2.2. MTT Assay
MTT solution: 15 mg MTT, 3 ml of culture medium used throughout the experiment. Solution is stable for at least 6 months when stored at −20°C.
MTT Solubilization solution: 10% Triton-X 100, anhydrous isopropanol, and 0.1 N HCl. For 50 ml, mix 5 ml Triton-X 100, 45 ml isopropanol, 1 drop of 12 M HCl.
Incubator appropriate for neuronal cell line.
Shaker and/or sonicator.
2.3. Neuronal Dissection and Maintenance of Cultures
Borate Buffer: 4.76 g (77 mM) boric acid, 2.54 g (6.66 mM) borax, 1,000 ml double-distilled H2O, pH to 8.4 with NaOH. Sterile filter and store at 4°C (see Note 1).
Poly-l-ornithine (PLO, Sigma P3655): 1 mg PLO for each 2 ml of borate buffer (see Note 2).
Embryonic day 18 pregnant rat (see Note 3).
Plating medium: 80% Dulbecco’s Modified Eagle’s Medium (DMEM), 10% Ham’s F12-nutrients, 10% bovine calf serum (iron-supplemented, Hyclone) with 24 U/ml penicillin, 24 g/ml streptomycin, and 2 mM l-glutamine. Store at 4°C.
Trypsin.
Culture medium: Neurobasal medium (Invitrogen) containing 50× B27 supplement (Gibco), 50× NS21 supplement (40), 24 U/ml penicillin, and 24 g/ml streptomycin. Store at 4°C (see Note 4).
Dissection tools: large scissors, fine tip dissecting scissors, and forceps.
500× Cytosine Arabinoside (AraC) Stock: 100 mg Ara-C, 6 ml double-distilled H2O. Sterile filter solution and store at −20°C in 0.5 ml aliquots.
Working AraC: 20 μl of 500× stock, 10 ml double-distilled H2O. Sterile filter solution and store at 4°C.
0.1N HCl: 248 ml double-distilled H2O, 2 ml 12N HCl.
95 and 70% Ethanol.
HBSS: 8.4 g NaCl, 224 mg KCl, 2.38 g HEPES, 1.0 g glucose, 1,000 ml double-distilled H2O, pH to 7.3 with NaOH. Sterile filter the medium in the tissue culture hood and store at 4°C.
Trypan blue, microscope and hemocytometer for cell counts.
Tissue culture water.
Large coverslips (25 mm) and 6-well tissue culture plates.
Anesthesia for rat, Nembutal sodium solution.
Sonicator and orbital shaker.
Sterile Tissue culture hood.
Sterile incubator at 37°C and 5% CO2.
35-mm Culture dishes (sterile).
Pasteur pipettes.
2.4. Oxygen Glucose Deprivation
MEM/BSA/HEPES: 487.5 ml clear MEM, 12.5 ml 1 M HEPES, and 50 mg bovine serum albumin. Sterile filter and store at 4°C. Make up fresh medium before each experiment.
MEM/BSA/HEPES/2× N2: 98 ml MEM/BSA/HEPES and 2 ml N2 supplement (Gibco) (see Note 5).
Glucose-free balanced salt solution (GBSS): 5.26 g NaCl, 0.125 g KCl, 66.6 mg CaCl2, 1.43 g HEPES, 600 ml double-distilled H2O. pH to 7.3 using NaOH and HCl. Sterile filter and store at 4°C.
NMDA toxicity medium: MEM/BSA/HEPES/2× N2, 10 μM glycine, and 100 μM NMDA.
Anaerobic gas mix (95% nitrogen and 5% CO2).
Hypoxic chamber.
Sterile Incubator at 37°C and 5% CO2.
35-mm Culture dishes (sterile).
N2 meter.
2.5. LDH and Lysis LDH Assay
LDH Assay kit (Sigma): LDH assay substrate, dye, enzyme, and lysis buffer. Enzyme is stored at -20°C. Lysis buffer is stored at room temperature, while all other components are stored at 4°C.
LDH assay mixture: Equal amounts of LDH assay substrate, dye and enzyme. Mix the appropriate volume to have 20 μl for each well that will be measured in a 96-well plate plus 10% extra. Both the enzyme and mixture is light sensitive so combine reagents in the appropriate lighting conditions. Make up fresh before each experiment.
Phosphate-buffered saline solution (PBS): 1 l double-distilled H2O, 80 g NaCl, 2 g KCl, 2 g KH2PO4, 115 g Na2HPO4. pH to 7.5 using NaOH or HCl. Dilute this 10× PBS to 1× using double-distilled H2O.
