

Abstract
Object
Although Chiari Type I (CM-I) and Type 0 (CM-0) malformations have been previously characterized clinically and radiologically, there have been no studies focusing on the possible genetic link between these disorders. The goal of this study was to identify families in whom CM-0 and CM-I co-occurred and to further assess the similarities between these disorders.
Methods
Families were ascertained through a proband with CM-I. Detailed family histories were obtained to identify first-degree relatives diagnosed with CM-0. Several criteria were used to exclude individuals with acquired forms of CM-I and/or syringomyelia. Individuals were excluded with syndromic, traumatic, infectious, or tumor-related syringomyelia, as well as CM-I due to a supratentorial mass, hydrocephalus, history of cervical or cranial surgery unrelated to CM-I, or development of symptoms following placement of a lumbar shunt. Medical records and MR images were used to characterize CM-I and CM-0 individuals clinically and radiologically.
Results
Five families were identified in which the CM-I proband had a first-degree relative with CM-0. Further assessment of affected individuals showed similar clinical and radiological features between CM-0 and CM-I individuals, although CM-I patients in general had more severe symptoms and skull base abnormalities than their CM-0 relatives. Overall, both groups showed improvement in symptoms and/or syrinx size following craniocervical decompression surgery.
Conclusions
There is accumulating evidence suggesting that CM-0 and CM-I may be caused by a common underlying developmental mechanism. The data in this study are consistent with this hypothesis, showing similar clinical and radiological features between CM-0 and CM-I individuals, as well as the occurrence of both disorders within families. Familial clustering of CM-0 and CM-I suggests that these disorders may share an underlying genetic basis, although additional epigenetic and/or environmental factors are likely to play an important role in the development of CM-0 versus CM-I.
Keywords: Chiari malformation, idiopathic syringomyelia, genetics
Chiari malformations encompass a broad spectrum of neurodevelopmental disorders, some of which are unlikely to share a common pathophysiology. Originally, 4 types of this malformation were described.6,7 Chiari malformation Type I is characterized by displacement of the cerebellar tonsils at least 3 mm below the foramen magnum. Chiari malformation Type II is characterized by the downward displacement of the cerebellar vermis, brainstem, and fourth ventricle, and is associated with a myelomeningocele. Chiari malformation Type III is characterized by herniation of the contents of the posterior cranial fossa into a cervical or occipital encephalocele. Chiari malformation Type IV is rare and is characterized by cerebellar hypoplasia. Over the years, additional groups of patients have been identified that do not fit the historical classification of CMs. This has given rise to additional subtypes, such as CM-0.
Chiari malformation Type 0 was originally described in 1998,8 when 5 cases with syringohydromyelia without cerebellar tonsillar herniation were reported. Craniocervical decompression surgery was performed on all patients, resulting in an improvement in the size of the syrinx in all 5 patients, and symptom resolution in all 4 symptomatic patients.8 In 2001, MRI was used to assess the bony compactness of the posterior cranial fossa in these same individuals, with the addition of a sixth patient.24 Based on their findings, Tubbs et al.24 suggested that the syringomyelia may be due to a crowded posterior cranial fossa and that tonsillar herniation was not necessary to disrupt normal CSF flow in these 6 patients. Another set of 4 cases of syringomyelia without tonsillar herniation was reported in 2002.9 All 4 cases showed improvement in their symptoms and a reduction in syrinx size following craniocervical decompression surgery, although the improvement in 1 of the cases was only temporary.9 Interestingly, the authors reported that the cerebellar tonsils effaced the cisterna magna in these patients. In a later study,2 clinical features and MRI measurements of the posterior cranial fossa were compared across “idiopathic” syringomyelia patients, patients with CM-I and syringomyelia, and controls. Bogdanov and colleagues2 found that patients with CM-I–type syringomyelia and idiopathic syringomyelia shared similar clinical features, as well as similar radiological features such as shortened posterior cranial fossa bones and narrowed CSF pathways. Finally, in 2011, the original Birmingham series was extended to 15 patients with similar results of idiopathic syringomyelia that resolved after posterior fossa decompression.5 An interesting observation made in that article is that while there was no tonsillar descent noted on the imaging studies, all patients were found to have “physical barriers” to CSF intraoperatively, including 8 patients with obstructive veils at the foramen of Magendie.5 Nonetheless, there are currently no clear clinical or imaging criteria that would distinguish between CM-0 and idiopathic syringomyelia. As such, the diagnosis of CM-0 can only be presumed (or suspected) preoperatively, and ascertained postoperatively, when improvement in syringomyelia occurs after posterior cranial fossa decompression.
