

Abstract
Reactive oxygen species (ROS) and the NADPH oxidases contribute to hypertension via mechanisms that remain undefined. ROS produced in the central nervous system have been proposed to promote sympathetic outflow, inflammation and hypertension, but the contribution of the NADPH oxidases to these processes in chronic hypertension is uncertain. We therefore sought to identify how NADPH oxidases in the subfornical organ (SFO) of the brain regulate blood pressure and vascular inflammation during sustained hypertension. We produced mice with loxP sites flanking the coding region of the NADPH oxidase docking subunit p22phox. SFO-targeted injections of an adenovirus encoding cre-recombinase (AdCre) markedly diminished p22phox, Nox2 and Nox4 mRNA in the SFO as compared to a control adenovirus (AdRFP) injection. Increased superoxide production in the SFO by chronic angiotensin II infusion (490 ng/kg/min × 2 weeks), was blunted in AdCre-treated mice as detected by dihydroethidium fluorescence. Deletion of p22phox in the SFO eliminated the hypertensive response observed at two weeks of angiotensin II infusion compared to AdRFP-treated mice (mean arterial pressures = 97±15 vs. 154±6 mmHg respectively, p = 0.0001). Angiotensin II-infusion also promoted marked vascular inflammation, as characterized by accumulation of activated T cells and other leukocytes, and this was prevented by deletion of the SFO p22phox. These experiments definitively identify the NADPH oxidases in the SFO as a critical determinant of the blood pressure and vascular inflammatory responses to chronic angiotensin II, and further support a role of ROS in central nervous system signaling in hypertension.
Keywords: NADPH oxidase, blood pressure, inflammation, vasculature, central nervous system
INTRODUCTION
It is now clear that reactive oxygen species (ROS) are involved in the genesis of experimental hypertension. Superoxide (O ·−2) production is increased in the vasculature, the kidney and the central nervous system in various models of hypertension, 1, 2 and scavenging O ·−2 using membrane-targeted forms of superoxide dismutase (SOD) or SOD mimetics blunts hypertension in these models,3, 4 The NADPH oxidases are major sources of ROS in hypertension. The catalytic subunits of these enzymes are the Nox proteins, which facilitate transfer of electrons from NADPH to molecular oxygen to form O ·2. There are 5 Nox proteins, which vary in their means of activation, subunit binding partners and output of O ·−2. Genetic deletion of the NADPH oxidase subunits Nox1, Nox2 and p47phox prevents many of the consequences of angiotensin II and renal artery stenosis, including elevation of blood pressure, increased renal vascular resistance and endothelial dysfunction.5-7
All Nox proteins, except Nox 5, which does not exist in rodents, require the small membrane subunit p22phox. Angiotensin II increases vascular p22phox expression in vivo and deletion of p22phox in vascular smooth muscle cells prevents ROS formation and the growth response to angiotensin II.8, 9 Conversely, transgenic overexpression of vascular smooth muscle p22phox enhances hypertension and vascular smooth muscle hypertrophy in response to angiotensin II.10
Recently, it has been recognized that ROS production is increased in several regions of the brain in hypertension, including the circumventricular organs (CVO),11 hypothalamic centers and the brainstem.12 ROS increases neuronal firing in these regions and ultimately increases sympathetic outflow. The subfornical organ SFO is particularly important in modulating these responses. This region is rich in angiotensin type 1 (AT1) receptors and is a major cardiovascular regulatory and dipsogenic center in the brain.13,14 Intracerebroventricular (ICV) administration of adenoviruses overexpressing superoxide dismutase blunts the acute pressor effects of angiotensin II in the SFO, and over the long-term prevents the hypertension caused by chronic angiotensin II infusion.11,15 Recent studies have shown that both Nox2 and Nox4 are involved in the acute pressor response to angiotensin II, while the dipsogenic response is dependent on Nox2.16 Likewise, ICV administration of an adenovirus expressing a dominant negative form of the small G-protein Rac1 prevents the acute pressor and water-drinking responses to angiotensin II.17 The roles of Nox2, Nox4 and Rac1 in chronic hypertension have not been defined in these acute studies.
In the present study we sought to definitively define the role of the NADPH oxidases in the SFO in hypertension caused by prolonged angiotensin II infusion. To accomplish this, we created mice with loxP sites flanking the coding region of p22phox and used Cre/Lox technology to delete this molecule in the SFO. Our findings show that p22phox in the SFO is essential for hypertension and vascular inflammation caused by angiotensin II.