PBS/lysis buffer: 150 μl of lysis buffer and 950 μl of PBS. Make 1.5 ml of buffer for each well of a 6-well tissue culture plate plus 40 μl for each blank well of the 96-well plate.
96-Well clear transparent plate.
Repeater pipette.
Spectrometer to measure absorbance at a wavelength of 492 nm.
2.6. ATP Bioluminescent Assay
Ice cold PBS.
ViaLight® Plus Kit (Lonza): Assay buffer, cell lysis reagent, ATP monitoring reagent (AMR). Store at 4°C.
AMR: Place 10 ml of assay buffer into lyophilized AMR. Mix to reconstitute the AMR and then add mixture to the assay buffer bottle. Store reagent in polypropylene tubes at -20°C for up to 2 months.
96-Well transparent plate with white walls.
Spectrometer to measure luminescence.
2.7. Lactate and Pyruvate Assays
Ice cold PBS.
Lactate assay kit (BioVision): Lactate assay buffer, standard, probe, and enzyme mix.
Pyruvate assay kit (BioVision): Pyruvate assay buffer, standard, probe, and enzyme mix.
Teflon cell scrapers.
Microcentrifuge at 4°C.
96-Well transparent plates.
1.5-ml Tubes.
Spectrometer to measure absorbance at 570 nm or fluorescence with an excitation of 530 nm and emission of 587 nm.
3. Methods
3.1. Galactose Challenge
In a sterile tissue culture hood, plate your cells in the appropriate size tissue culture plates containing the galactose/ glucose medium at various percentages or control medium for comparison heated to the necessary temperature for the cell line.
Cells can be maintained in the galactose/glucose medium percentages for the appropriate time depending on your cell type. At a minimum, expose cells to this medium for 24–48 h (see Note 6).
Proceed with the MTT assay (Subheading 3.2) to evaluate mitochondrial respiration and compare with control medium exposed cells.
Alternatively, cells can be exposed to increasing percentages of galactose over time. Upon splitting cells, place half of the cells in a higher galactose percentage medium. MTT assays should be performed at similar exposure times for each galactose/glucose percentage medium to determine differences in mitochondrial respiration.
3.2. MTT Assay
Add MTT solution in an amount equal to 10% of culture medium volume to each well in a sterile tissue culture hood. For example, for a 6-well tissue culture plate which contains 1.5 ml of medium in each well, add 150 μl of MTT solution (see Note 7).
Incubate the culture plate for 2 h depending on cell density. Longer incubation times (2–4 h) may be necessary to compensate for low cell densities and metabolic activity.
Remove the plate and add an amount of MTT solubilization solution equal to the original medium volume to dissolve the formazan crystals. For 6-well tissue culture plates, add 1.5 ml to each well.
Gently shake, pipette up and down, or use a sonicator to completely dissolve the crystals.
Using the spectrometer, measure absorbance of the samples directly in the plate using a wavelength of 570 nm (Fig. 2).
Fig. 2.
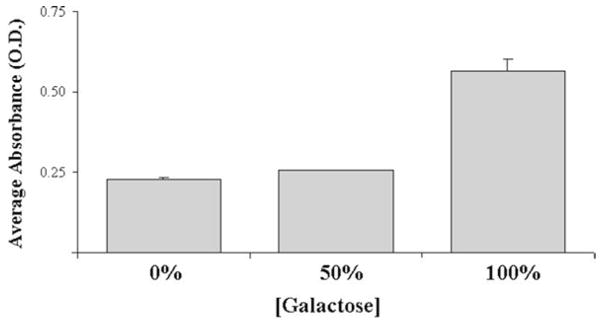
The HT-22 cell line exhibits an increase in mitochondrial activity upon exposure to galactose. The HT22 mouse neuroblastoma cell line was exposed to varying concentrations of galactose for 48 h and reducing capacity of the cells was assessed using an MTT assay. Complete replacement of glucose with galactose (100%) led to an increase in MTT absorbance due to enhanced mitochondria activity. Slow metabolism of galactose altering viability and ATP production reveals underlying dependence upon oxidative phosphorylation. This data suggests that even with half the normal amount of glucose, HT-22 cells can successfully produce sufficient ATP for survival and proliferation.
3.3. Neuronal Dissection
3.3.1. Coverslip Preparation
Place one large box of coverslips and 250 ml of double-distilled H2O into a 500 ml beaker and swirl the contents gently for 2 min. Decant the water into the sink making sure no coverslips pour out.
Add 250 ml of new double-distilled H2O and sonicate for 15 min.
Repeat steps 1–2. Following the last step, decant the water, and add 250 ml of 0.1N HCl to the coverslips. Cover the beaker with parafilm and place on the orbital shaker for 1 h.