As suggested by the studies above, CM-0 and CM-I may be caused by a similar underlying disease mechanism. One of the theories behind the genesis of CM-I is a developmental problem that commonly results in an underdeveloped occipital bone, and less frequently in abnormalities of craniocervical bone alignment.10,16 According to this theory, herniation of the cerebellar tonsils and an upward shift of the tentorium cerebelli are believed to occur secondarily.16 Although CM-0 patients have no radiological findings of cerebellar tonsillar herniation, there is evidence that at least a subset of these patients have a compressed or distorted posterior cranial fossa, consistent with the developmental theory.2,24
The primary distinguishing feature between CM-0 and CM-I with syringomyelia is the presence or absence of tonsillar herniation. However, tonsillar herniation may not be the best criterion to use for diagnosis, because it does not correlate well with CM symptoms. For example, in a large retrospective study that examined 22,591 individuals with MR images, 25 (14.3%) of the 175 individuals diagnosed with CM-I (tonsillar herniation > 5 mm) were asymptomatic.12 A later study focused on individuals presenting with CM-I–like symptoms without significant tonsillar herniation (< 3 mm).19 In that study, patients with CM-I–like symptoms had a significantly shorter clivus, basisphenoid, basiocciput, and an increased tentorial angle as compared with controls, findings that are consistent with previous reports of patients with CM-I.19 Additional evidence suggesting that cerebellar tonsillar herniation may not be necessary for the development of syringomyelia was also noted in an earlier study in which only 10 (59%) of the 17 patients with syringomyelia had tonsillar herniation, 3 of whom had tonsils that extended less than 5 mm below the foramen magnum.11 In addition, Masur and colleagues11 reported no correlation between the extent of tonsillar herniation and various dimensions of the syrinx.
To establish the possible connection between CM-I and CM-0, particularly with respect to a common underlying genetic mechanism, we identified families showing familial aggregation of CM-I and idiopathic syringomyelia, which were either proven (2 cases) or presumed (3 cases) to represent CM-0, from a collection of families ascertained through a CM-I–affected proband. Clinical and radiological features were compared between affected family members. The similarities between these disorders, as well as the notion of a possible genetic link, are discussed.
Methods
Study Population
Families were ascertained through a proband with MRI-confirmed CM-I with or without syringomyelia. Family histories were obtained to identify additional affected family members. Several criteria were used to exclude individuals believed to have an acquired form of syringomyelia, including individuals with syndromic, traumatic, or tumor-related syringomyelia. There was also no evidence of ventriculomegaly or tethered cord in any patient with CM-0. In addition, individuals were excluded if the CM-I was associated with a supratentorial mass, hydrocephalus, history of cervical or cranial surgery unrelated to CM, or development of symptoms following placement of a lumbar shunt. Detailed family histories, medical records, and preoperative MR images were collected from all family members when possible. We identified 5 families that contained a CM-I proband who had a first-degree relative with syringomyelia without tonsillar ectopia (Figs. 1–3). Two of the syringes proved to be related to CM-0 as they improved following posterior cranial fossa decompression, while the other 3 had no operative intervention on the posterior cranial fossa. All participating family members provided written informed consent that had been approved by Duke University’s institutional review board.
Fig. 1.

Familial clustering of CM-0 and CM-I. Symbols: circle = female; square = male; diagonal line = deceased; fully shaded = CM-I; half-shaded = CM-0; small diamond = miscarriage; angled connecting lines (9509-0001 and 9509-0100) = monozygotic twins.
Fig. 3.
