

Abstract
Congenital melanocytic nevi (CMN) can be associated with neurological abnormalities and an increased risk of melanoma. Mutations in NRAS, BRAF, and Tp53 have been described in individual CMN samples; however, their role in the pathogenesis of multiple CMN within the same subject and development of associated features has not been clear. We hypothesized that a single postzygotic mutation in NRAS could be responsible for multiple CMN in the same individual, as well as for melanocytic and nonmelanocytic central nervous system (CNS) lesions. From 15 patients, 55 samples with multiple CMN were sequenced after site-directed mutagenesis and enzymatic digestion of the wild-type allele. Oncogenic missense mutations in codon 61 of NRAS were found in affected neurological and cutaneous tissues of 12 out of 15 patients, but were absent from unaffected tissues and blood, consistent with NRAS mutation mosaicism. In 10 patients, the mutation was consistently c.181C>A, p.Q61K, and in 2 patients c.182A>G, p.Q61R. All 11 non-melanocytic and melanocytic CNS samples from 5 patients were mutation positive, despite NRAS rarely being reported as mutated in CNS tumors. Loss of heterozygosity was associated with the onset of melanoma in two cases, implying a multistep progression to malignancy. These results suggest that single postzygotic NRAS mutations are responsible for multiple CMN and associated neurological lesions in the majority of cases.
INTRODUCTION
Congenital melanocytic nevi (CMN) can cover up to 80% of the body surface area and large CMN, which occur in 1 in 20,000 births (Castilla et al., 1981), are usually associated with multiple smaller nevi (Figure 1). CMN can be associated with neurological abnormalities, sometimes termed neurocutaneous melanosis, although many of the abnormalities are not melanocytic. The commonest finding is foci of melanin-producing cells within the brain parenchyma, found on magnetic resonance imaging in ∼20% of affected children. Other neurological associations comprise communicating hydrocephalus, arachnoid cysts, syringomyelia, tumors (including astrocytoma, choroid plexus papilloma, ependymoma, and pineal germinoma), and malformations such as Dandy–Walker or Arnold–Chiari (Frieden et al., 1994; Foster et al., 2001; Agero et al., 2005; Kinsler et al., 2008; Ramaswamy et al., 2012). Leptomeningeal melanocytosis is a diagnosis that was previously made only at post-mortem but now can be made radiologically, and is a description of leptomeningeal deposits with a characteristic signal for melanin in a discrete or diffuse pattern. These particular lesions can be stable and benign in behavior, but are frequently rapidly progressive and capable of metastasis. Histology is not always informative, and clinical and radiological progressions are the best indicators of prognosis currently available. Neurological symptoms in patients with CMN can be present without radiological abnormality (Ruiz-Maldonado et al., 1997), and this disconnect is likely to be due to developmental intraparenchymal lesions below the resolution of current imaging (Kinsler et al., 2012c). Overall, the prevalence of radiological neurological abnormalities increases with the severity of cutaneous phenotype (i.e., size of the largest CMN and total number of nevi), and in males (Kadonaga and Frieden, 1991; Kinsler, 2008; Ramaswamy et al., 2012).
Figure 1.

Clinical and radiological images of congenital melanocytic nevus (CMN) syndrome. (From left to right) An example of the cutaneous phenotype of multiple CMNs (written consent for publication was obtained); magnetic resonance (MR) images showing two thoracic spinal tumors (neurocristic hamartomata), diffuse leptomeningeal melanocytosis, and frontal lobe meningioma.
In addition to the developmental abnormalities, CMN are a known risk factor for malignant melanoma in postnatal life. The absolute risk is associated with the severity of the cutaneous phenotype, estimated as a 1–2% lifetime risk for all individuals with CMN, but rising to 10–15% in those with the largest nevi. For those with severe cutaneous phenotypes, the risk peaks in childhood, at an age when melanoma is otherwise extremely rare (Krengel et al., 2006). Importantly, in cases where melanoma does arise, the primary tumor is not necessarily within the skin, but often within the central nervous system (CNS), and occasionally elsewhere (Hale et al., 2005; Krengel et al., 2006; Kinsler et al., 2009). Melanoma in these children is usually highly aggressive and refractory to therapy, being rapidly fatal in all cases that we have treated over the past 20 years (including unpublished cases), and in the vast majority of adequately documented cases reported in the literature (Krengel et al., 2006; Kinsler et al., 2009). The median age for the onset of melanoma in children with CMN is estimated at 7 years from a review of the literature (Krengel et al., 2006).
The phenotype of multiple CMN does not follow a Mendelian pattern of inheritance with only a few familial cases reported (de Frieden and Williams, 1994; Wijn et al., 2010). This, and the description of multiple CMN in one twin of a monozygotic pair (Amir et al., 1982), led us to consider a causative somatic mutation for multiple CMN and associated neurological lesions. Mutations already described in single samples of CMN were deemed good candidates in this model, namely those in NRAS (Papp et al., 1999; Bauer et al., 2007; Dessars et al., 2009; Phadke et al., 2011; Wu et al., 2011), BRAF (Papp et al., 1999; Pollock et al., 2003; Kumar et al., 2004; Papp et al., 2005; Ichii-Nakato et al., 2006; Dessars et al., 2007; Adjei et al., 2008), and TP53 (Papp et al., 1999), and polymorphisms in melanocortin 1 receptor (MC1R) (Papp et al., 1999; Kinsler et al., 2012b). We have previously reported a role for germline MC1R genotype in CMN, and had shown at that time consistency of germline and somatic genotype, dismissing MC1R as a somatic mosaic candidate (Kinsler et al., 2012b). Of the other candidates, NRAS was the strongest for larger CMN, although mutation rates in studies of single samples were variable. Furthermore, NRAS mutations have been described in many other types of tumors (COSMIC, 2012), consistent with the 25% rate of RAS family mutations in all human tumors (Castellano and Downward, 2011). In an attempt to uncover a causal mutation, we therefore postulated that neurological tumors in patients with CMN may also harbor activating NRAS mutations, and that lesions from a single individual may have a consistent genotype with skin lesions as a consequence of a single neuroectodermal mutation in the developing embryo, leading to both neurological and cutaneous features. Consistent with this, our results demonstrate that different CMN lesions from patients with multiple CMN contain identical codon 61 NRAS mutations and that neurological lesions from these patients also contain mutations at codon 61 of NRAS.