Decant the 0.1N HCl into the appropriate hazardous waste drum and wash the coverslips three times with double-distilled H2O.
Add 250 ml of 95% ethanol to the coverslips and place the beaker on the orbital shaker for an additional hour.
Decant the 95% ethanol into the appropriate hazardous waste drum and wash the coverslips twice with double-distilled H2O.
Add 250 ml of 95% ethanol to the coverslips, place parafilm over the beaker, and store the cleaned coverslips until future use.
3.3.2. Embryonic Dissection
At least 2 days before dissection, in a sterile tissue culture hood place cleaned coverslips into 6-well tissue culture plates (one coverslip per well). Parafilm the tissue culture plate and store until future use.
One day before dissection, place culture plates containing coverslips into sterile tissue culture hood. Remove the lid of each plate and expose to ultraviolet (UV) light for 1 h (see Note 8).
Place 2 ml of freshly made PLO/borate buffer solution into each well following UV treatment. After a minimum of 1 h, wash each well twice with 2 ml of tissue culture water. Remove the water and lids from each plate and allow the plate to dry in the hood for 2–4 h. Add 1.5 ml of plating medium to each well and place plates in the incubator until dissection.
For dissection preparation, weigh out 3 mg of trypsin and dissolve in 5 ml of HBSS. Sterile filter the solution into a 35-mm culture dish and label. Add 3 ml of HBSS to seven of the 35-mm culture dishes. Add 450 μl of trypan blue to a 1.5 ml Eppendorf tube. Clean dissection tools with 70% alcohol. Aspirate the coating medium from the culture plates. Add 5 ml of plating medium to two 15-ml tubes. Set out three more tubes for tituration procedure. Place plating medium in hood to bring to room temperature.
Inject the rat with the sedative, Nembutal, by intraperitoneal injection (~2 ml), then wait a few minutes for her to relax and lay down. Check her “blink” reaction and squeeze her foot to determine if she is properly sedated.
Place the rat on the absorbent pad belly up and spray the belly with 70% alcohol. Use the large forceps to lift the belly skin and cut open the rat with the heavy-duty dissecting scissors along the midline up to the diaphragm area.
Next, cut the muscle along the midline to expose the embryos and the diaphragm and cut both sides of the rat’s diaphragm to stop respiration.
Use the large forceps to pull all the embryos out of the rat. Cut the embryos open with fine-tip dissecting scissors and cut off the head of the embryo into the HBSS culture dish.
Transfer one embryo head into a new dish of HBSS that is now under the microscope. Hold the head straight up, puncture the skin and membrane outside of the brain with the dissecting forceps. Peel the skin and membrane toward the front of the head until the brain pops out. Turn the head around and peel toward the back of the head until the brain stem is exposed. Put the dissecting forceps under the brain stem and pull toward the back of the head until the brain is separated with the skull. Pinch cut the brain stem to separate the brain completely.
Place the brain in a new HBSS dish. With the dorsal of the brain facing up, hold the brain stem gently with forceps and separate the hemispheres with the dissecting forceps. Pull the cerebral cortex away from the brain stem.
Place one cortex in a new dish of HBSS. Flip the cerebral cortex so that the outside is facing up. Pinch cut the olfactory bulb with the dissecting forceps and peel the meninges from the front toward the back. Try to peel it off in one piece (see Note 9).
Pinch cut any mid-brain structures underlying the cortex and transfer the cortex to a new dish with HBSS. Repeat for the other cortices (see Note 10).
Transfer the dish with all the cortices to the hood and place the cortices into the trypsin and HBSS solution with the forceps, making sure to take as little HBSS as possible with them. Allow cortices to sit in the solution for 30 min in the hood at room temperature.
Transfer the cortices from the trypsin to a new dish with HBSS with a Pasteur pipette, one at a time. Do not break the cortices. Gently swish them around to wash off the trypsin. Repeat once more.
Transfer the cortices with a new Pasteur pipette to the first tube of plating medium. Slowly triturate the cortices with the Pasteur pipette to maximize the total cell number and viability. Let the chunks settle down to the bottom of the tube.
Transfer only the cell suspension (not the larger pieces) to a new 15-ml tube with a Pasteur pipette. Again, slowly triturate with cortices. Continue this process until the suspension looks even (see Note 11).
When triturations are complete, add 50 μl of the cell suspension to the Eppendorf tube with trypan blue (a 1:10 dilution). Mix well by flipping the tube upside-down several times. Remove 50 μl of trypan blue mixture, and add it to the hemo-cytometer (it takes ~15–20 μl to fill each side).