MATERIALS AND METHODS
Creation of p22phox/flox mice
A targeting vector was created in which a neomycin cassette flanked by loxP sites was cloned into an Apo1 site 7.1 kb upstream of the p22phox transcription start site. An additional loxP site was placed in intron 1. This was microinjected into ES cells and proper homologous recombination as confirmed using Southern blots. Mice containing this targeted mutation were produced using standard techniques and backcrossed to C57Bl/6 mice for >11 generations. These mice were viable, developed normally and had normal growth. All studies were performed according to a protocol approved by Emory and Cornell Universities Institutional Animal Care and Use Committees. For detailed Material and Methods, please see the online Data Supplement at http://hyper.ahajournals.org.
RESULTS
Effect of AdCre on p22phox mRNA and superoxide production in the SFO
In prior studies, we and others found that uptake of adenovirus after SFO targeted ICV injection is greatest in the SFO, with uptake in other CVO to be limited to ependymal cells.18 We therefore focused on the SFO for measurements of p22phox mRNA and superoxide production in the present studies. Quantitative real-time PCR on punch biopsies of the SFO showed markedly reduced p22phox mRNA after injection of adenovirus encoding Cre-recombinase (AdCre) as compared to adenovirus red-fluorescent protein (AdRFP) control injection, confirming successful targeting (Figure 1A). Interestingly, this was accompanied by a marked downregulation of Nox2 and Nox4 mRNA at baseline and after 14 days angiotensin II infusion (Figure 1A, Figure S1, available only on the on-line data supplement). Real-time PCR also revealed very low levels of Nox1 in the SFO after 14d angiotensin II in mice injected with AdRFP and this was unchanged by AdCre injection (data not shown).
Figure 1.

Selective deletion of p22phox in the SFO and its effect on ROS. Real-time PCR confirmed diminished p22phox mRNA in the SFO after 14 d angiotensin II infusion in mice that were injected with an adenovirus encoding for Cre-recombinase (AdCre). Levels of p22phox mRNA were normalized to 18S RNA (A, n = 5, 2 brains per n). Mice injected with AdCre showed blunted DHE staining after 14 days angiotensin II infusion compared to control mice (B, n = 5). Densitometry is shown in Panel C.
To investigate whether deletion of p22phox reduces O ·−2production in the SFO, we performed DHE staining using confocal microscopy and measured fluorescence at 405 nm. In keeping with prior studies,11 angiotensin II increased dihydroethidium (DHE) fluorescence in the SFO of AdRFP injected mice and this was markedly reduced in mice that had received ICV AdCre (Figures 1B and 1C).
Effect of SFO p22phox deletion on hemodynamics
In mice treated with AdRFP angiotensin II infusion caused a progressive increase in blood pressure to a value of 154 ± 6 mmHg. In striking contrast, mice with SFO deletion of p22phox, had blood pressure that rose transiently at days 1-4 of angiotensin II infusion and then returned to normal values (Figure 2A).
Figure 2.

The role of SFO p22phox in regulation of blood pressure. Radiotelemeters were implanted in p22phox/flox mice and adenoviruses encoding either Cre-recombinase or Red-fluorescent protein were injected ICV. Osmotic minipumps were inserted on day five for infusion of angiotensin II (490 ng/kg/min). Panel A shows mean arterial pressures recorded during the two weeks of angiotensin II infusion (n = 5). Results from power spectral analysis of mean arterial pressure and heart rate are depicted in Panel B. Baseline data was average from day -3 until day 0 of blood pressure recordings. Angiotensin II represents day 13 and 14 of angiotensin II infusion to reflect the maximal blood pressure response (n = 5). Heart rate before and during angiotensin II treatment is shown in Panel C (n = 5) and power spectral analysis of heart rate variability is depicted in Panel D (n = 5).