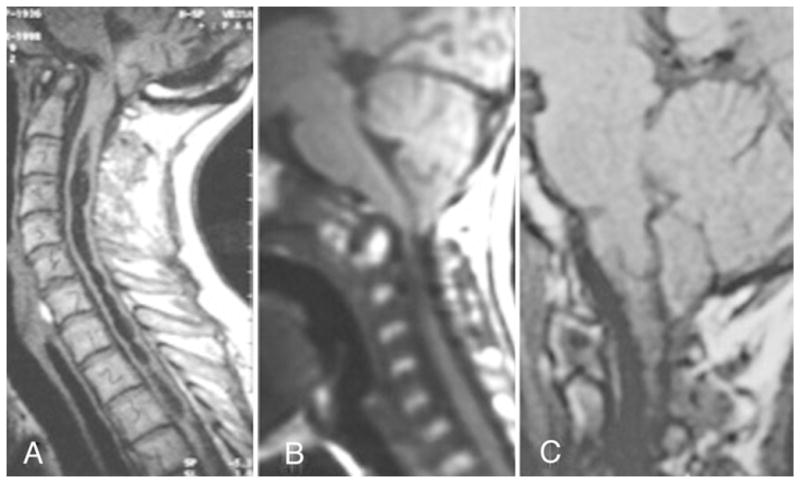
Representative sagittal MR images from 3 of the patients with CM-I. The suboptimal quality of the images is a reflection of data acquisition over many years from multiple surgeons and centers. A: 9453-1001. B: 9509-0001. C: 9448-0001.
Magnetic Resonance Imaging Measurements
Magnetic resonance imaging measurements have been described previously.24 All measurements were performed using midline sagittal T1-weighted MRI. The distance between the basion and the opisthion was used to define the foramen magnum. To evaluate caudal displacement of the brainstem, the distance between the tip of the obex and a midpoint on a line drawn from the basion to the opisthion was determined. The AP width of the fourth ventricle was measured from the fastigium to the floor of the fourth ventricle. The prepontine space at the sphenooccipital synchondrosis was assessed using the distance between the basis pontis and the sphenooccipital synchondrosis. In addition, the tentorial angle was measured as the angle of the tentorium cerebelli from a horizontal line extending from the internal occipital protuberance to the posterior edge of the hard palate. The statistical software package SAS version 9.2 (SAS Institute) was used to calculate summary statistics and to perform a t-test. A p value < 0.05 was considered statistically significant.
Results
Clinical Characterization
Clinical in Table 1. All individuals diagnosed with CM-0 were matched to their first-degree relative with CM-I, creating 5 pairs for comparison. Clinical information was unavailable for family 9448, so only 4 pairs were used in the analysis, which focused on symptom presentation, syrinx location, additional radiological findings, and surgical outcome. For 3 of the 4 pairs, symptom presentation was almost identical between the CM-0 and CM-I relatives. For family 9453 (individual 0001 compared with 1001), symptoms were more severe in the individual diagnosed with CM-I. Syrinx location was also the same in 3 of the 4 pairs. In family 9453, the individual diagnosed with CM-I had a more extensive syrinx, extending from the cervical to the thoracic region, whereas the individual diagnosed with CM-0 had only a cervical syrinx. In all 4 pairs, additional radiological findings such as basilar invagination, bone fusions, and tethering of the hindbrain and medulla, were present more often in individuals diagnosed with CM-I. There were only 2 pairs of individuals in whom both members underwent decompression surgery and had outcome information available. All 4 individuals had a positive surgical outcome, as determined by significant improvement in syrinx and/or symptoms/signs with several years of reported follow-up. Taking all individuals into account who had decompression surgery and available outcome information, 3 of the 4 CM-I individuals and both CM-0 individuals responded well to surgery.
TABLE 1.
