

Abstract
Cardiac dysfunction is a major cause of morbidity and mortality in patients with end-stage liver disease, yet the mechanisms remain largely unknown. We hypothesized that the complex interrelated impairments in cardiac structure and function secondary to progression of liver diseases involve alterations in signaling pathways engaged in cardiac energy metabolism and hypertrophy, augmented by direct effects of high circulating levels of bile acids. Biliary fibrosis was induced in male C57BL/6J mice by feeding a 0.1% 3, 5-diethoxycarbonyl-1, 4-dihydroxychollidine (DDC) supplemented diet. After 3 weeks, mice underwent live imaging (DEXA scanning, 2DEcho, EKG, cardiac MRI), exercise treadmill testing and histological and biochemical analyses of livers and hearts. Compared to chow-fed mice, DDC-fed mice fatigued earlier on the treadmill, with reduced VO2. Marked changes were identified electrophysiologically (bradycardia and prolonged QTc) and functionally (hyperdynamic left ventricular (LV) contractility along with increased LV thickness). Hearts of DDC-fed mice showed hypertrophic signaling (activation of AKT, inhibition of GSK3β and a 20-fold upregulation of β myosin heavy chain RNA) and elevated Gsα/Giα ratio. Genes regulating cardiac fatty acid oxidation pathways were suppressed, along with a 3-fold increase in myocardial glycogen content. Treatment of mouse cardiomyocytes (which express the membrane bile acid receptor TGR5) with potent natural TGR5 agonists, taurochenodeoxycholic acid and lithocholic acid, activated AKT and inhibited GSK3β, similar to the changes seen in DDC-fed mouse hearts. This provides support for a novel mechanism whereby circulating natural bile acids can induce signaling pathways in heart associated with hypertrophy. In conclusion, 3 weeks of DDC feeding-induced biliary fibrosis leads to multiple functional, metabolic, electrophysiological and hypertrophic adaptations in the mouse heart recapitulating some of the features of human cirrhotic cardiomyopathy.
Keywords: TGR5, metabolism, bile acids, fatty acid oxidation, glycogen
Cardiac dysfunction in cirrhosis (cirrhotic cardiomyopathy) is characterized by multiple electrical, physiological and structural responses in the heart including left ventricular (LV) hypertrophy with impaired relaxation, baseline hyperdynamic contractility, conduction abnormalities and an attenuated cardiovascular response to stress(1). The development of cirrhotic cardiomyopathy increases the risk of death in patients with cirrhosis and is associated with increased risks of hepato-renal syndrome, serious post-operative complications and dysrhythmias (2, 3). In addition to increased mortality, cardiomyopathy is a co-morbid contributor to exercise fatigue in patients with cirrhosis (4, 5).
Rodent models of liver injury (mainly common bile duct ligation [CBDL] and carbon tetrachloride treatment) have revealed critical insights into some of the molecular mechanisms underlying cirrhotic cardiomyopathy. Increased cholesterol/phospholipid ratio within the cardiomyocyte cell membrane, impaired β-adrenergic receptor (β-AR) function and abnormal gene expression of β- adrenergic signaling pathway members contributes to attenuated contractile response to inotropic stress in cirrhosis (6, 7). In addition, carbon monoxide, hemoxygenase, nitric oxide, endogenous cannabinoids, bile acids (BA) and endotoxins have been implicated in the pathophysiology of diminished cardiac function in cirrhotic rodents(8). Though these mediators help explain some of the attenuated cardiac contractility in cirrhosis, the causes of hyperdynamic contractility, hypertrophy and rhythm disturbances, key features of baseline cardiac phenotype in cirrhosis in adults and children, remain essentially unknown(9, 10).
Alteration in cardiac energy metabolism, especially substrate utilization, plays a major role in the physiology of cardiac function in health, disease and hypertrophy. Often metabolic changes precede structural and functional changes in the heart (11, 12). Dysregulation in cardiac energetics has been proposed as one of the potential mechanisms underlying cirrhotic cardiomyopathy.(13) Detailed investigations of whole body energetics, characterization of cardiac hypertrophy and altered metabolism in human and animal models have been limited and could provide a critical insight into the pathophysiology of cirrhotic cardiomyopathy. In addition, it is likely that several pathways are engaged prior to the development of cirrhosis, although the timeline and mediators remain to be determined.
The goal of this study was to characterize the live mouse cardiac phenotype and explore the extrahepatic molecular consequences in the heart of a recently described mouse model of biliary fibrosis (14). We found multiple impairments in cardiac function and structure in mice with biliary fibrosis, along with distinct changes in cell signaling pathways leading to hypertrophy and altered metabolism. Finally, evidence is provided that supports a contributing role for elevated circulating levels of bile acids to altered cardiac signaling pathways in biliary fibrosis.
MATERIALS AND METHODS
Animals and Diet
Biliary fibrosis was induced by feeding 6-8 week old male C57BL6J mice (Jackson Labs; Bar Harbor, ME.) with 0.1% 3, 5-diethoxycarbonyl-1, 4-dihydroxychollidine (DDC) (Sigma-Aldrich. St. Louis, MO.) supplemented chow (Harlan-Teklad Inc, Madison, WI) for 3 weeks. Age matched male C57BL6J mice fed isocaloric chow were used as controls for all experiments. Mice were weighed twice a week. Feeds and bedding were weighed and changed twice a week to provide an estimate of food intake. Mice were fed ad libitum and had free access to water. Food was withdrawn 4 hours prior to experiments and all experiments were done in accordance with IACUC approved protocols at Baylor College of Medicine (BCM).
Anthropometric measurements
After 3 weeks DDC and control mice underwent dual energy x-ray absorptiometry (DEXA) scanning (GE-Lunar PIXImus, Madison, WI.) to evaluate lean mass, fat mass, bone mineral density, bone mineral content and percent fat content (Mouse Phenotype Core facility (MPC), BCM, Houston).
Exercise Tolerance and Energy Expenditure
After 3 weeks mice were exposed to a standard exercise protocol on a treadmill integrated with metabolic chamber (Oxymax Deluxe VO2/VCO2 System Columbus Instruments, Columbus, OH) (See Supplement). Indirect calorimetry was performed at baseline, during exercise and for 5 minutes after treadmill was stopped. Oxygen consumption (VO2), carbon dioxide production (VCO2) and Respiratory Exchange Ratio (RER) were analyzed from the gas sampled (MPC at BCM, Houston).
Cardiac Parameters
Continuous electrocardiograms (EC Genie (Mouse Systems Inc, Quincy MA) and tail-cuff blood pressures ( 6 channel NIBP system with BPMONWIN software [IITC Life Science Inc. CA, USA]) were recorded noninvasively in unsedated mice. Two-dimensional echocardiography (2DE) was performed in MPC (BCM) on sedated mice (Vevo 770 Digital RF, VisualSonic Inc. Toronto, CN). Cardiac MRI (Bruker Biospin, Billerica, MA) was performed in sedated mice and analyzed by a blinded investigator as described previously (15) (See Supplement).
Serum analyses
Sera and anti-coagulated blood were collected from the inferior vena cava (IVC) and analyzed for alanine aminotransferase (ALT), aspartate aminotransferase (AST), Lactate Dehydrogenase (LDH), total and conjugated bilirubin levels and Complete Blood Count (CBC) respectively (Cobas Integra 400+; Roche) at Center of Comparative Medicine (BCM). Serum bile acid levels and non-esterified fatty acid levels (NEFA) were evaluated by colorimetric methods (BioQuant Inc, San Diego, CA. & Roche). Serum leptin and TNFα levels were measured using commercial ELISA kits (RayBiotech Inc. Norcross, GA).