-
Count the number of live cells and dead cells in three of the 4 × 4 quadrants (one on one side and two on the other side). Use this series of formulas to determine the needed cell to medium dilution.
(Live cells)/(three squares) = y
y × 10 = z (for 1:10 Dilution in Trypan blue)
z × 10,000 = (hemocytometer volume correction)
Dilute the cell suspension to the desire plating density with plating medium (350,000 cell/ml). Plate 2 ml/well, and change the pipette after every two plates. Make sure to gently swirl medium bottle to insure proper homogeneity of cell suspension in between plates. Carefully return dishes to the incubator, making sure not to splash medium on lids or disrupt cells.
Add 1–2 μM working Ara-C solution directly into each culture well 2 days after dissociation to inhibit glial proliferation. On the following day, replace the plating medium with 2 ml growth medium. Medium will need to be partially replaced every 2–3 days. All neuronal experiments should be conducted 3 weeks after dissociation (Fig. 3).
Fig. 3.

Healthy neuronal cultures following dissection. Neuronal architecture was monitored following initial plating until 3 weeks following dissection. Healthy neurons display phase bright somas early on following the dissection with many neurites. Older neurons demonstrate increasing well-defined processes along with phase bright somas.
3.4. Oxygen Glucose Deprivation
Warm MEM/BSA/HEPES, MEM/BSA/HEPES/2× N2 and GBSS to 37°C. Place the hypoxic chamber with lid off into sterile tissue culture hood and treat with ultraviolet light for 10 min.
Place appropriately labeled 35-mm dishes into the chamber with their lids off. By adjusting the N2 meter in the hood to 5 PSI, degas the chamber for 5 min with the anaerobic mixture by connecting the N2 tube to the entrance tube of the chamber. Check the exit tube on the chamber to ensure that the anaerobic mix is flowing properly by covering up the exit tube on the chamber for ~5 s. When released, there will be a hissing sound and a slight rise and fall in the lid of the chamber. Close the clasp on the exit tube followed by closing the entrance tube and place the chamber in the incubator for a minimum of 10 min (see Note 12).
Place the GBSS medium into the hood and degas it for 5 min by placing a 2-ml pipette in the bottle with the anaerobic mix flowing through it. Make sure to check for proper flow (5 PSI).
Return the chamber from the incubator to the hood and add 3 ml GBSS medium to each dish. Carefully add one coverslip of neurons to each dish using the forceps. Make sure that the medium covers the coverslips. Replace the chamber lid and allow the anaerobic mixture to flow for 5 min. Close bath clasps and return to the incubator for the appropriate incubation time.
Following the incubation, return the chamber to the hood, remove the lid and using a 25-ml pipette, remove the GBSS medium from each dish. Gently wash each dish with 2 ml of MEM/BSA/HEPES. Add 1.5 ml MEM/BSA/HEPES/2× N2 to each dish, place the lid on the dish, and return to incubator for the remaining post time.
For normalizing to total kill, add NMDA toxicity medium to wells containing 1–2 coverslips.
3.5. LDH and Lysis LDH Assay
In a 96-well transparent plate, fill all wells in column A with 40 μL of control medium. In the case of our primary neuronal culture OGD experiments, we use MEM/BSA/ HEPES/2× N2.
Pipette 40 μl of each sample from your toxicity experiment in triplicate into the 96-well plate. Using a repeater, add 20 μl of the LDH assay mix to each well. The reaction is light sensitive so cover the plate with paper towel and take to the spectrometer (see Note 13).
-
Shake the plate for 5 s to mix components and store the plate in a dark location for 20–30 min at room temperature. Measure absorbance at a wavelength of 490 nm using the spectrometer.
Alternatively or in combination with the LDH assay, a lysis LDH assay can be performed. Lysis LDH is a modification of the assay which allows you to wash off the “released” LDH, then lyse healthy cells to determine cell viability.
Wash each well twice with 2 ml of PBS. Add 1.5 ml of PBS/ lysis buffer to each well and incubate the plate for 45 min at 37°C. Following the lysis, proceed with the LDH protocol at step 1. The PBS/lysis buffer will be used for the blank wells instead of MEM/BSA/HEPES/2× N2.
Data can be normalized to LDH and lysis LDH values from NMDA-treated cells to obtain a percentage of death or viability (Fig. 4).
Fig. 4.