To investigate whether deletion of p22phox in the SFO affects sympathetic activity we performed power spectral analysis of mean arterial pressure. Blood pressure oscillations are influenced by sympathetic and parasympathetic neurons, the renin-angiotensin system and vasoactive factors like nitric oxide. Power spectral analysis was utilized to detect which factors are influencing the blood pressure. Sympathetic outflow modulates low frequency oscillations (0.015 to 0.6 Hz), thus absolute values divided by the total power of low frequency provides an indirect measurement of sympathetic outflow.19 The power of low frequency blood pressure oscillations was significantly increased following 14 days angiotensin II infusion in AdRFP injected mice but not in mice with SFO deletion of p22phox by AdCre (Figure 2B). Heart rate was diminished by SFO p22phox deletion before and during angiotensin II infusion. Power spectral analysis of heart rate variability showed that angiotensin II increased the low frequency/high frequency ratio in AdRFP-treated but not AdCre treated mice.
Effect of p22phox deletion in the SFO on vascular inflammation
Flow cytometric analysis indicated that angiotensin II infusion promoted vascular inflammation as reflected by infiltration CD45+ (Figure S2A) and CD3+ (Figure 3A) cells detected in single cell suspensions of vascular homogenates from AdRFP-treated mice. Moreover, the infiltrating T cells expressed the early activation marker CD69 (Figure 3B) and CD44high (Figure S2B), characteristic of effector memory T cells. Deletion of p22phox in the SFO virtually eliminated the increase in vascular leukocytes, including CD69+ and CD44high T cells.
Figure 3.
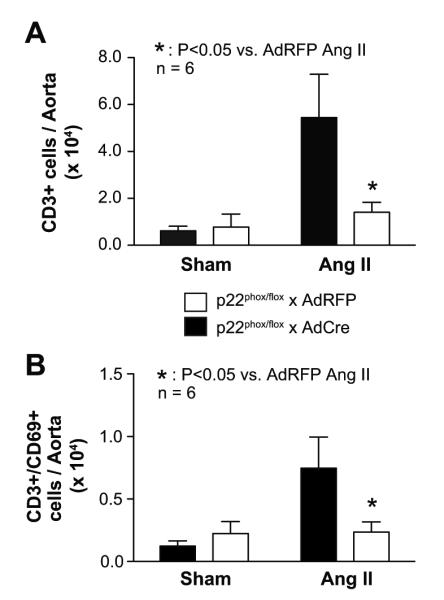
The role of p22phox in the circumventricular organ on vascular inflammation in p22phox/flox mice. Panel A shows CD3+ cell infiltration in aortic tissue in p22phox/flox mice that received intracerebroventricular injection of adenoviruses encoding either cre-recombinase or red-fluorescent protein. Osmotic minipumps to deliver angiotensin II (490 ng/kg/min) or vehicle were inserted 10 days later. Panel B shows the T cell activation marker CD69+ in aortic tissue. n = 6, *: P < 0.05.
Effect of p22phox deletion in the SFO on vascular NADPH oxidase expression
To determine if deletion of p22phox or angiotensin II in the SFO affects vascular NADPH oxidase subunits, we measured levels of p22phox and Nox2 mRNA in the aorta. There was no a change in p22phox or Nox2 expression in vehicle infused mice (Figure 4A and S3A). Vascular p22phox and Nox2 mRNA expression was upregulated in the aortas of both AdCre and AdRFP injected mice after 14 days angiotensin II infusion. As in other studies,20, 21 we observed a trend of reduced aortic Nox4 mRNA expression in response to angiotensin II; however, this did not reach statistical significance (Figure S3B).
Figure 4.
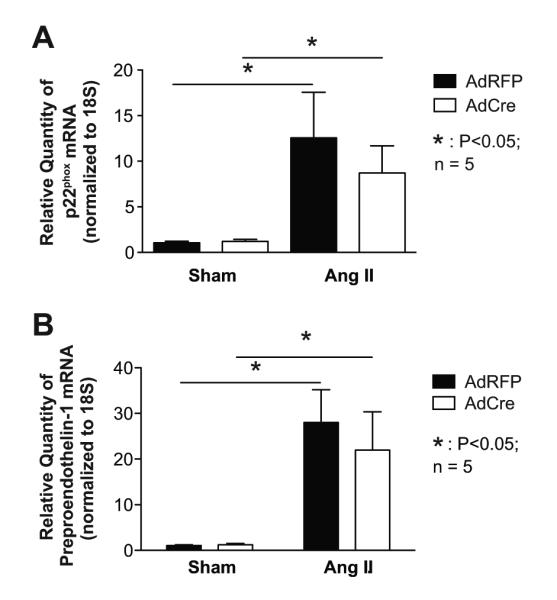
The effects of p22phox in the SFO on vascular NADPH oxidase and preproendothelin-1 expression. Panels A shows mRNA expression of p22phox in aortas of p22phox/flox mice treated with AdCre or AdRFP and 14 days of angiotensin II or vehicle infusion (n = 4 -5). Panel B shows mRNA expression levels of preproendothelin-1 in aortas of the same mice. n = 5, *: P<0.05.