Clinical characterization of the patients used in the analysis*
| Family No.† | Individual No. | Sex | Diagnosis | Symptoms | Syrinx Location | Radiological Findings | Good Surgical Outcome‡ |
|---|---|---|---|---|---|---|---|
| 9320 | 0001 | M | CM-0 | spastic quadriparesis | C-T | scoliosis | NA |
| 9320 | 0100 | M | CM-I/S | spastic quadriparesis | C-T | scoliosis & occipitocervical fusion | no |
|
| |||||||
| 9448 | 0001 | F | CM-I/S | unknown | T | unknown | unknown |
| 9448 | 1001 | F | CM-0 | unknown | unknown | unknown | unknown |
|
| |||||||
| 9453 | 0001 | F | CM-0 | asymptomatic | C | NA | NA |
| 9453 | 1001 | F | CM-I/S | lt arm weakness, dysphagia, unstable gait, choking | C-T | basilar invagination | NA |
| 9453 | 1002 | F | CM-I/S | bilat LE weakness, swallowing difficulties, nystagmus | Me-C | NA | yes |
| 9453 | 2002 | F | CM-I/S | headache, dizziness, trigeminal neuralgia | C-T | NA | NA |
|
| |||||||
| 9495 | 0001 | M | CM-I/S | rt claw hand & general hyperreflexia | C | tethering of hindbrain & medulla | yes |
| 9495 | 0100 | M | CM-0 | rt claw hand & general hyperreflexia | C | NA | yes |
|
| |||||||
| 9509 | 0001 | M | CM-I/S | LE hyperreflexia & clonus | T | fusion anomaly in C-spine, C-rib, scoliosis | yes |
| 9509 | 100 | M | CM-0 | LE hyperreflexia & clonus | T | NA | yes |
C = cervical; LE = lower extremity; Me = medulla; NA = not applicable; S = syringomyelia; T = thoracic.
Family and individual numbers correspond to numbers assigned in Fig. 1.
Determined by resolution of the syrinx.
Magnetic Resonance Imaging Measurements
Measurements were obtained from preoperative MR images as described above (Table 2). For analysis, we selected same-sex relative pairs (2 parent-offspring pairs from the 5 families are and 1 monozygotic twin pair) from families 9448, 9453, and 9509, which were discordant with respect to their CM diagnosis. Summary statistics for all measurements are provided in Table 3. There were no statistically significant differences for any of the measurements between disease groups (p < 0.05, paired t-test). Using CM-I as the reference, the average percentage difference exceeded 15% for only 1 measurement, the position of the obex (+225%).
TABLE 2.
Magnetic resonance imaging measurements*
| Family No. | Individual No. | Sex | Age (yrs) at MRI | Diagnosis | Prepontine Space (mm)† | Obex (mm)‡ | AP Width of 4th Ventricle (mm) | Tentorial Angle (°) | AP Width of FM (mm) |
|---|---|---|---|---|---|---|---|---|---|
| 9320 | 0001 | M | 46 | CM-0 | 9 | 0 | 10 | 95 | 30 |
| 9320 | 0100 | M | unknown | CM-I/S | unknown | unknown | unknown | unknown | unknown |
|
| |||||||||
| 9448 | 0001 | F | 12 | CM-I/S | 2 | 1 | 10 | 85 | 40 |
| 9448 | 1001 | F | 40 | CM-0 | 1.5 | 0 | 10 | 80 | 39 |
|
| |||||||||
| 9453 | 0001 | F | 33 | CM-0 | 2 | 5 | 10 | 65 | 40 |
| 9453 | 1001 | F | 62 | CM-I/S | 1 | −7 | 12 | 77 | 31 |
| 9453 | 1002 | F | unknown | CM-I/S | unknown | unknown | unknown | unknown | unknown |
| 9453 | 2002 | F | unknown | CM-I/S | unknown | unknown | unknown | unknown | unknown |
|
| |||||||||
| 9495 | 0001 | M | unknown | CM-I/S | unknown | unknown | unknown | unknown | unknown |
| 9495 | 0100 | M | 20 | CM-0 | 5 | 4 | 10 | 84 | 35 |
|
| |||||||||
| 9509 | 0001 | M | 12 | CM-I/S | 2 | 2 | 10 | 75 | 32 |
| 9509 | 0100 | M | 13 | CM-0 | 1 | 0 | 9 | 72 | 35 |
FM = foramen magnum.
Prepontine space at sphenooccipital synchondrosis.
Tip of obex to a midpoint on the basion-opisthion line.
TABLE 3.