Neurons cannot survive long periods of OGD or NMDA toxicity. (a) Primary neuronal cultures were either naïve, exposed to 90 min of OGD (90′OGD) or NMDA (100 μM for 60 min) and cell death was assessed 24 h later by measuring LDH release. The higher LDH values signify a release of this enzyme into the medium from ruptured cells observed with 90 min OGD or NMDA exposure. Data represent the mean ± SEM from six-independent experiments. Asterisk denotes statistical significance by two-tailed t test with p < 0.05. (b) Representative photomicrographs taken 24 h after the exposures illustrate the effect of OGD or NMDA exposure on neuronal architecture. Neurons from control conditions have phase bright somas with well-defined processes, whereas the 90 min OGD and NMDA exposed neurons show signs of cell death with small debris evident and widespread neurite beading and retraction.
3.6. ATP Bioluminescent Assay
Remove culture plate from the incubator for 5 min to allow plate to cool to room temperature. Set up new 6-well tissue culture plates by adding 300 μL of cell lysis reagent to each well. After cooling, remove coverslips from initial plate and place in the plate containing the cell lysis reagent (see Note 14).
Allow cells to lyse for 10 min. During that time, remove an AMR aliquot from the -20°C refrigerator to thaw, and program the plate reader with the following parameters: measurement mode luminescence, integration time of 1,000 ms, and gain of 150.
Transfer 80 μl of cell lysate from each well to a 96-well transparent white plate. Add 100 μl of AMR to each well using a repeater and let the plate sit at room temperature for 2 min before measuring on the spectrometer. Use cellular lysate to determine protein concentrations of each well. It is necessary to normalize ATP values to protein concentrations to account for differences between cultures (see Note 15, Fig. 5).
Fig. 5.
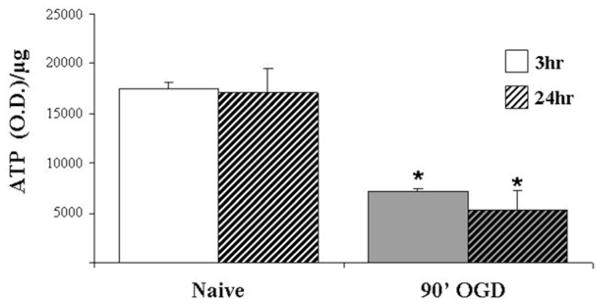
ATP generation collapses following 90 min of OGD. ATP levels from neuronal cultures were measured 3 or 24 h after exposure to 90 min of OGD (90′OGD) or control medium (Naïve). ATP levels are significantly reduced 3 h following OGD exposure and do not recover even 24 h later. Data were normalized for total protein and are expressed as ATP/μg of protein ± SEM from three-independent experiments. Statistical significance compared with control as determined by two-tailed t test with *p < 0.05.
3.7. Lactate and Pyruvate Assays
The procedure for both the lactate and pyruvate assays is similar except for using the corresponding appropriate enzyme, probe, standard, and assay buffer within the kit.
Wash each well twice with ice cold PBS and harvest in 300 μl of assay buffer, transferring the same buffer to each well and scraping to combine the contents of all the wells into a 1.5-ml tube.
To fully lyse the cells, pipette cells up and down several times. Save 50 μl of each lysate in a separate tube for protein assay. The protein assay sample can be stored at -80°C.
Spin the cellular lysate at 700× g in a microcentrifuge for 10 min at 4°C to clear extract.
During the spin, prepare the standard curve by diluting the appropriate standard 100-fold to 1 nmol/μl in assay buffer. In the first column of the 96-well plate, add 0, 2, 4, 6, 8, and 10 μl of standard into each well individually. Adjust the volume of each well to 50 μl with the appropriate assay buffer to generate 0, 2, 4, 6, 8, and 10 nmol/well of the standard. Repeat this in the second column of the 96-well plate for a duplicate standard.
Add the appropriate volume of each cleared extract in duplicate to the 96-well plate. To determine the volume that is within the standard curve, use two or three initial volumes of 50 μl or less. Bring the volume of each sample well to 50 μl using the assay buffer.
For each well, prepare a 50 μl total volume reaction mix consisting of 46 μl of assay buffer, 2 μl of probe, and 2 μl of enzyme mix. Add 50 μl of the reaction mix to each well containing the standard or test samples. Protect plate from light as reaction is light sensitive.
At the spectrometer, shake the plate for 5 s to mix well. Incubate the samples at room temperature for 30 min protected from light. Measure the absorbance at 570 nm or fluorescence with an excitation of 535 nm and emission of 587 nm. Normalize values to protein concentrations as determined using a protein assay (Fig. 6).
Fig. 6.