Prior studies from our group and others have shown that angiotensin II stimulates vascular preproendothelin-1 mRNA expression.22 We confirmed this in the present study, and found that this was unaffected by deletion of SFO p22phox (Figure 4D). These data suggest that angiotensin II directly affects vascular p22phox, Nox2 and endothelin-1 expression, independent of sympathetic outflow or pressure increases.
DISCUSSION
In the present study we used cre-lox methodology to specifically delete p22phox in the SFO and showed that this reduced O ·−2 in this region. Moreover, deletion of p22phox in this brain region attenuated the hypertensive response and eliminated the vascular inflammation caused by this angiotensin II. This study definitively identifies a critical role of the NADPH oxidases in the brain SFO in this form of experimental hypertension.
Our findings are in agreement with prior reports showing that augmenting SOD or inhibiting Rac-1 activity in the SFO reduces the acute pressor and dipsogenic responses to ICV injections of angiotensin II.17,18 Prior studies by Peterson et al. showed that silencing either Nox2 or Nox4 blocks the acute pressor response to angiotensin II. Silencing Nox2 also prevented the dipsogenic response to angiotensin II, but silencing Nox4 did not.16 While it has been shown that augmenting SOD in the SFO inhibits chronic angiotensin II hypertension,11 our current study extends these prior observations by specifically identifying a critical role of the NADPH oxidases in the SFO in the long-term effect of angiotensin II.
In these experiments, we specifically targeted p22phox for selective deletion in the SFO. While this subunit of the NADPH oxidase is not catalytically active, it is critical for functioning of all Nox isoforms. In the case of Nox2 activation, the proline rich area in the C-terminal region of p22phox serves as a docking unit for activated p47phox.23 This region also binds p40phox, which activates Nox2 and completes for p47phox binding.24 Moreover, p22phox associates with and stabilizes the Nox2 during processing and transport to the cell membrane.25 A similar role for Nox1 likely exists, in that we previously found overexpression of p22phox in transgenic mice markedly increased Nox1 expression.26 Nox1 associates with p22phox,27 and point mutations in the proline-rich region of p22phox inhibit Nox1 function.28 Likewise, p22phox binds Poldip2, a newly recognized activator of Nox4.29 Thus, our deletion of p22phox in the SFO should have functionally disabled all Nox isoforms.
In a prior study, we showed that ICV injection of AdCre in mice with floxed extracellular superoxide dismutase (SOD3flox mice) specifically deleted SOD3 in the SFO, but had no effect on SOD3 in peripheral tissues, suggesting that the adenovirus does not gain access to peripheral sites when injected ICV.18 We further showed that even intraperitoneal injection of the small amount of AdCre used for ICV injection had no effect on vascular SOD3 levels in this study. It is therefore unlikely that injection of AdCre in the present study had any direct effect on p22phox expression in tissues outside of the CNS.
In the same study, we also found that the adenovirus expression following ICV injection was limited to the periventricular tissues, and predominantly in the SFO, and did not reach deeper regions of the brain.18 It is therefore likely that in the present study, SFO-targeted ICV injection of AdCre caused excision of the p22phox gene predominantly in the SFO and not at other sites within the CNS. Deletion of p22phox in the SFO corresponded with a loss of superoxide production, as reflected by DHE staining, blunting of the hypertensive response to angiotensin II and a marked attenuation in sympathetic outflow, as characterized by the low frequency blood pressure variations. These findings are thus compatible with a critical role of ROS in modulation of neuronal signaling in SFO in response to stimuli such as angiotensin II.
A striking finding in our study was that deletion of p22phox in the SFO decreased mRNA levels of Nox4 and Nox2. The precise reason for this is unclear. It is possible that mRNA transcription or stability is redox regulated in neuronal cells. While previously uninvestigated, it is also possible that p22phox has functions other than simply acting as a docking subunit for the Nox subunits that could modulate expression of other genes. Additional studies of examining transcriptional and post-transcriptional regulation of Nox2 and Noxs4 mRNA would be necessary to address this issue.