Cardiac and liver histology
Hearts and liver were rapidly isolated and flash frozen in liquid nitrogen, or used for subsequent histological analyses - routine H/E and Mason–Trichrome (for collagen). OCT embedded frozen sections were stained with Oil Red-O (for lipids) and Periodic Acid Schiff (PAS) for glycogen. Immunohistochemistry for TGR5 was performed on frozen hearts using rabbit polyclonal antibody (Sigma-Aldrich; St. Louis, MO.) at 1:200 dilution. Liver sections were stained with routine H/E. All histological studies were performed by the Texas Gulf Coast Digestive Disease Center. Myocardial glycogen content was compared enzymatically as described previously (16) and histologically using image analysis (see Supple1ment) .
Quantitative real time PCR (qRTPCR)
RNA levels were analyzed by qRTPCR (primer sequences in supplement Table 1.s), as described before (17) using SYBR® Green™ (Applied Biosystems, Foster city, CA.). Relative RNA expression was calculated by delta Ct method. Expression of target genes were normalized to internal standard (GAPDH) and reported as fold change compared to chow.
Immunoblotting
Proteins from homogenized whole hearts and membranes were extracted using standard procedures(17). After gel electrophoresis and immunoblotting, the gels were analyzed for expression of various proteins with specific antibodies (Supplement). Equal protein loading was confirmed by α-tubulin (Sigma-Aldrich, St. Louis, MO) for whole hearts or Sodium-Potassium ATPase plasma membrane antibody (Abcam, Cambridge, MA) and Ponceau staining for membrane extracts. Results were analyzed by densitometry (Kodak software, Scientific Imaging Systems; New Haven, CT.) and reported as fold change compared to chow.
Isolation of neonatal cardiomyocytes and bile acid treatments
Cardiomyocytes were isolated from 2-3 day old neonatal C57BL6J mouse pups (Cellutron Life Sciences, Baltimore, MD) as described previously(18). (Supplement) Isolated cardiomyocytes (0.5×106cells/well) were incubated with 100μM sodium taurochenodeoxycholic acid (TCDCA), 10μM sodium lithocholic acid (LCA) (Sigma-Aldrich, St. Louis, MO) or vehicle (DMSO). After 4 hours, experiments were terminated with ice-cold PBS, cells were lysed and proteins extracted and analyzed by immunoblotting. Each experiment was performed in duplicate and repeated three times (n=3). For each set of experiment, cardiomyocytes were isolated and pooled from 15-20 neonatal hearts.
Statistical Analysis
All data are presented as Means ± SD (unless specified). Data was analyzed using non-parametric Mann-Whitney test (unless specified). All statistical calculations were done using the PRISM 5.0 software program (Graph-Pad Prism, San Diego, CA, USA). P < 0.05 was considered significant.
RESULTS
DDC-fed mice exhibit early fatigue, decreased whole body oxygen consumption (VO2) and increased Respiratory Exchange Ratio (RER)
Three weeks of DDC-feeding led to biliary fibrosis and changes in serum indices of liver disease consistent with those noted by other investigators(14) (Supplement Fig.2s). DDC-fed C57BL/6J mice lost weight and demonstrated changes in body composition mainly ~ 30% decrease in lean and fat mass, without a change in percent fat or bone mineral content (Supplement Fig 1s). On the metabolic treadmill, DDC-fed mice were exhausted earlier than their chow-fed counterparts (Fig.1A). In addition, indirect calorimetry performed continuously during exercise showed that DDC-fed mice have decreased VO2, decreased peak VO2, and an increased RER (RER = VCO2/VO2) (Fig.1B and D). Thus, for whole body composition and exercise parameters, DDC-fed mice have several key similarities to those seen in patients with biliary cirrhosis (4, 5).
Fig.1. DDC-fed mice have limited tolerance for exercise and altered oxygen utilization.
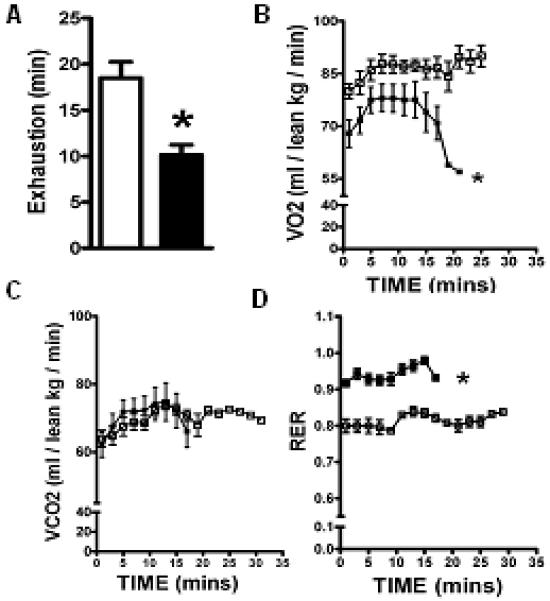
Mice were challenged with acute maximal exercise on a treadmill. Time to exhaustion, (VO2), (VCO2) and (RER) were measured by indirect calorimetry as reported in Methods. DDC fed mice demonstrated early fatigue (A), lower VO2 (B) and high RER (D) when compared to chow fed mice at each time point. VCO2 did not did not differ between the groups. (*p<0.05; Results: Mean±SEM; n=6; Mann-Whitney for each time point). Note: Chow (□) and DDC (■)
The hearts of DDC-fed mice are hypertrophic and exhibit altered electrical and functional parameters
Unsedated DDC-fed mice exhibited bradycardia, prolonged QT intervals (QTc) and higher systolic BP (Fig.2B). Two-dimensional echocardiography (2DEcho) revealed hyperdynamic contractility evidenced by ~ 25% increase in both ejection fractions (%EF) and shortening fractions (%FS) (Fig.2A and B), similar to those seen in patients with cirrhosis or in infants with biliary atresia and portal hypertension (5, 9, 10). Cardiac MRI of DDC-fed mice showed ~ 25% increase in both LV and RV ejection fractions compared to chow-fed hearts (Table 1). Cardiac mass index, LV free wall and inter-ventricular septal thickness were significantly increased in DDC-fed mouse hearts (Fig. 3A and Table 1). LV stroke volume index tended to be higher in the DDC fed mice (Table.1).
Fig.2. DDC-fed mouse hearts have altered electrocardiographic, echocardiographic and hemodynamic parameters.
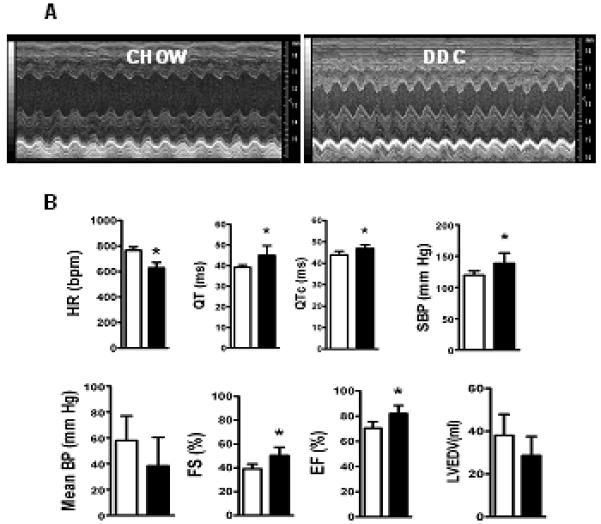
(A) denotes representative M-mode cardiac 2DEcho pictures of chow fed and DDC fed mice showing hyperdynamic LV in DDC fed mice. Bar graph in (B) denotes heart rate (HR), QT interval, corrected QT interval (QTc) as analyzed by rhythm strips, systolic (SBP) and mean (MBP) blood pressures as calculated in unsedated mice using tail-cuff, shortening fractions (%FS), ejection fractions (%EF) and calculated end diastolic volume (LVEDV) of the left ventricle by 2DEcho. (* p<0.05; n=6). Note: Chow (□) and DDC (■)
Table. 1.