Neurons are able to metabolically adapt following a brief OGD exposure. Lactate and pyruvate levels were measured at various times following an exposure to 5 or 90 min of OGD. The nonlethal 5 min OGD exposure did not significantly impact the production of lactate and pyruvate. In contrast, a lethal 90 min exposure results in a dramatic increase in lactate production and a loss of pyruvate over time indicating an inability for these neurons to adapt metabolically. Data were normalized to control cells and represent the mean from four-independent experiments ± SEM.
Footnotes
Borate buffer should be made up ahead of time as it takes approximately 8 h with gentle mixing for all components to go into solution.
PLO: borate buffer should be made up fresh before use.
The use of older embryos results in less neuronal plasticity and higher cell death following dissections.
We have found that a combination of the commercially available B27 supplement and a lab made NS21 supplement provides enhanced long-term health of neuronal cultures (40).
The N2 supplement is a chemically defined, serum-free supplement necessary for the growth and maintenance of primary neurons. Without the N2 supplement, the neurons will die within 24 h.
Longer exposures may allow cells to make proper adaptations necessary for aerobic pathways. If longer exposure times are used, make sure to feed cells in a similar manner as if cultures were in the control medium.
Phenol red and microbial contamination can lead to erroneous measurements during the MTT assay.
Longer ultraviolet exposures can lead to the leaching of chemicals from the plate into the culture medium and result in cellular toxicity.
If not removed, cells of the meninges will proliferate and overtake the neurons within the culture.
All cortices should be in trypsin within 30 min from the start of the dissection due to activity of cellular proteases compromising neuronal membrane integrity and culture health.
Trituration steps are critical. If too harsh, neuronal health will be compromised.
This warming procedure is done to minimize any stress to neurons due to temperature differentiations during the OGD experiment.
Pop any bubbles with a needle or forceps as they will interfere with absorbance readings.
The transfer of the cover slips to a new plate is due to ATP released into the current medium. The new well contains the cell lysis reagent and takes only the cells on the cover slip into account for the ATP, instead of ATP released into the medium.
The ATP lysis reagent is not compatible with the BioRad protein assay kit. Rather, we use the Pierce BCA protein assay kit to determine protein concentrations.
References
- 1.Zhang DW, Shao J, Lin J, Zhang N, Lu BJ, Lin SC, Dong MQ, Han J. RIP3, an energy metabolism regulator that switches TNF-induced cell death from apoptosis to necrosis. Science. 2009;325:332–336. doi: 10.1126/science.1172308. [DOI] [PubMed] [Google Scholar]
- 2.Dhar SS, Wong-Riley MT. Coupling of energy metabolism and synaptic transmission at the transcriptional level: role of nuclear respiratory factor 1 in regulating both cytochrome c oxidase and NMDA glutamate receptor subunit genes. Journal of Neuroscience. 2009;29:483–492. doi: 10.1523/JNEUROSCI.3704-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sonoda J, Mehl IR, Chong L-W, Nofsinger RR, Evans RM. PGC-1beta controls mitochondrial metabolism to modulate circadian activity, adaptive thermogenesis, and hepatic steatosis. Proceedings of the National Academy of Sciences. 2007;104:5223–5228. doi: 10.1073/pnas.0611623104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rossi DJ, Brady JD, Mohr C. Astrocyte metabolism and signaling during brain ischemia. Nature Neuroscience. 2007;10:1377. doi: 10.1038/nn2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Erecinska M, Cherian S, Silver IA. Energy metabolism in mammalian brain during development. Progress in Neurobiology. 2004;73:397–445. doi: 10.1016/j.pneurobio.2004.06.003. [DOI] [PubMed] [Google Scholar]
- 6.Erecinska M, Dagani F. Relationships between the neuronal sodium/ potassium pump and energy metabolism. Effects of K+, Na+, and adenosine triphos-phate in isolated brain synaptosomes. Journal of General Physiology. 1990;95:591–616. doi: 10.1085/jgp.95.4.591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Magistretti PJ, Pellerin L, Rothman DL, Shulman RG. Energy on demand. Science. 1999;283:496–497. doi: 10.1126/science.283.5401.496. [DOI] [PubMed] [Google Scholar]