Summary statistics of MRI measurements*
| Diagnosis | Age (yrs) at MRI | Prepontine Space (mm)† | Obex (mm)‡ | AP Width of 4th Ventricle (mm) | Tentorial Angle (°) | AP Width of FM (mm) |
|---|---|---|---|---|---|---|
| CM-I | ||||||
|
| ||||||
| mean | 28.67 | 1.67 | −1.33 | 10.67 | 79 | 34.33 |
| median | 12.00 | 2 | 1 | 10 | 77 | 32 |
| SD | 28.87 | 0.58 | 4.93 | 1.15 | 5.29 | 4.93 |
|
| ||||||
| CM-0 | ||||||
|
| ||||||
| mean | 28.67 | 1.5 | 1.67 | 9.67 | 72.33 | 38 |
| median | 33.00 | 1.5 | 0 | 10 | 72 | 39 |
| SD | 14.01 | 0.5 | 2.89 | 0.58 | 7.51 | 2.65 |
Individuals 9448-0001, 9448-1001, 9453-0001, 9453-1001, 9509-0001, and 9509-0100 were included.
Prepontine space at sphenooccipital synchondrosis.
Tip of obex to a midpoint on the basion-opisthion line.
Discussion
In this study we describe 5 families with co-occurrences of CM-0 and CM-I. Clinical characteristics and cranial morphology measurements were presented for all affected individuals when available. When comparing individuals within relative pairs with discordant CM diagnoses, we found that, in general, CM-I and CM-0 individuals shared very similar clinical features. When different, the CM-I patients appeared to have additional abnormalities, more severe symptoms, and/or a more extensive syrinx. In addition, 5 presurgical MRI measurements were examined. We found no significant differences for any of the MRI measurements across groups, and only 1 measurement differed by more than 15% across groups. Specifically, caudal displacement of the brainstem (obex) was less in CM-0 individuals as compared with CM-I individuals. The large difference in the distance between the obex and a midpoint on a line drawn from the basion to the opisthion is most likely due to an outlier (9453-1001).
Although we found many similarities between CM-I and CM-0 patients within these families, we acknowledge that our study has several limitations, including limited power to detect differences due to our small sample size, and the inability to completely control for age in the radiological analysis. Age might be expected to be a confounder, but this is not likely to be a concern as neither the mean age at MRI nor any measurements were significantly different across disease groups. However, we cannot rule out the possibility that we were unable to detect subtle differences in cranial morphology measurements between groups due to age. For example, a difference may only be observed when comparing old CM-0 versus old CM-I patients, as opposed to young CM-0 versus young CM-I patients. In addition, our analysis used relative pairs (CM-0/CM-I); thus, our findings cannot be directly compared with previous studies that investigated MRI measurements between unrelated CM-0 and CM-I individuals.
Despite these limitations, we are encouraged by our findings because this study further supports the hypothesis that CM-0 and CM-I occur due to a similar underlying mechanism, but may represent different sides of the disease spectrum.2,8,24,25 Consistent with previous reports, we show that CM-I and CM-0 individuals share similar clinical and radiological characteristics, and both respond well to decompression surgery. Additionally, our familial clustering of CM-0 and CM-I suggests that there may be a common genetic link between these disorders. While familial clustering can be due to other factors, such as chance or the environment, we have already ruled out many of the known environmental causes for CM and syringomyelia such as trauma, tumor, surgery, and lumbar shunt, as we detailed in the study eligibility description in the methods section. The ascertainment of CM-I families has been ongoing since before 1995. Because of this we do not have the exact date on which ascertainment for this particular substudy ceased. However, if we use the date on which the last CM-0/CM-I family was ascertained as a lower-bound estimate of our ascertainment completion date, we would have collected roughly 90 families that met our inclusion criteria. Based on this estimate, approximately 5% of our families showed a co-occurrence of CM-0 and CM-I. Given that the estimated prevalence of idiopathic syringomyelia is less than 1%20 (CM-0 likely representing a portion of this), we would expect CM-0 to occur in CM-I families very infrequently if these were independent disorders (< 5/500 families). Additional evidence for a genetic component includes significant heritability estimates for components of the posterior fossa,3 which is compromised in many CM-I (see Noudel et al.17) and CM-02,24 patients. Our data are consistent with this, as related individuals would be expected to have similar posterior cranial fossa measurements if there was a heritable component. There has also been at least 1 additional report of familial syringomyelia with discordant CM-I.18 Furthermore, there are several lines of evidence that suggest a genetic component in at least a subset of nonsyndromic cases with CM-I with or without syringomyelia, including twin studies,1,4,20,22,23 familial clustering,14,21,23 and cosegregation with known genetic syndromes.13,20 There have also been several reports of familial syringomyelia, some of which have CM-I (see Yabe et al.26).