Cardiac parameters from MRI: Table depicts values obtained for the respective cardiac parameters after quantitative analysis of cardiac MR. (Statistics: Students t test)
| CARDIAC PARAMETERS |
CHOW (n=5)
(Results±SD) |
DDC (n=5)
(Results±SD) |
P value |
|---|---|---|---|
| LV Ejection Fraction (%) | 48.03±4.7 | 66.9±3.7 | 0.0002 |
| RV Ejection Fraction (%) | 49.7±1.35 | 62.12±4.5 | 0.007 |
| Posterior Wall thickness (mm) | 0.71±0.04 | 0.88.±0.035 | 0.0001 |
| Septal thickness (mm) | 0.92±0.06 | 1.168±0.036 | 0.0001 |
|
Cardiac Mass (Normalized to body wt) |
0.0021±0.0003 | 0.0037±0.0007 | 0.008 |
| LVEDV (ml) | 0.054±0.004 | 0.043±0.009 | 0.0547 |
| LVESV (ml) | 0.025±0.006 | 0.014±0.004 | 0.009 |
| LV Stroke Volume Index (ml/g) | 1.44±0.1 | 1.75±0.3 | 0.07 |
| RV Stroke Volume Index (ml/g) | 1.10±0.14 | 1.4±0.34 | 0.13 |
Fig.3. Cardiac hypertrophy and hypertrophic signaling in the DDC-fed heart.
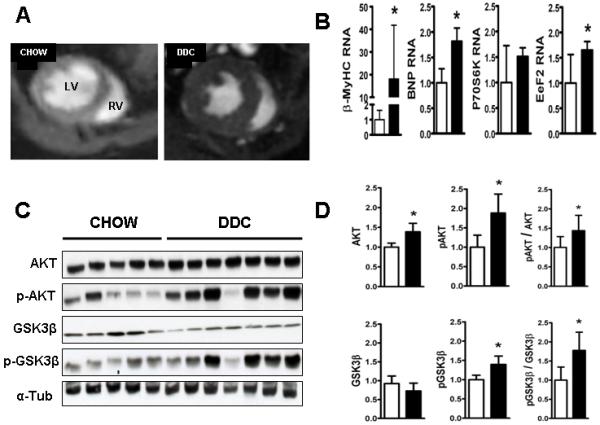
(A) Representative pictures of cardiac MR short axis mid-ventricular slices in diastole shows chow fed hearts (CH) on the left and DDC fed hearts on the right. Concentric LV hypertrophy in DDC hearts is evident on these images showing increased LV posterior wall and septal thickness. This was verified by quantitative analysis of the posterior wall, septal thickness and LV mass normalized to the body weight (see Table.1). (B) shows qRTPCR analysis of key genes involved in cardiac hypertrophy. Note upregulation of β-MyHC, BNP and eEF2. (C) shows representative immunoblots for AKT, Ser473-phospho-AKT, GSK3β and Ser9-phospho-GSK3β with α-tubulin (loading control). (D) shows densitometric analysis of the respective bands normalized to α-tubulin and analyses of the degree of phosphorylation (pAKT/AKT and pGSK3b/GSK3b). (*p<0.05; n=5-7) Chow (□) and DDC (■)
DDC-induced biliary cirrhosis leads to multiple alterations in cardiac hypertrophy signaling pathways and expression of relevant target genes
Central among the many changes associated with cardiac hypertrophy in a variety of mouse models are activation of v-akt murine thymoma viral oncogene/Protein kinase B (AKT) and inhibition of Glycogen Synthase Kinase- 3β (GSK3β) (19). DDC feeding resulted in a 1.5 fold increase in cardiac levels of phospho-Thr308-AKT levels (activation) and a 1.75 fold increase in phospho-Ser9-GSK3β (inhibition) (Fig.3C and D), both associated with increased expression of genes responsible for cardiac hypertrophy(20). Expression of several, but not all key genes involved in cardiac hypertrophy were upregulated: notably β myosin heavy chain (β MyHC; 20-fold), brain natriuretic peptide (BNP; 2-fold) and elongation factor eEF2 (1.8-fold) (Fig. 3B and Supplement Fig. 3s A).
β-adrenergic receptor signaling
Several researchers have identified altered β-adrenergic signaling in cirrhosis, linked to reductions in either β-adrenergic receptor (βAR) expression (7) or impairments in post-receptor signaling pathways (6). Myocardial contractility is governed in large part by signaling through βAR-1 and βAR-2 and their associated co-regulatory Gsα and Giα subunits. DDC-feeding did not change RNA or protein expression of either β-ARs (Fig.4). However, a significant suppression of Giα, with a modest activation of Gsα, led to a 2-fold increase in the Gsα/Giα ratio.
Fig.4. Elevated Gsα/ Giα ratios in the hearts of DDC-fed mice.
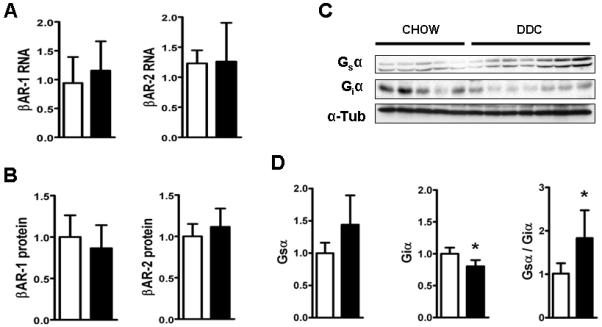
(A) shows QPCR results of βAR-1 and βAR-2 RNA standardized to GAPDH and (B) depicts densitometric analysis of βAR-1and βAR-2 protein expression normalized to α-tubulin (C) Immunoblot pictures of whole heart incubated with antibodies for Gsα, Giα and α-tubulin. (D) shows densitometric analysis of the respective bands normalized to α-tubulin. (*p<0.05; n=5-7) Note: Chow (□) and DDC (■)
Glycogen accumulation and altered fatty acid metabolism in the hearts of DDC-fed mice
Cardiac histology revealed increased staining with PAS (Fig.5A). Quantitative image analysis and biochemical analysis of cardiac tissue showed a 3-fold increase in myocardial glycogen content (Fig 5B and C) after DDC feeding. Both GLUT-1 and GLUT-4 membrane protein levels, which are responsible for the majority of cellular glucose uptake, were increased in DDC-fed mouse hearts. RNA levels of GLUT-1, but not GLUT-4 were elevated (Fig. 5D and E). Serum free fatty acids (a major energy substrate for a healthy resting heart), and leptin levels (a key regulator of cardiac fatty acid oxidation [FAO]) (21) were, respectively, 60% and 80% reduced in DDC-fed mice (Fig. 6A). Expression of heart-type fatty acid binding protein (h-FABP/FABP-3), mitochondrial-carnitine palmitoyl transferase-2 (mCPT-2), short chain acyl-coA-dehydrogenase (s-ACAD) and uncoupling protein-3 (UCP-3) - key genes responsible for cardiac FAO, were suppressed in DDC-fed mice (Fig.6B). CD36, mCPT-1, medium-ACAD, long-ACAD and very long-ACAD RNA levels did not significantly change with DDC feeding (data not shown). DDC-feeding induces activation of cardiac acetyl-CoA carboxylase (ACC, via a ~50% decrease in phospho-ACC, Fig 6D) which has been shown to contribute to suppression of FAO (22). AMPKα protein levels were unchanged by DDC feeding (Fig.6C). Finally, UCP-3 which is involved in cardiac FAO (23) and remodeling (24), was ~ 50% suppressed after DDC-feeding at both RNA and protein levels (Fig. 6B and D). This reduction in FAO was not linked to changes in myocardial lipid accumulation (Fig. 6E).