There is ample evidence that oxidative signaling in central cardiovascular control centers modulates sympathetic outflow.11, 14, 30, 31 Increased oxidative stress in the SFO increases sympathetic outflow and moderately elevates blood pressure.18 In elegant studies Chan et al. showed that angiotensin II in the rostral venterolateral medulla (RVLM) increases O ·−2 via NADPH oxidase activation and causes increased neural firing.32 In spontaneous hypertensive rats, a model of neurogenic hypertension characterized by increased sympathetic nervous activity, reduction of O ·−2 in the RVLM blunts hypertension.31 In keeping with these data, power spectral analysis indicated that sympathetic outflow in response to angiotensin II is significantly diminished by deletion of p22phox in the SFO, supporting the concept that NADPH oxidase derived O ·−2 enhances neuronal firing in these centers.
Recent studies from our laboratory have shown that T cells are essential for the development of various forms of experimental hypertension.33, 34 In these studies, we found that hypertension promotes accumulation of activated T cells and macrophages in the perivascular fat of both large and small arteries. Our current findings are also compatible with a recent study in which anteroventral third ventricle (AV3V) lesions prevent T cell activation and vascular inflammation. The virtual elimination of vascular inflammation after central deletion of p22phox might be a consequence of simply preventing hypertension. In this regard, we have previously shown that lowering blood pressure with hydralazine also prevents vascular inflammation in response to either angiotensin II or norepinephrine infusion.34 It is also possible that disruption of sympathetic stimulation reduces T cell activation and vascular inflammation. In keeping with this concept, Ganta et al. have shown that angiotensin II stimulates cytokine production by splenocytes via sympathetic activation.35 Innate and adaptive immune cells possess both and -adrenergic receptors,36 and modulate immune functions such as antigen presentation by dendritic cells, clonal expansion of lymphocytes, migration and cell trafficking.
Interestingly, the increase in vascular p22phox, Nox2 and preproendothelin-1 caused by angiotensin II infusion was not affected by p22phox deletion in the SFO. These results indicate that p22phox, Nox2 and preproendothelin-1 are regulated by direct actions of angiotensin II on vascular cells, independent of blood pressure and sympathetic outflow, and are in keeping with studies in which angiotensin II induces expression of these genes in cultured vascular cells. These findings also suggest that blockade of angiotensin II production its receptors could have additional benefit beyond blockade of sympathetic outflow.
Supplementary Material
PERSPECTIVES.
This study definitively identifies a critical role of the NADPH oxidases, and by inference, a role for ROS derived from the NADPH oxidases, in the SFO in modulation of hypertension and the systemic inflammation caused by angiotensin II. Furthermore, our study suggests, that therapeutic interventions that target this central site could prevent the untoward effects of this common disease.
Novelty and Significance.
What is new?
This study confirms that NADPH oxidases in the subfornical organ are major sources of oxidative stress during angiotensin II-induced hypertension. The study is the first to make use of a mouse model in which p22phox, necessary for function of all rodent Nox subunits, is selectively deleted using Cre-Lox technology. We also provide the first evidence that angiotensin II-induced changes in the vasculature, such as expression of preproendothelin and Nox2 are independent of blood pressure or sympathetic outflow.
Why is it relevant?
This study is relevant because it confirms that NADPH oxidases in the circumventricular organs of the brain are the major ROS producing enzymes involved in angiotensin II-induced hypertension. These studies aid in understanding the mechanisms by which angiotensin II activates the subfornical organ.
Summary
In this study we definitively identified a critical role of the NADPH oxidases in the SFO in modulation of hypertension and the systemic inflammation caused by angiotensin II. Furthermore, our study suggests, that vascular effects of angiotensin II are independent of blood pressure. Therapeutic interventions that target the SFO could prevent the untoward effects of this common disease.
Acknowledgments
Sources of support This work was supported by 12SDG9160010 (HEL), NIH R01HL063387 (RLD) and NIH R01HL039006, P01HL058000, P01HL095070, P01GM015431 and R01HL105294 (DGH).
Footnotes
Conflict of Interest None.