We hypothesize that a possible mechanism for CM involves genetic factors that influence the development of the posterior cranial fossa and craniocervical junction, leading to a small posterior cranial fossa and abnormal craniocervical angles, which in turn define one’s risk for the development of CM-I and CM-0. Additional genetic, epigenetic, and/or environmental factors may also contribute to this risk, explaining the phenotypic variation observed among CM patients and allowing for the possibility that CM-0 and CM-I are part of a continuum of CM phenotypes. In this study, we are only considering CM-I patients consistent with the “disordered craniocervical development” mechanism, as opposed to other mechanisms such as spinal cord tethering, intracranial hypertension, and intraspinal hypotension.15 Although these were originally proposed as mechanisms for cerebellar tonsillar herniation, there is accumulating evidence suggesting that CM-0 may also follow a similar mechanism. The observation that obstructive veils are present at the fourth ventricular outlet foramina of some CM-0 and CM-I patients adds an interesting twist to the pathophysiology of these disorders. However, it is impossible to determine at this time whether these adhesions represent primary or secondary processes.
Conclusions
Our results provide further support for the idea that CM-0 and CM-I share a common pathophysiological mechanism, which may be due to an underlying genetic or epigenetic problem. Currently, the extent of tonsillar herniation is the gold standard for diagnosing someone with CM-I; however, there is increasing evidence suggesting that tonsillar herniation is not essential to produce a CM-I–like clinical phenotype. Furthermore, we have shown that individuals from the same family but with disparate diagnoses can present with similar craniocervical morphology and clinical symptoms. Thus, craniocervical morphology may be more useful for identifying a CM disease mechanism, and CM-I and CM-0 may simply represent a continuum of the same condition. It is becoming more evident that idiopathic syringomyelia may just be a collection of disorders with origins that are yet unidentifiable with current diagnostic techniques. Chiari malformation Type 0, or posterior fossa abnormalities without obvious MRI correlates, which responds to craniocervical decompression, probably explains a subset of those cases. It is likely that other currently unknown origins exist as well.
Fig. 2.

Representative sagittal MR images from all 5 CM-0 patients. Note the absence of tonsillar descent and the presence of syringomyelia. The suboptimal quality of the images is a reflection of data acquisition over many years from multiple surgeons and centers. A: 9320-0001. B and C: 9448-1001. D and E: 9453-0001. F and G: 9509-0100. H and I: 9495-0100.
Acknowledgments
This work was supported in part by grants from the National Institutes of Health (grant no. NS063273 to A.A.K. and S.G.G.) and the Chiari and Syringomyelia Patient Foundation (to S.G.G.).
The authors especially acknowledge that this manuscript was born out of discussions with Dr. Marcy C. Speer, who died on August 4, 2007.
Abbreviations used in this paper
- AP
anteroposterior
- CM
Chiari malformation
- CM-I
CM Type I
- CM-0
CM Type 0
Footnotes
Disclosure
Author contributions to the study and manuscript preparation include the following. Conception and design: Iskandar, Speer. Acquisition of data: Iskandar, Tubbs, Moftakhar, Oakes, Speer. Analysis and interpretation of data: Iskandar, Markunas, Tubbs, Ashley-Koch, Gregory, Speer. Drafting the article: Iskandar, Markunas, Tubbs. Critically revising the article: all authors. Reviewed submitted version of manuscript: all authors. Approved the final version of the manuscript on behalf of all authors: Iskandar. Statistical analysis: Iskandar, Markunas. Study supervision: Iskandar.
Portions of this work were presented in 2004 at the AANS Scientific Meeting in San Francisco, California, as well as in 2004 at the American Syringomyelia Alliance Project patient conference in Miami, Florida.