Fig.5. Increased glucose transporter expression and glycogen deposition in hearts of DDC-fed mice.
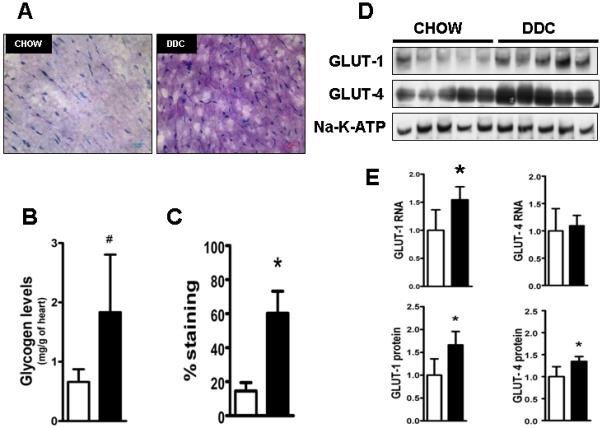
Representative frozen sections (A) of heart (n=5/group) after PAS staining shows increased glycogen deposition in DDC hearts compared with chow fed hearts. Myocardial glycogen content was quantified enzymatically (B) and by blinded colorimetric image analysis to quantify percent staining (C) by PAS. (D) denotes representative immunoblot results of equally loaded isolated membrane protein samples probed with antibodies for GLUT-1 and GLUT-4 proteins. (E) shows gene expressions of GLUT-1 and GLUT-4 standardized to GAPDH and (lower row) densitometric analysis of GLUT-1 and GLUT-4 bands. Equal loading confirmed by Sodium-Potassium-ATPase membrane protein and Ponceau staining. (*p<0.05; n=5; and # p<0.05; n=3, Students t test.) Note: Chow (□) and DDC (■)
Fig. 6. Reduced fatty acid oxidation in hearts of DDC-fed mice.
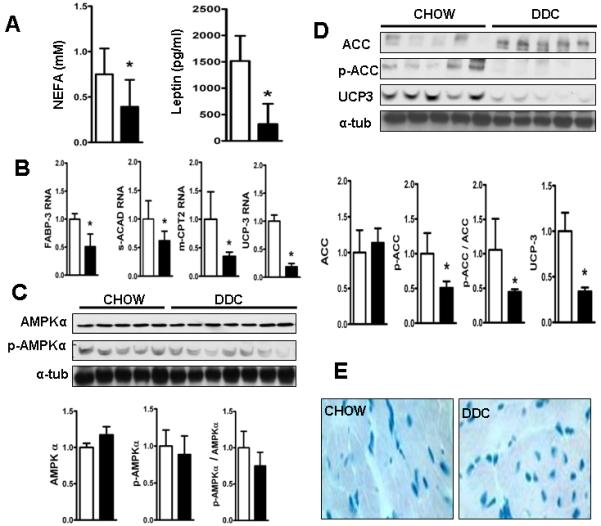
(A) shows decreased levels of serum NEFA levels and leptin levels in DDC fed mice. (B) shows QPCR results of key genes regulating fatty acid oxidation. (C) shows immunoblots of membranes incubated with antibodies for AMPKα and Thr172-phospho-AMPKα with bar graph below showing densitometric analysis of the respective bands standardized to α-tubulin and analysis of the degree of phosphorylation of AMPKα. (D) shows membranes incubated with antibodies for ACC, Ser79-phospho-ACC and UCP-3 and α-tubulin as loading control with bar graph showing densitometric analysis of the respective bands standardized to α-tubulin and analysis of the degree of phosphorylation of ACC. (*p<0.01; n=5) Note: Chow (□) and DDC (■). (E) shows representative pictures of Oil-Red-O stained frozen sections of the hearts showing no evidence of lipid accumulation in the DDC fed cardiac muscle.
TGR5 and bile acid signaling in mouse heart
As a first step to identify the molecular mediators of altered cardiac myocyte function in biliary fibrosis, we focused upon exploring roles for bile acids (BA) and the recently discovered G protein coupled receptor (GPCR) for BAs -TGR5(25). DDC-fed mice have ~50-fold elevated circulating levels of serum bile acids compared to chow-fed mice (Fig 7E), at concentrations capable of activating TGR5(26). When activated by extracellular BAs, including taurine-conjugated primary BAs, this GPCR initiates G protein containing signaling pathways in several metabolically-active tissues(27). However, little is known for a role for cardiac TGR5. Immunoblot and qRTPCR analysis of RNA indicates that TGR5 is present in mouse heart and isolated cardiomyocytes (Fig.7A and B). Immunohistochemistry indicates its presence in both endothelium and cardiomyocytes (Fig 7C). Cardiac TGR5 RNA and protein expression did not change in DDC fed mouse hearts (Fig.7 D). To study if there are direct effects of BAs (natural TGR5 agonists) on cardiomyocytes, we exposed neonatal mouse cardiomyocyte cultures to the TCDCA (EC50 for TGR5 of 1.9 μM) and LCA (EC50 for TGR5 of 0.58μM)(26). After incubation for 4 hours, TCDCA (100μM) as well as LCA (10μM) activated AKT and inhibited GSK3β, similar to the changes in-vivo after DDC feeding (compare Fig 7G to Fig 3).
Fig. 7. TGR5 and role for bile acids.