- 8.Grunewald T, Beal MF. Bioenergetics in Huntington’s disease. Annals of the New York Academy of Sciences. 1999;893:203–213. doi: 10.1111/j.1749-6632.1999.tb07827.x. [DOI] [PubMed] [Google Scholar]
- 9.Blandini F, Braunewell KH, Manahan-Vaughan D, Orzi F, Sarti P. Neurodegeneration and energy metabolism: from chemistry to clinics. Cell Death & Differentiation. 2004;11:479–484. doi: 10.1038/sj.cdd.4401323. [DOI] [PubMed] [Google Scholar]
- 10.Chan PH. Mitochondrial Dysfunction and Oxidative Stress as Determinants of Cell Death/Survival in Stroke. Annals of the New York Academy of Sciences. 2005;1042:203–209. doi: 10.1196/annals.1338.022. [DOI] [PubMed] [Google Scholar]
- 11.Andersen JK. Oxidative stress in neurodegeneration: cause or consequence? Nature Medicine. 2004;10(Suppl):S18–25. doi: 10.1038/nrn1434. [DOI] [PubMed] [Google Scholar]
- 12.Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443:787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- 13.Aschner M, Erikson KM, Hernandez EH, Tjalkens R. Manganese and its role in Parkinson’s disease: from transport to neuropathology. Neuromolecular Medicine. 2009;11 :252–266. doi: 10.1007/s12017-009-8083-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Planel E, Miyasaka T, Launey T, Chui D-H, Tanemura K, Sato S, Murayama O, Ishiguro K, Tatebayashi Y, Takashima A. Alterations in Glucose Metabolism Induce Hypothermia Leading to Tau Hyper-phosphorylation through Differential Inhibition of Kinase and Phosphatase Activities: Implications for Alzheimer’s Disease. Journal of Neuroscience. 2004;24:2401–2411. doi: 10.1523/JNEUROSCI.5561-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sherer TB, Betarbet R, Stout AK, Lund S, Baptista M, Panov AV, Cookson MR, Greenamyre JT. An In Vitro Model of Parkinson’s Disease: Linking Mitochondrial Impairment to Altered alpha -Synuclein Metabolism and Oxidative Damage. Journal of Neuroscience. 2002;22 :7006–7015. doi: 10.1523/JNEUROSCI.22-16-07006.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dykens JA, Will Y. The significance of mitochondrial toxicity testing in drug development. Drug Discovery Today. 2007;12:777–785. doi: 10.1016/j.drudis.2007.07.013. [DOI] [PubMed] [Google Scholar]
- 17.Petrozzi L, Ricci G, Giglioli NJ, Siciliano G, Mancuso M. Mitochondria and neurodegeneration. Bioscience Reports. 2007;27:87–104. doi: 10.1007/s10540-007-9038-z. [DOI] [PubMed] [Google Scholar]
- 18.Nicotera P, Leist M. Energy supply and the shape of death in neurons and lymphoid cells. Cell Death & Differentiation. 1997;4:435–442. doi: 10.1038/sj.cdd.4400265. [DOI] [PubMed] [Google Scholar]
- 19.Nicholls DG, Budd SL. Mitochondria and neuronal survival. Physiology Reviews. 2000;80:315–360. doi: 10.1152/physrev.2000.80.1.315. [DOI] [PubMed] [Google Scholar]
- 20.Hertz L, Drejer J, Schousboe A. Energy metabolism in glutamatergic neurons, GABAergic neurons and astrocytes in primary cultures. Neurochemical Research. 1988;13 :605–610. doi: 10.1007/BF00973275. [DOI] [PubMed] [Google Scholar]
- 21.Rodriguez-Enriquez S, Juarez O, Rodriguez-Zavala JS, Moreno-Sanchez R. Multisite control of the Crabtree effect in ascites hepatoma cells. European Journal of Biochemistry. 2001;268:2512–2519. doi: 10.1046/j.1432-1327.2001.02140.x. [DOI] [PubMed] [Google Scholar]
- 22.Yan H, Parsons DW, Jin G, McLendon R, Rasheed BA, Yuan W, Kos I, Batinic-Haberle I, Jones S, Riggins GJ, Friedman H, Friedman A, Reardon D, Herndon J, Kinzler KW, Velculescu VE, Vogelstein B, Bigner DD. IDH1 and IDH2 mutations in gliomas. New England Journal of Medicine. 2009;360:765–773. doi: 10.1056/NEJMoa0808710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Marroquin LD, Hynes J, Dykens JA, Jamieson JD, Will Y. Circumventing the Crabtree effect: replacing medium glucose with galactose increases susceptibility of HepG2 cells to mitochondrial toxicants. Toxicological Sciences. 2007;97:539–547. doi: 10.1093/toxsci/kfm052. [DOI] [PubMed] [Google Scholar]
- 24.Amacher DE. Drug-associated mitochondrial toxicity and its detection. Current Medicinal Chemistry. 2005;12:1829–1839. doi: 10.2174/0929867054546663. [DOI] [PubMed] [Google Scholar]