References
- 1.Atkinson JL, Kokmen E, Miller GM. Evidence of posterior fossa hypoplasia in the familial variant of adult Chiari I malformation: case report. Neurosurgery. 1998;42:401–404. doi: 10.1097/00006123-199802000-00129. [DOI] [PubMed] [Google Scholar]
- 2.Bogdanov EI, Heiss JD, Mendelevich EG, Mikhaylov IM, Haass A. Clinical and neuroimaging features of “idiopathic” syringomyelia. Neurology. 2004;62:791–794. doi: 10.1212/01.wnl.0000113746.47997.ce. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Boyles AL, Enterline DS, Hammock PH, Siegel DG, Slifer SH, Mehltretter L, et al. Phenotypic definition of Chiari type I malformation coupled with high-density SNP genome screen shows significant evidence for linkage to regions on chromosomes 9 and 15. Am J Med Genet A. 2006;140:2776–2785. doi: 10.1002/ajmg.a.31546. [DOI] [PubMed] [Google Scholar]
- 4.Cavender RK, Schmidt JH., III Tonsillar ectopia and Chiari malformations: monozygotic triplets. Case report. J Neurosurg. 1995;82:497–500. doi: 10.3171/jns.1995.82.3.0497. [DOI] [PubMed] [Google Scholar]
- 5.Chern JJ, Gordon AJ, Mortazavi MM, Tubbs RS, Oakes WJ. Pediatric Chiari malformation Type 0: a 12-year institutional experience. Clinical article. J Neurosurg Pediatr. 2011;8:1–5. doi: 10.3171/2011.4.PEDS10528. [DOI] [PubMed] [Google Scholar]
- 6.Chiari H. Über Veränderungen des Kleinhirns, des Pons und der Medulla Oblangata in Folge von kongenitaler Hydrocephalie des Grosshirns. Denkschriften Akad Wiss Wien. 1896;63:71–116. [Google Scholar]
- 7.Chiari H. Über veränderungen des kleinhirns in folge von hydrocephales des grosshirns. Dtsch Med Wochenschr. 1891;17:1172–1175. [Google Scholar]
- 8.Iskandar BJ, Hedlund GL, Grabb PA, Oakes WJ. The resolution of syringohydromyelia without hindbrain herniation after posterior fossa decompression. J Neurosurg. 1998;89:212–216. doi: 10.3171/jns.1998.89.2.0212. [DOI] [PubMed] [Google Scholar]
- 9.Kyoshima K, Kuroyanagi T, Oya F, Kamijo Y, El-Noamany H, Kobayashi S. Syringomyelia without hindbrain herniation: tight cisterna magna. Report of four cases and a review of the literature. J Neurosurg. 2002;96 (2 Suppl):239–249. doi: 10.3171/spi.2002.96.2.0239. [DOI] [PubMed] [Google Scholar]
- 10.Marin-Padilla M, Marin-Padilla TM. Morphogenesis of experimentally induced Arnold–Chiari malformation. J Neurol Sci. 1981;50:29–55. doi: 10.1016/0022-510x(81)90040-x. [DOI] [PubMed] [Google Scholar]
- 11.Masur H, Oberwittler C, Reuther G, Heyen P. Cerebellar herniation in syringomyelia: relation between tonsillar herniation and the dimensions of the syrinx and the remaining spinal cord. A quantitative MRI study. Eur Neurol. 1995;35:162–167. doi: 10.1159/000117114. [DOI] [PubMed] [Google Scholar]
- 12.Meadows J, Kraut M, Guarnieri M, Haroun RI, Carson BS. Asymptomatic Chiari Type I malformations identified on magnetic resonance imaging. J Neurosurg. 2000;92:920–926. doi: 10.3171/jns.2000.92.6.0920. [DOI] [PubMed] [Google Scholar]
- 13.Milhorat TH, Bolognese PA, Nishikawa M, McDonnell NB, Francomano CA. Syndrome of occipitoatlantoaxial hypermobility, cranial settling, and chiari malformation type I in patients with hereditary disorders of connective tissue. J Neurosurg Spine. 2007;7:601–609. doi: 10.3171/SPI-07/12/601. [DOI] [PubMed] [Google Scholar]