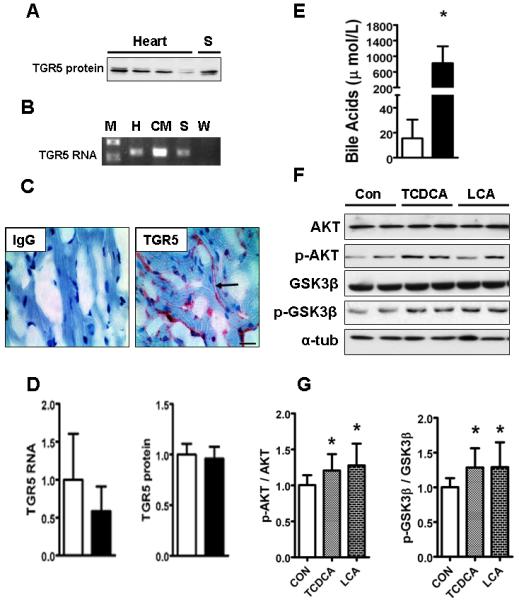
(A) shows immunoblot of 4 whole heart tissues [H] showing a band detected at 37kDa using TGR5 antibody with spleen (S) as a positive control. (B) shows PCR analysis of flash frozen whole heart (H), isolated neonatal cardiomyocytes (CM), spleen [S] as positive control and water [W] used as a negative control. Note TGR5 band at 126 base pair besides molecular weight marker (M). (C) shows presence of TGR5 (red staining) in the OCT fixed flash frozen hearts by immunohistochemistry using rabbit polyclonal antibody for TGR5 (Bar=20 μm thick). No staining seen in the isotype control stained heart. (D) shows bar graph comparing QPCR results of TGR5 RNA (standardized to GAPDH) and TGR5 protein expression (normalized to α-tubulin) between chow (□) and DDC fed (■) groups and expressed as fold change compared to chow group. (E) depicts circulating serum bile acids in DDC fed mice. (*p<0.05; Results: Mean±SD; (n=5) Note: Chow (□) and DDC (■)) (F) shows representative immunoblot results from n=3 experiments of equally loaded protein samples from isolated neonatal cardiomyocytes (0.5×106 cells/well) incubated with DMSO as vehicle (Con), TCDCA (100μM) and LCA (10μM) for 4 hours. Membranes were incubated with antibodies for AKT, Ser473-phospho-AKT, GSK3β and Ser9-phospho-GSK3β with α-tubulin as the loading control. (G) depicts bar graphs showing densitometric analysis of the degree of activation (phosphorylation) of AKT (p-AKT/AKT) and inhibition (phosphorylation) of GSK3β (p-GSK3β/GSK3β). (*p<0.05; Statistics:Mann-Whitney as compared with DMSO control; n=3).
DISCUSSION
Extra-hepatic consequences of cirrhosis such as cirrhotic cardiomyopathy strongly impact upon morbidity and mortality(8). In this study, we report that the DDC-fed model of biliary fibrosis, cholestasis and liver injury, recapitulates many of the cardiac abnormalities seen in human cirrhotic cardiomyopathy. In addition, changes were identified in multiple signaling pathways which could potentially provide insight into the development of cirrhotic cardiomyopathy. DDC-fed mice have multiple alterations in exercise physiology (early fatigability, decreased VO2 and increased RER), cardiac rhythm (bradycardia and prolonged QT interval), cardiac morphology (LV and septal hypertrophy) and cardiac function (increased LV ejection and shortening fractions) which model most of the essential elements of overt cardiac dysfunction in cirrhotic patients(5, 8, 9, 28, 29). At the molecular level, these hearts exhibit activation of AKT and inhibition of GSK3β pathways: both of which contribute to hypertrophic and hyperdynamic changes in the heart(30-35). Though cardiac expression of β-ARs did not change, Giα was significantly reduced with a 2-fold increase in Gsα/Giα ratio; altering βAR signal transduction. (36)
DDC-feeding leads to suppression of key regulators of cardiac FAO, enhanced expression of membrane glucose transporters and a 3 fold increase in glycogen content suggesting a novel dysregulation in cardiac energetics and metabolism. Finally, a direct activation of AKT and inhibition of GSK3β is seen in isolated cardiomyocytes cultures by TCDCA and LCA (potent natural TGR5 ligands), supporting a potential direct role for high levels of circulating BAs.
2DE and cardiac MRI of DDC-fed mice show increased thickness of the LV free wall and interventricular septum, consistent with findings in hearts of adults (37), as well as infants (9, 10) with biliary cirrhosis. Several animal models of liver injury (carbon tetrachloride(38), CBDL (39), and now the DDC feeding) lead to LV hypertrophy, which, in turn, can lead to diastolic dysfunction and the inability to compensate for increased demands (e.g., systolic dysfunction when stressed). From a cell signaling perspective, several critical mediators have emerged as central to cardiac hypertrophy--AKT activation and GSK3β inhibition.(40). Chronic activation of AKT leads to myocardial hypertrophy in mice via mechanisms including either inhibition of GSK3β(19, 20) or activation of p70S6K via mTOR(35). Transgenic mice overexpressing cardiac AKT demonstrate hypertrophy, hypercontractility and an attenuated contractile response to dobutamine (30) which mirror the cardiac impairments seen in cirrhotic patients. In the DDC-fed mouse model, the heart displays several phenotypic and molecular features of physiologic hypertrophy such as: hypercontractile LV, AKT activation and upregulation of eEF2(41) as well as characteristics of pathologic hypertrophy such as “re-expression” of fetal growth genes - β-MyHC and BNP(42). Whether these changes reflect adaptive or maladaptive responses to liver injury/biliary fibrosis, or response to vascular changes remain to be seen. Importantly, whether or not the remodeling is reversible in the DDC model as seen in patients with cirrhotic cardiomyopathy after liver transplantation, remains to be determined.
Cardiopulmonary exercise testing of cirrhotic patients revealed early fatigue, lower VO2, and lower maximal VO2 (4, 5). DDC-fed mice showed similar physiological changes to humans throughout the course of exercise treadmill testing. Though not statistically significant, these mice demonstrated higher VCO2 which also contributed for higher RER. High RER is often seen when carbohydrates are oxidized preferentially over lipids(43), suggesting that substrate utilization by metabolically active tissues, including the heart, is impaired in these mice.
A resting heart utilizes circulating fatty acids for 60-70% of its energy needs while glucose and lactate supply the remainder. The heart switches its substrate preference from fatty acids to glucose as a compensatory mechanism under stressors like congestive heart failure, pathologic hypertrophy, and cardiomyopathy(11, 12). Histologic and biochemical evidence suggest a switch in cardiac substrate utilization favoring glycogen accumulation and reduced fatty acid oxidation in DDC fed mice(Fig. 5 and 6). The presence of glycogen in the hearts of this model is surprising and intriguing, and may be a contributor to the structural and functional alterations, rather than an innocent bystander. It is possible that in the DDC model, cardiomyocytes store glycogen as endogenous fuel in the face of shortage of exogenous fuel (fatty acids) to maintain vital functions as a survival mechanism. Energy efficiency (ATP/mole O2) improves when heart “switches” from fats to glycogen as preferred fuel for respiration(44). This could explain the hypercontractility in an “un-stressed state”. During catecholamine stress as in exercise, this endogenous glycogen is rapidly depleted leading to decompensation--a hallmark of cirrhotic cardiomyopathy. Accumulation of minimal amounts of glycogen as seen in mice over-expressing PRKAG2 leads to hypertrophy and increased contractility, while excessive glycogen deposition leads to severe cardiac dysfunction(45-47). At a molecular level, accumulation of glycogen can be explained by activation of AKT with resultant inactivation of GSK3β(44). At a physiologic level, myocardial glycogen content is increased in fasting or as an adaptive response to stress “hibernation”(44). . Overall, detailed mechanisms and consequences underlying the marked alterations in cardiac metabolism, along with changes in cell signaling pathways and their physiologic relevance in the development of cardiomyopathy in this liver injury model await further study.