This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
LITERATURE CITED
- 1.Rajagopalan S, Kurz S, Munzel T, Tarpey M, Freeman BA, Griendling KK, Harrison DG. Angiotensin II-mediated hypertension in the rat increases vascular superoxide production via membrane NADH/NADPH oxidase activation. Contribution to alterations of vasomotor tone. J Clin Invest. 1996;97:1916–1923. doi: 10.1172/JCI118623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Harrison DG, Gongora MC. Oxidative stress and hypertension. Med Clin North Am. 2009;93:621–635. doi: 10.1016/j.mcna.2009.02.015. [DOI] [PubMed] [Google Scholar]
- 3.Laursen JB, Rajagopalan S, Galis Z, Tarpey M, Freeman BA, Harrison DG. Role of superoxide in angiotensin II-induced but not catecholamine-induced hypertension. Circulation. 1997;95:588–593. doi: 10.1161/01.cir.95.3.588. [DOI] [PubMed] [Google Scholar]
- 4.Schnackenberg CG, Welch WJ, Wilcox CS. Normalization of blood pressure and renal vascular resistance in SHR with a membrane-permeable superoxide dismutase mimetic: role of nitric oxide. Hypertension. 1998;32:59–64. doi: 10.1161/01.hyp.32.1.59. [DOI] [PubMed] [Google Scholar]
- 5.Matsuno K, Yamada H, Iwata K, Jin D, Katsuyama M, Matsuki M, Takai S, Yamanishi K, Miyazaki M, Matsubara H, Yabe-Nishimura C. Nox1 is involved in angiotensin II-mediated hypertension: a study in Nox1-deficient mice. Circulation. 2005;112:2677–2685. doi: 10.1161/CIRCULATIONAHA.105.573709. [DOI] [PubMed] [Google Scholar]
- 6.Landmesser U, Cai H, Dikalov S, McCann L, Hwang J, Jo H, Holland SM, Harrison DG. Role of p47(phox) in vascular oxidative stress and hypertension caused by angiotensin II. Hypertension. 2002;40:511–515. doi: 10.1161/01.hyp.0000032100.23772.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Landmesser U, Dikalov S, Price SR, McCann L, Fukai T, Holland SM, Mitch WE, Harrison DG. Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J Clin Invest. 2003;111:1201–1209. doi: 10.1172/JCI14172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fukui T, Ishizaka N, Rajagopalan S, Laursen JB, Capers Qt, Taylor WR, Harrison DG, deLeon H, Wilcox JN, Griendling KK. p22phox mRNA expression and NADPH oxidase activity are increased in aortas from hypertensive rats. Circ Res. 1997;80:45–51. doi: 10.1161/01.res.80.1.45. [DOI] [PubMed] [Google Scholar]
- 9.Zafari AM, Ushio-Fukai M, Akers M, Yin Q, Shah A, Harrison DG, Taylor WR, Griendling KK. Role of NADH/NADPH oxidase-derived H2O2 in angiotensin II-induced vascular hypertrophy. Hypertension. 1998;32:488–495. doi: 10.1161/01.hyp.32.3.488. [DOI] [PubMed] [Google Scholar]
- 10.Weber DS, Rocic P, Mellis AM, Laude K, Lyle AN, Harrison DG, Griendling KK. Angiotensin II-induced hypertrophy is potentiated in mice overexpressing p22phox in vascular smooth muscle. Am J Physiol Heart Circ Physiol. 2005;288:37–42. doi: 10.1152/ajpheart.00638.2004. [DOI] [PubMed] [Google Scholar]
- 11.Zimmerman MC, Lazartigues E, Sharma RV, Davisson RL. Hypertension caused by angiotensin II infusion involves increased superoxide production in the central nervous system. Circ Res. 2004;95:210–216. doi: 10.1161/01.RES.0000135483.12297.e4. [DOI] [PubMed] [Google Scholar]
- 12.Oliveira-Sales EB, Nishi EE, Carillo BA, Boim MA, Dolnikoff MS, Bergamaschi CT, Campos RR. Oxidative stress in the sympathetic premotor neurons contributes to sympathetic activation in renovascular hypertension. Am J Hypertens. 2009;22:484–492. doi: 10.1038/ajh.2009.17. [DOI] [PubMed] [Google Scholar]