- 14.Milhorat TH, Chou MW, Trinidad EM, Kula RW, Mandell M, Wolpert C, et al. Chiari I malformationre defined: clinical and radiographic findings for 364. Neurosurgery. 1999;44:1005–1017. doi: 10.1097/00006123-199905000-00042. [DOI] [PubMed] [Google Scholar]
- 15.Milhorat TH, Nishikawa M, Kula RW, Dlugacz YD. Mechanisms of cerebellar tonsil herniation in patients with Chiari malformations as guide to clinical management. Acta Neurochir (Wien) 2010;152:1117–1127. doi: 10.1007/s00701-010-0636-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Nishikawa M, Sakamoto H, Hakuba A, Nakanishi N, Inoue Y. Pathogenesis of Chiari malformation: a morphometric study of the posterior cranial fossa. J Neurosurg. 1997;86:40–47. doi: 10.3171/jns.1997.86.1.0040. [DOI] [PubMed] [Google Scholar]
- 17.Noudel R, Jovenin N, Eap C, Scherpereel B, Pierot L, Rousseaux P. Incidence of basioccipital hypoplasia in Chiari malformation type I: comparative morphometric study of the posterior cranial fossa. Clinical article. J Neurosurg. 2009;111:1046–1052. doi: 10.3171/2009.2.JNS08284. [DOI] [PubMed] [Google Scholar]
- 18.Robenek M, Kloska SP, Husstedt IW. Evidence of familial syringomyelia in discordant association with Chiari type I malformation. Eur J Neurol. 2006;13:783–785. doi: 10.1111/j.1468-1331.2006.01285.x. [DOI] [PubMed] [Google Scholar]
- 19.Sekula RF, Jr, Jannetta PJ, Casey KF, Marchan EM, Sekula LK, McCrady CS. Dimensions of the posterior fossa in patients symptomatic for Chiari I malformation but without cerebellar tonsillar descent. Cerebrospinal Fluid Res. 2005;2:11. doi: 10.1186/1743-8454-2-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Speer MC, Enterline DS, Mehltretter L, Hammock P, Joseph J, Dickerson M, et al. Chiari type I malformation with or without syringomyelia: prevalence and genetics. J Genet Couns. 2003;12:297–311. doi: 10.1023/A:1023948921381. [DOI] [PubMed] [Google Scholar]
- 21.Speer MC, George TM, Enterline DS, Franklin A, Wolpert CM, Milhorat TH. A genetic hypothesis for Chiari I malformation with or without syringomyelia. Neurosurg Focus. 2000;8(3):E12. doi: 10.3171/foc.2000.8.3.12. [DOI] [PubMed] [Google Scholar]
- 22.Stovner LJ, Cappelen J, Nilsen G, Sjaastad O. The Chiari type I malformation in two monozygotic twins and relatives. Ann Neurol. 1992;31:220–222. doi: 10.1002/ana.410310213. [DOI] [PubMed] [Google Scholar]
- 23.Szewka AJ, Walsh LE, Boaz JC, Carvalho KS, Golomb MR. Chiari in the family: inheritance of the Chiari I malformation. Pediatr Neurol. 2006;34:481–485. doi: 10.1016/j.pediatrneurol.2005.09.008. [DOI] [PubMed] [Google Scholar]
- 24.Tubbs RS, Elton S, Grabb P, Dockery SE, Bartolucci AA, Oakes WJ. Analysis of the posterior fossa in children with the Chiari 0 malformation. Neurosurgery. 2001;48:1050–1055. doi: 10.1097/00006123-200105000-00016. [DOI] [PubMed] [Google Scholar]
- 25.Tubbs RS, Wellons JC, III, Blount JP, Oakes WJ. Syringomyelia in twin brothers discordant for Chiari I malformation: case report. J Child Neurol. 2004;19:459–462. doi: 10.1177/088307380401900613. [DOI] [PubMed] [Google Scholar]
- 26.Yabe I, Kikuchi S, Tashiro K. Familial syringomyelia: the Japanese case and review of the literature. Clin Neurol Neurosurg. 2002;105:69–71. doi: 10.1016/s0303-8467(02)00091-4. [DOI] [PubMed] [Google Scholar]