High circulating levels of BAs, in the order of fifty to hundred-fold higher than normal (48) are one of the central components of cirrhosis, and biliary cirrhosis in particular. The primary BAs such as TCDCA and LCA, whose levels are elevated in cirrhosis, activate AKT and inhibits GSK3β in isolated cardiomyocyte cultures at concentrations seen in cirrhosis (Fig 7), supporting a potential novel role in mediating hypertrophy, hypercontractility and metabolic changes. BAs are multifunctional molecules, that have recently been identified as true hormones(49), with intracellular (cell signaling cascades, nuclear receptors) and extracellular functions targeting cell surface receptors (TGR5) (25). BA nuclear receptors and BA transporters are poorly expressed in the myocardium, but TGR5 is present (fig 7)(50, 51). Thus, there is likely a pathway for circulating BAs to signal to cardiomyocytes extracellularly, through TGR5 (52). These data along with others (53, 54) are among the first to identify cardiomyocytes as a direct target for BA signaling, suggesting a strong role for BAs in the pathophysiology of cirrhotic cardiomyopathy. It is likely that the hypertrophy, hypercontractility, electrocardiographic disturbance and altered cardiac metabolism are interlinked and the “cross-talk” needs further analysis. It is also likely that BAs do not act alone—rather they act in concert with other circulating molecules (cytokines, endotoxin, chemokines, lipids, etc) to contribute to the ultimate changes seen in the cirrhotic heart.
Though head to head comparison between cardiac findings in the mouse DDC model and rat CBDL and rat carbon tetrachloride (CCl4) models are difficult secondary to distinct modes of injury, timelines and species differences, there are some similarities of note. All three models demonstrate cardiac hypertrophy(38, 55). Attenuated contractility to inotropic stress in CBDL and CCl4 models has been shown to be secondary to downstream effects of decreased β-adrenergic signaling and upregulated cannabinoid receptor signaling likely mediated by endotoxemia, high circulating TNFα, NFκB, high levels of HO-1 and increased NO production. In contrast, cardiac physiology in the DDC mouse model is mostly hyperdynamic, with a robust up-regulation of cardiac HO-1 RNA, similar to CBDL rat hearts ((56) and Supplemental Fig.4s). There are other differences as well, and direct comparisons of the energetic, metabolic, and molecular phenotypes between these models in mice are needed to properly compare the consequences of these distinct models.
Among the more relevant issues is whether or not the mouse DDC model develops portal hypertension and systemic hemodynamic changes seen in some of the other models. There is some indirect evidence for this, via an increase in spleen weight index (Supp. Fig.2) but direct determination of portal pressures and hemodynamics will be needed to be certain. In fact, it may be even more significant that such changes in the hearts of these mice can develop with biliary fibrosis alone. Further studies are needed to properly define these important issues.
In conclusion, multiple inter-related impairments in cardiac cell biology and function are evident in the DDC-induced model of biliary fibrosis which involve multiple metabolites and signaling pathways, including those engaged by high circulating levels of BAs. Investigating the mechanisms of the multiple metabolic, functional, and molecular signaling alterations induced by liver failure on cardiac function, along with analyzing in detail the contributions of systemic bile acids on cardiomyocyte biology, will help design rational targets to counteract the serious clinical cardiac consequences of cirrhosis.
Supplementary Material
Acknowledgements
We thank David Durgan PhD and Martin Young PhD for enzymatic glycogen analysis, Milton Finegold MD and the Texas Gulf Coast Digestive Disease Center for histology and Mary Michele Mariscalco MD for critical review of the manuscript.
Funding: Support for this study comes from the Texas Gulf Coast Digestive Disease Center (NIH DK58338) and NIH DK56239.
Abbreviations
- 2DEcho
Two-dimensional echocardiography
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- ACC
acetyl-CoA carboxylase
- AKT
v-akt murine thymoma viral oncogene/Protein kinase B
- βAR
β-adrenergic receptor
- BA
bile acids
- β MyHC
β myosin heavy chain
- BNP
brain natriuretic peptide
- CBDL
common bile duct ligation
- DDC
0.1% 3, 5-diethoxycarbonyl-1, 4-dihydroxychollidine
- DEXA
dual energy x-ray absorptiometry
- %EF
ejection fractions
- eEF2
eukaryotic elongation factor-2
- %FS
shortening fractions
- FAO
fatty acid oxidation
- GLUT
Glucose transporter proteins
- GPCR
G protein coupled receptor
- GSK3β
Glycogen Synthase Kinase- 3β
- h-FABP/FABP-3
heart-type fatty acid binding protein
- LDH
Lactate Dehydrogenase
- mCPT-2
mitochondrial-carnitine palmitoyl transferase-2
- NEFA
non-esterified fatty acid levels
- PAS
Periodic Acid Schiff
- RER
Respiratory Exchange Ratio
- s-ACAD
short chain acyl-coA-dehydrogenase
- TCDCA
taurochenodeoxycholic acid
- LCA
lithocholic acid
- UCP-3
uncoupling protein-3
- iNOS
inducible nitric oxide synthase
- eNOS
endothelial nitric oxide synthase
- HO-1
hemoxygenase-1
- VO2
Oxygen consumption
- VCO2
Carbon dioxide production
Reference List
- 1.Gaskari SA, Honar H, Lee SS. Therapy insight: Cirrhotic cardiomyopathy. Nat Clin Pract Gastroenterol Hepatol. 2006;3(6):329–337. doi: 10.1038/ncpgasthep0498. [DOI] [PubMed] [Google Scholar]
- 2.Henriksen JH, Moller S. Cardiac and systemic haemodynamic complications of liver cirrhosis. Scand Cardiovasc J. 2009:1–8. doi: 10.1080/14017430802691528. [DOI] [PubMed] [Google Scholar]
- 3.Krag A, Bendtsen F, Henriksen JH, Moller S. Low cardiac output predicts development of hepatorenal syndrome and survival in patients with cirrhosis and ascites. Gut. 2009 doi: 10.1136/gut.2009.180570. [DOI] [PubMed] [Google Scholar]
- 4.Epstein SK, Ciubotaru RL, Zilberberg MD, Kaplan LM, Jacoby C, Freeman R, et al. Analysis of impaired exercise capacity in patients with cirrhosis. Dig Dis Sci. 1998;43(8):1701–1707. doi: 10.1023/a:1018867232562. [DOI] [PubMed] [Google Scholar]