- 13.Mangiapane ML, Simpson JB. Subfornical organ: forebrain site of pressor and dipsogenic action of angiotensin II. Am J Physiol. 1980;239:R382–389. doi: 10.1152/ajpregu.1980.239.5.R382. [DOI] [PubMed] [Google Scholar]
- 14.Hendel MD, Collister JP. Contribution of the subfornical organ to angiotensin II-induced hypertension. Am J Physiol Heart Circ Physiol. 2005;288:H680–685. doi: 10.1152/ajpheart.00823.2004. [DOI] [PubMed] [Google Scholar]
- 15.Zimmerman MC, Lazartigues E, Lang JA, Sinnayah P, Ahmad IM, Spitz DR, Davisson RL. Superoxide mediates the actions of angiotensin II in the central nervous system. Circ Res. 2002;91:1038–1045. doi: 10.1161/01.res.0000043501.47934.fa. [DOI] [PubMed] [Google Scholar]
- 16.Peterson JR, Burmeister MA, Tian X, Zhou Y, Guruju MR, Stupinski JA, Sharma RV, Davisson RL. Genetic silencing of Nox2 and Nox4 reveals differential roles of these NADPH oxidase homologues in the vasopressor and dipsogenic effects of brain angiotensin II. Hypertension. 2009;54:1106–1114. doi: 10.1161/HYPERTENSIONAHA.109.140087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zimmerman MC, Dunlay RP, Lazartigues E, Zhang Y, Sharma RV, Engelhardt JF, Davisson RL. Requirement for Rac1-dependent NADPH oxidase in the cardiovascular and dipsogenic actions of angiotensin II in the brain. Circ Res. 2004;95:532–539. doi: 10.1161/01.RES.0000139957.22530.b9. [DOI] [PubMed] [Google Scholar]
- 18.Lob HE, Marvar PJ, Guzik TJ, Sharma S, McCann LA, Weyand C, Gordon FJ, Harrison DG. Induction of hypertension and peripheral inflammation by reduction of extracellular superoxide dismutase in the central nervous system. Hypertension. 2010;55:277–283. doi: 10.1161/HYPERTENSIONAHA.109.142646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stauss HM. Power spectral analysis in mice: What are the appropriate frequency bands? Am J Physiol Regul Integr Comp Physiol. 2007;292:R902–903. doi: 10.1152/ajpregu.00716.2006. [DOI] [PubMed] [Google Scholar]
- 20.Lassegue B, Sorescu D, Szocs K, Yin Q, Akers M, Zhang Y, Grant SL, Lambeth JD, Griendling KK. Novel gp91(phox) homologues in vascular smooth muscle cells : nox1 mediates angiotensin II-induced superoxide formation and redox-sensitive signaling pathways. Circ Res. 2001;88:888–894. doi: 10.1161/hh0901.090299. [DOI] [PubMed] [Google Scholar]
- 21.Alvarez A, Cerda-Nicolas M, Naim Abu Nabah Y, Mata M, Issekutz AC, Panes J, Lobb RR, Sanz MJ. Direct evidence of leukocyte adhesion in arterioles by angiotensin II. Blood. 2004;104:402–408. doi: 10.1182/blood-2003-08-2974. [DOI] [PubMed] [Google Scholar]
- 22.Rajagopalan S, Laursen JB, Borthayre A, Kurz S, Keiser J, Haleen S, Giaid A, Harrison DG. Role for endothelin-1 in angiotensin II-mediated hypertension. Hypertension. 1997;30:29–34. doi: 10.1161/01.hyp.30.1.29. [DOI] [PubMed] [Google Scholar]
- 23.Ago T, Kuribayashi F, Hiroaki H, Takeya R, Ito T, Kohda D, Sumimoto H. Phosphorylation of p47phox directs phox homology domain from SH3 domain toward phosphoinositides, leading to phagocyte NADPH oxidase activation. Proc Natl Acad Sci U S A. 2003;100:4474–4479. doi: 10.1073/pnas.0735712100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tamura M, Shiozaki I, Ono S, Miyano K, Kunihiro S, Sasaki T. p40phox as an alternative organizer to p47phox in Nox2 activation: a new mechanism involving an interaction with p22phox. FEBS Lett. 2007;581:4533–4538. doi: 10.1016/j.febslet.2007.08.040. [DOI] [PubMed] [Google Scholar]