- 25.McKee EE, Ferguson M, Bentley AT, Marks TA. Inhibition of mammalian mitochondrial protein synthesis by oxazolidinones. Antimicrobial Agents and Chemotherapy. 2006;50:2042–2049. doi: 10.1128/AAC.01411-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wallace KB, Starkov AA. Mitochondrial targets of drug toxicity. Annual Review of Pharmacology and Toxicology. 2000;40:353–388. doi: 10.1146/annurev.pharmtox.40.1.353. [DOI] [PubMed] [Google Scholar]
- 27.Crabtree HG. The differential effect of radium radiation on the carbohydrate metabolism of normal and tumour tissues irradiated at low temperature. Biochemical Journal. 1935;29:2334–2343. doi: 10.1042/bj0292334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Al-Nasiry S, Geusens N, Hanssens M, Luyten C, Pijnenborg R. The use of Alamar Blue assay for quantitative analysis of viability, migration and invasion of choriocarcinoma cells. Human Reproduction. 2007;22 :1304–1309. doi: 10.1093/humrep/dem011. [DOI] [PubMed] [Google Scholar]
- 29.Klein CL, Wagner M, Kirkpatrick CJ, Van Kooten TG. A new quantitative test method for cell proliferation based on detection of the Ki-67 protein. Journal of Materials Science−Materials in Medicine. 1999;11:125–132. doi: 10.1023/a:1008953319485. [DOI] [PubMed] [Google Scholar]
- 30.McLaughlin B, Levitt P. Molecular basis of neurological disease. Hanley and Belfus Inc; Philadelphia: 2000. [Google Scholar]
- 31.Zhang Y, Lipton P. Cytosolic Ca2+ Changes during In Vitro Ischemia in Rat Hippocampal Slices: Major Roles for Glutamate and Na+-Dependent Ca2+ Release from Mitochondria. Journal of Neuroscience. 1999;19 :3307–3315. doi: 10.1523/JNEUROSCI.19-09-03307.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nicholls DG. Mitochondrial dysfunction and glutamate excitotoxicity studied in primary neuronal cultures. Current Molecular Medicine. 2004;4:149–177. doi: 10.2174/1566524043479239. [DOI] [PubMed] [Google Scholar]
- 33.McLaughlin BA, Hartnett KA, Erhardt JA, Legos JJ, White RF, Barone FC, Aizenman E. Caspase 3 activation is essential for neuroprotection in ischemic preconditioning. Proceedings of the National Academy of Sciences of the United States of America. 2003;100:715–720. doi: 10.1073/pnas.0232966100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sinor JD, Du S, Venneti S, Blitzblau RC, Leszkiewicz DN, Rosenberg PA, Aizenman E. NMDA and Glutamate Evoke Excitotoxicity at Distinct Cellular Locations in Rat Cortical Neurons In Vitro. Journal of Neuroscience. 2000;20:8831–8837. doi: 10.1523/JNEUROSCI.20-23-08831.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Czyzyk-Krzeska MF, Furnari BA, Lawson EE, Millhorn DE. Hypoxia increases rate of transcription and stability of tyrosine hydroxylase mRNA in pheochromocytoma (PC12) cells. Journal of Biological Chemistry. 1994;269:760–764. [PubMed] [Google Scholar]
- 36.Maurer BJ, Metelitsa LS, Seeger RC, Cabot MC, Reynolds CP. Increase of ceramide and induction of mixed apoptosis/necrosis by N-(4-hydroxyphenyl)- retinamide in neuroblastoma cell lines. Journal of National Cancer Institute. 1999;91:1138–1146. doi: 10.1093/jnci/91.13.1138. [DOI] [PubMed] [Google Scholar]
- 37.Jin KL, Mao XO, Greenberg DA. Vascular endothelial growth factor: direct neuroprotective effect in in Vitro ischemia. Proceedings of the National Academy of Sciences of the United States of America. 2000;97:10242–10247. doi: 10.1073/pnas.97.18.10242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gerencser AA, Neilson A, Choi SW, Edman U, Yadava N, Oh RJ, Ferrick DA, Nicholls DG, Brand MD. Quantitative Microplate-Based Respirometry with Correction for Oxygen Diffusion. Analytical Chemistry. 2009 doi: 10.1021/ac900881z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Eklund SE, Snider RM, Wikswo J, Baudenbacher F, Prokop A, Cliffel DE. Multianalyte microphysiometry as a tool in metabolomics and systems biology. Journal of Electroanalytical Chemistry. 2006;587:333. [Google Scholar]
- 40.Chen Y, Stevens B, Chang J, Milbrandt J, Barres BA, Hell JW. NS21: Re-defined and modified supplement B27 for neuronal cultures. Journal of Neuroscience Methods. 2008;171:239. doi: 10.1016/j.jneumeth.2008.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]