- 5.Wong F, Girgrah N, Graba J, Allidina Y, Liu P, Blendis L. The cardiac response to exercise in cirrhosis. Gut. 2001;49(2):268–275. doi: 10.1136/gut.49.2.268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ceolotto G, Papparella I, Sticca A, Bova S, Cavalli M, Cargnelli G, et al. An abnormal gene expression of the beta-adrenergic system contributes to the pathogenesis of cardiomyopathy in cirrhotic rats. Hepatology. 2008;48(6):1913–1923. doi: 10.1002/hep.22533. [DOI] [PubMed] [Google Scholar]
- 7.Ma Z, Miyamoto A, Lee SS. Role of altered beta-adrenoceptor signal transduction in the pathogenesis of cirrhotic cardiomyopathy in rats. Gastroenterology. 1996;110(4):1191–1198. doi: 10.1053/gast.1996.v110.pm8613009. [DOI] [PubMed] [Google Scholar]
- 8.Alqahtani SA, Fouad TR, Lee SS. Cirrhotic cardiomyopathy. Semin Liver Dis. 2008;28(1):59–69. doi: 10.1055/s-2008-1040321. [DOI] [PubMed] [Google Scholar]
- 9.Arikan C, Kilic M, Tumgor G, Levent E, Yuksekkaya HA, Yagci RV, et al. Impact of liver transplantation on rate-corrected QT interval and myocardial function in children with chronic liver disease*. Pediatr Transplant. 2009;13(3):300–306. doi: 10.1111/j.1399-3046.2008.00909.x. [DOI] [PubMed] [Google Scholar]
- 10.Arnon R, Srivastava S, Fahey M, Kerkar N, Shneider BL. Hyperdynamic circulation in infants with biliary atresia and portal hypertension. Hepatology. 2007;46(4):720A. [Google Scholar]
- 11.Taegtmeyer H. Switching metabolic genes to build a better heart. Circulation. 2002;106(16):2043–2045. doi: 10.1161/01.cir.0000036760.42319.3f. [DOI] [PubMed] [Google Scholar]
- 12.Taha M, Lopaschuk GD. Alterations in energy metabolism in cardiomyopathies 21. Ann Med. 2007;39(8):594–607. doi: 10.1080/07853890701618305. [DOI] [PubMed] [Google Scholar]
- 13.Fukazawa K, Gologorsky E, Manmohansingh V, Nishida S, Vigoda MM, Pretto EA., Jr. Is the immediate reversal of diastolic dysfunction of cirrhotic cardiomyopathy after liver transplantation a sign of the metabolic etiology? Liver Transpl. 2009;15(11):1417–1419. doi: 10.1002/lt.21861. [DOI] [PubMed] [Google Scholar]
- 14.Fickert P, Stoger U, Fuchsbichler A, Moustafa T, Marschall HU, Weiglein AH, et al. A new xenobiotic-induced mouse model of sclerosing cholangitis and biliary fibrosis. Am J Pathol. 2007;171(2):525–536. doi: 10.2353/ajpath.2007.061133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yang Z, Bowles NE, Scherer SE, Taylor MD, Kearney DL, Ge S, et al. Desmosomal dysfunction due to mutations in desmoplakin causes arrhythmogenic right ventricular dysplasia/cardiomyopathy. Circ Res. 2006;99(6):646–655. doi: 10.1161/01.RES.0000241482.19382.c6. [DOI] [PubMed] [Google Scholar]
- 16.Durgan DJ, Moore MW, Ha NP, Egbejimi O, Fields A, Mbawuike U, et al. Circadian rhythms in myocardial metabolism and contractile function: influence of workload and oleate. Am J Physiol Heart Circ Physiol. 2007;293(4):H2385–H2393. doi: 10.1152/ajpheart.01361.2006. [DOI] [PubMed] [Google Scholar]
- 17.Ghose R, Zimmerman TL, Thevananther S, Karpen SJ. Endotoxin leads to rapid subcellular re-localization of hepatic RXRalpha: A novel mechanism for reduced hepatic gene expression in inflammation 20. Nucl Recept. 2004;2(1):4. doi: 10.1186/1478-1336-2-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vallentin A, Mochly-Rosen D. RBCK1, a protein kinase CbetaI (PKCbetaI)-interacting protein, regulates PKCbeta-dependent function 6. J Biol Chem. 2007;282(3):1650–1657. doi: 10.1074/jbc.M601710200. [DOI] [PubMed] [Google Scholar]
- 19.Heineke J, Molkentin JD. Regulation of cardiac hypertrophy by intracellular signalling pathways. Nat Rev Mol Cell Biol. 2006;7(8):589–600. doi: 10.1038/nrm1983. [DOI] [PubMed] [Google Scholar]
- 20.Hardt SE, Sadoshima J. Glycogen synthase kinase-3beta: a novel regulator of cardiac hypertrophy and development. Circ Res. 2002;90(10):1055–1063. doi: 10.1161/01.res.0000018952.70505.f1. [DOI] [PubMed] [Google Scholar]
- 21.Atkinson LL, Fischer MA, Lopaschuk GD. Leptin activates cardiac fatty acid oxidation independent of changes in the AMP-activated protein kinase-acetyl-CoA carboxylase-malonyl-CoA axis 74. J Biol Chem. 2002;277(33):29424–29430. doi: 10.1074/jbc.M203813200. [DOI] [PubMed] [Google Scholar]
- 22.Arad M, Seidman CE, Seidman JG. AMP-activated protein kinase in the heart: role during health and disease. Circ Res. 2007;100(4):474–488. doi: 10.1161/01.RES.0000258446.23525.37. [DOI] [PubMed] [Google Scholar]
- 23.Nabben M, Hoeks J. Mitochondrial uncoupling protein 3 and its role in cardiac- and skeletal muscle metabolism. Physiol Behav. 2008;94(2):259–269. doi: 10.1016/j.physbeh.2007.11.039. [DOI] [PubMed] [Google Scholar]
- 24.Taegtmeyer H, Razeghi P, Young ME. Mitochondrial proteins in hypertrophy and atrophy: a transcript analysis in rat heart. Clin Exp Pharmacol Physiol. 2002;29(4):346–350. doi: 10.1046/j.1440-1681.2002.03656.x. [DOI] [PubMed] [Google Scholar]
- 25.Kawamata Y, Fujii R, Hosoya M, Harada M, Yoshida H, Miwa M, et al. A G protein-coupled receptor responsive to bile acids. J Biol Chem. 2003;278(11):9435–9440. doi: 10.1074/jbc.M209706200. [DOI] [PubMed] [Google Scholar]
- 26.Sato H, Macchiarulo A, Thomas C, Gioiello A, Une M, Hofmann AF, et al. Novel potent and selective bile acid derivatives as TGR5 agonists: biological screening, structure-activity relationships, and molecular modeling studies. J Med Chem. 2008;51(6):1831–1841. doi: 10.1021/jm7015864. [DOI] [PubMed] [Google Scholar]
- 27.Nguyen A, Bouscarel B. Bile acids and signal transduction: role in glucose homeostasis. Cell Signal. 2008;20(12):2180–2197. doi: 10.1016/j.cellsig.2008.06.014. [DOI] [PubMed] [Google Scholar]
- 28.Lunseth JH, OLMSTEAD EG, ABBOUD F. A study of heart disease in one hundred eight hospitalized patients dying with portal cirrhosis. AMA Arch Intern Med. 1958;102(3):405–413. doi: 10.1001/archinte.1958.00030010405009. [DOI] [PubMed] [Google Scholar]
- 29.Zambruni A, Trevisani F, Caraceni P, Bernardi M. Cardiac electrophysiological abnormalities in patients with cirrhosis. J Hepatol. 2006;44(5):994–1002. doi: 10.1016/j.jhep.2005.10.034. [DOI] [PubMed] [Google Scholar]
- 30.Condorelli G, Drusco A, Stassi G, Bellacosa A, Roncarati R, Iaccarino G, et al. At induces enhanced myocardial contractility and cell size in vivo in transgenic mice 20. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(19):12333–12338. doi: 10.1073/pnas.172376399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Morisco C, Seta K, Hardt SE, Lee Y, Vatner SF, Sadoshima J. Glycogen synthase kinase 3beta regulates GATA4 in cardiac myocytes. J Biol Chem. 2001;276(30):28586–28597. doi: 10.1074/jbc.M103166200. [DOI] [PubMed] [Google Scholar]