- 25.DeLeo FR, Burritt JB, Yu L, Jesaitis AJ, Dinauer MC, Nauseef WM. Processing and maturation of flavocytochrome b558 include incorporation of heme as a prerequisite for heterodimer assembly. J Biol Chem. 2000;275:13986–13993. doi: 10.1074/jbc.275.18.13986. [DOI] [PubMed] [Google Scholar]
- 26.Laude K, Cai H, Fink B, Hoch N, Weber DS, McCann L, Kojda G, Fukai T, Schmidt HH, Dikalov S, Ramasamy S, Gamez G, Griendling KK, Harrison DG. Hemodynamic and biochemical adaptations to vascular smooth muscle overexpression of p22phox in mice. Am J Physiol Heart Circ Physiol. 2005;288:H7–12. doi: 10.1152/ajpheart.00637.2004. [DOI] [PubMed] [Google Scholar]
- 27.Hanna IR, Hilenski LL, Dikalova A, Taniyama Y, Dikalov S, Lyle A, Quinn MT, Lassegue B, Griendling KK. Functional association of nox1 with p22phox in vascular smooth muscle cells. Free Radic Biol Med. 2004;37:1542–1549. doi: 10.1016/j.freeradbiomed.2004.08.011. [DOI] [PubMed] [Google Scholar]
- 28.Kawahara T, Ritsick D, Cheng G, Lambeth JD. Point mutations in the proline-rich region of p22phox are dominant inhibitors of Nox1- and Nox2-dependent reactive oxygen generation. J Biol Chem. 2005;280:31859–31869. doi: 10.1074/jbc.M501882200. [DOI] [PubMed] [Google Scholar]
- 29.Lyle AN, Deshpande NN, Taniyama Y, Seidel-Rogol B, Pounkova L, Du P, Papaharalambus C, Lassegue B, Griendling KK. Poldip2, a novel regulator of Nox4 and cytoskeletal integrity in vascular smooth muscle cells. Circ Res. 2009;105:249–259. doi: 10.1161/CIRCRESAHA.109.193722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Oliveira-Sales EB, Dugaich AP, Carillo BA, Abreu NP, Boim MA, Martins PJ, D’Almeida V, Dolnikoff MS, Bergamaschi CT, Campos RR. Oxidative stress contributes to renovascular hypertension. Am J Hypertens. 2008;2:98–104. doi: 10.1038/ajh.2007.12. [DOI] [PubMed] [Google Scholar]
- 31.Kishi T, Hirooka Y, Kimura Y, Ito K, Shimokawa H, Takeshita A. Increased reactive oxygen species in rostral ventrolateral medulla contribute to neural mechanisms of hypertension in stroke-prone spontaneously hypertensive rats. Circulation. 2004;109:2357–2362. doi: 10.1161/01.CIR.0000128695.49900.12. [DOI] [PubMed] [Google Scholar]
- 32.Chan SH, Hsu KS, Huang CC, Wang LL, Ou CC, Chan JY. NADPH oxidase-derived superoxide anion mediates angiotensin II-induced pressor effect via activation of p38 mitogen-activated protein kinase in the rostral ventrolateral medulla. Circ Res. 2005;97:772–780. doi: 10.1161/01.RES.0000185804.79157.C0. [DOI] [PubMed] [Google Scholar]
- 33.Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S, Goronzy J, Weyand C, Harrison DG. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J Exp Med. 2007;204:2449–2460. doi: 10.1084/jem.20070657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Marvar PJ, Thabet SR, Guzik TJ, Lob HE, McCann LA, Weyand C, Gordon FJ, Harrison DG. Central and peripheral mechanisms of T-lymphocyte activation and vascular inflammation produced by angiotensin II-induced hypertension. Circ Res. 2010;107:263–270. doi: 10.1161/CIRCRESAHA.110.217299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ganta CK, Lu N, Helwig BG, Blecha F, Ganta RR, Zheng L, Ross CR, Musch TI, Fels RJ, Kenney MJ. Central angiotensin II-enhanced splenic cytokine gene expression is mediated by the sympathetic nervous system. Am J Physiol Heart Circ Physiol. 2005;289:H1683–1691. doi: 10.1152/ajpheart.00125.2005. [DOI] [PubMed] [Google Scholar]
- 36.Swanson MA, Lee WT, Sanders VM. IFN-gamma production by Th1 cells generated from naive CD4+ T cells exposed to norepinephrine. J Immunol. 2001;166:232–240. doi: 10.4049/jimmunol.166.1.232. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.