- 32.Rota M, Boni A, Urbanek K, Padin-Iruegas ME, Kajstura TJ, Fiore G, et al. Nuclear targeting of Akt enhances ventricular function and myocyte contractility 1211. Circulation Research. 2005;97(12):1332–1341. doi: 10.1161/01.RES.0000196568.11624.ae. [DOI] [PubMed] [Google Scholar]
- 33.Schiekofer S, Shiojima I, Sato K, Galasso G, Oshima Y, Walsh K. Microarray analysis of Akt1 activation in transgenic mouse hearts reveals transcript expression profiles associated with compensatory hypertrophy and failure 1210. Physiological Genomics. 2006;27(2):156–170. doi: 10.1152/physiolgenomics.00234.2005. [DOI] [PubMed] [Google Scholar]
- 34.Shiojima I, Yefremashvili M, Luo Z, Kureishi Y, Takahashi A, Tao J, et al. Akt signaling mediates postnatal heart growth in response to insulin and nutritional status. J Biol Chem. 2002;277(40):37670–37677. doi: 10.1074/jbc.M204572200. [DOI] [PubMed] [Google Scholar]
- 35.Shiojima I, Walsh K. Regulation of cardiac growth and coronary angiogenesis by the Akt/PKB signaling pathway. Genes Dev. 2006;20(24):3347–3365. doi: 10.1101/gad.1492806. [DOI] [PubMed] [Google Scholar]
- 36.Xiao RP, Avdonin P, Zhou YY, Cheng H, Akhter SA, Eschenhagen T, et al. Coupling of beta2-adrenoceptor to Gi proteins and its physiological relevance in murine cardiac myocytes. Circ Res. 1999;84(1):43–52. doi: 10.1161/01.res.84.1.43. [DOI] [PubMed] [Google Scholar]
- 37.Pozzi M, Carugo S, Boari G, Pecci V, de CS, Maggiolini S, et al. Evidence of functional and structural cardiac abnormalities in cirrhotic patients with and without ascites. Hepatology. 1997;26(5):1131–1137. doi: 10.1002/hep.510260507. [DOI] [PubMed] [Google Scholar]
- 38.Lunseth JH. CARDIAC HYPERTROPHY IN RATS WITH CARBON TETRACHLORIDE CIRRHOSIS. Arch Pathol. 1965;79:644–646. [PubMed] [Google Scholar]
- 39.Liu H, Ma Z, Lee SS. Contribution of nitric oxide to the pathogenesis of cirrhotic cardiomyopathy in bile duct-ligated rats. Gastroenterology. 2000;118(5):937–944. doi: 10.1016/s0016-5085(00)70180-6. [DOI] [PubMed] [Google Scholar]
- 40.Dorn GW. Adrenergic pathways and left ventricular remodeling 73. J Card Fail. 2002;8(6 Suppl):S370–S373. doi: 10.1054/jcaf.2002.129267. [DOI] [PubMed] [Google Scholar]
- 41.Kemi OJ, Ceci M, Wisloff U, Grimaldi S, Gallo P, Smith GL, et al. Activation or inactivation of cardiac Akt/mTOR signaling diverges physiological from pathological hypertrophy 8. J Cell Physiol. 2008;214(2):316–321. doi: 10.1002/jcp.21197. [DOI] [PubMed] [Google Scholar]
- 42.Molkentin JD, Lu JR, Antos CL, Markham B, Richardson J, Robbins J, et al. A calcineurin-dependent transcriptional pathway for cardiac hypertrophy. Cell. 1998;93(2):215–228. doi: 10.1016/s0092-8674(00)81573-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Faldt J, Wernstedt I, Fitzgerald SM, Wallenius K, Bergstrom G, Jansson JO. Reduced exercise endurance in interleukin-6-deficient mice. Endocrinology. 2004;145(6):2680–2686. doi: 10.1210/en.2003-1319. [DOI] [PubMed] [Google Scholar]
- 44.Taegtmeyer H. Glycogen in the heart--an expanded view. J Mol Cell Cardiol. 2004;37(1):7–10. doi: 10.1016/j.yjmcc.2004.05.001. [DOI] [PubMed] [Google Scholar]
- 45.Arad M, Moskowitz IP, Patel VV, Ahmad F, Perez-Atayde AR, Sawyer DB, et al. Transgenic mice overexpressing mutant PRKAG2 define the cause of Wolff-Parkinson-White syndrome in glycogen storage cardiomyopathy 75. Circulation. 2003;107(22):2850–2856. doi: 10.1161/01.CIR.0000075270.13497.2B. [DOI] [PubMed] [Google Scholar]
- 46.Davis MK, Weinstein DA. Liver transplantation in children with glycogen storage disease: controversies and evaluation of the risk/benefit of this procedure 2. Pediatr Transplant. 2008;12(2):137–145. doi: 10.1111/j.1399-3046.2007.00803.x. [DOI] [PubMed] [Google Scholar]
- 47.Wolf CM, Arad M, Ahmad F, Sanbe A, Bernstein SA, Toka O, et al. Reversibility of PRKAG2 glycogen-storage cardiomyopathy and electrophysiological manifestations 26. Circulation. 2008;117(2):144–154. doi: 10.1161/CIRCULATIONAHA.107.726752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Fickert P, Trauner M, Fuchsbichler A, Stumptner C, Zatloukal K, Denk H. Bile acid-induced Mallory body formation in drug-primed mouse liver. Am J Pathol. 2002;161(6):2019–2026. doi: 10.1016/S0002-9440(10)64480-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Groen AK. The emerging role of bile acids as integrators of intermediary metabolism 19. J Hepatol. 2006;45(2):337–338. doi: 10.1016/j.jhep.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 50.Bookout AL, Jeong Y, Downes M, Yu RT, Evans RM, Mangelsdorf DJ. Anatomical profiling of nuclear receptor expression reveals a hierarchical transcriptional network. Cell. 2006;126(4):789–799. doi: 10.1016/j.cell.2006.06.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gorelik J, Patel P, Ng’andwe C, Vodyanoy I, Diakonov I, Lab M, et al. Genes encoding bile acid, phospholipid and anion transporters are expressed in a human fetal cardiomyocyte culture 68. BJOG. 2006;113(5):552–558. doi: 10.1111/j.1471-0528.2006.00918.x. [DOI] [PubMed] [Google Scholar]
- 52.Watanabe M, Houten SM, Mataki C, Christoffolete MA, Kim BW, Sato H, et al. Bile acids induce energy expenditure by promoting intracellular thyroid hormone activation. Nature. 2006;439(7075):484–489. doi: 10.1038/nature04330. [DOI] [PubMed] [Google Scholar]
- 53.Gazawi H, Ljubuncic P, Cogan U, Hochgraff E, Ben-Shachar D, Bomzon A. The effects of bile acids on beta-adrenoceptors, fluidity, and the extent of lipid peroxidation in rat cardiac membranes 5. Biochem Pharmacol. 2000;59(12):1623–1628. doi: 10.1016/s0006-2952(00)00259-8. [DOI] [PubMed] [Google Scholar]
- 54.Gorelik J, Harding SE, Shevchuk AI, Koralage D, Lab M, de SM, et al. Taurocholate induces changes in rat cardiomyocyte contraction and calcium dynamics. Clin Sci (Lond) 2002;103(2):191–200. doi: 10.1042/cs1030191. [DOI] [PubMed] [Google Scholar]
- 55.Inserte J, Perello A, Agullo L, Ruiz-Meana M, Schluter KD, Escalona N, et al. Left ventricular hypertrophy in rats with biliary cirrhosis 14. Hepatology. 2003;38(3):589–598. doi: 10.1053/jhep.2003.50369. [DOI] [PubMed] [Google Scholar]
- 56.Liu H, Song D, Lee SS. Role of heme oxygenase-carbon monoxide pathway in pathogenesis of cirrhotic cardiomyopathy in the rat. Am J Physiol Gastrointest Liver Physiol. 2001;280(1):G68–G74. doi: 10.1152/ajpgi.2001.280.1.G68. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.