RESULTS
NRAS mutations in CMN
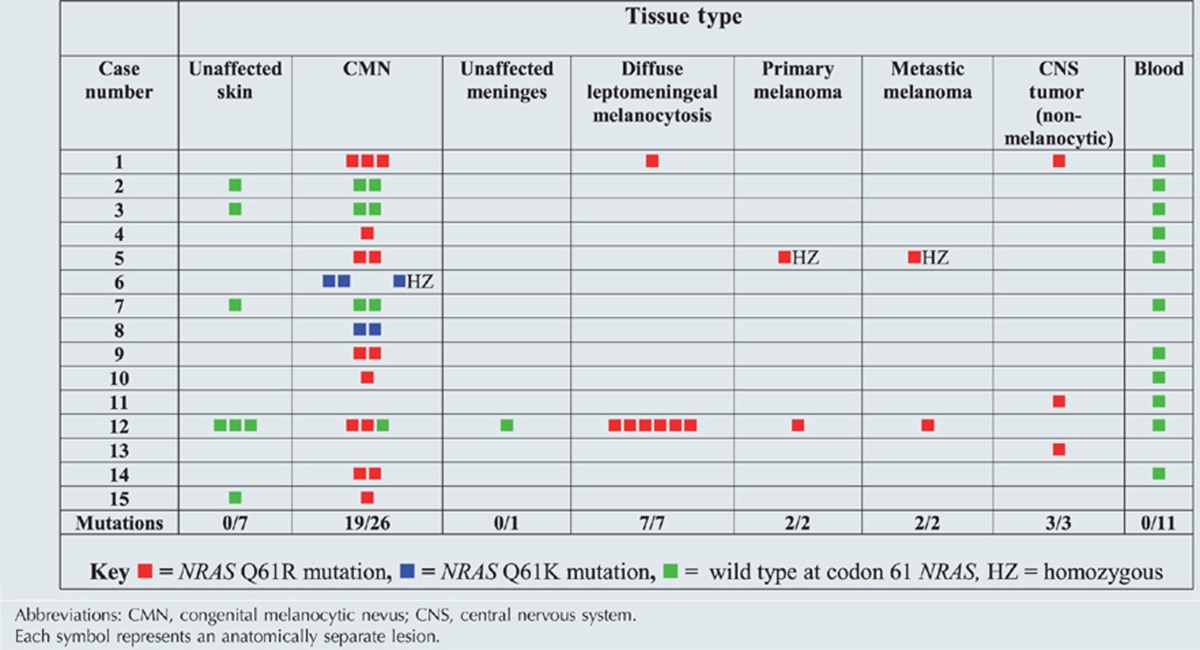
The proportion of nevus cells in a biopsy of CMN differs between lesions, and failure to identify mutations in CMN could result from this mosaicism of nevus and non-nevus cells within the lesions. The percentage of NRAS codon 61 mutant alleles measured on direct sequencing in cutaneous lesions varied from 7 to 48%, which may reflect the proportion of nevus cells in the biopsy. Therefore, in order to improve the detection of NRAS mutation in the samples, NRAS was sequenced after a site-directed mutagenesis approach that allowed enzymatic digestion of the wild-type allele. Samples with percentages <20% were enzymatically digested, and the percentage of mutant alleles in those rose to 25–63% after a single cycle of digestion. Measurement of the percentage mosaicism before digestion was validated using samples of known percentage heterozygosity (Supplementary Figure S1 online), and accuracy was found to be high. Using this approach, NRAS codon 61 mutations were identified in the CMN of 10 of the 13 subjects whose cutaneous tissue was available for sequencing (Table 1), with c.181C>A, p.Q61K in 8 subjects and c.182A>G, p.Q61R in 2 subjects. The same NRAS codon 61 mutation was seen in each of the anatomically separate CMN from the same subject, except for case 12 where one of three CMN failed to show a mutation.
Table 1. Genotype of samples of 15 patients who provided tissue.
NRAS mutations in neurological lesions from patients with CMN
All 10 neurological samples from 5 patients (or 11 from 5 patients if the sample of primary CNS melanoma is included), melanocytic and nonmelanocytic, were positive for the oncogenic NRAS missense mutation c.181C>A p.Q61K (Table 1). These included one choroid plexus papilloma, one neurocristic hamartoma, one meningioma, and two cases of leptomeningeal melanocytosis, where the same NRAS mutation was observed in six separate anatomical samples of affected meninges taken at post-mortem from one individual. No mutation was noted in the sample of normal meninges from this subject. In both cases where neurological tissues and CMN were available, the patients with CNS mutations also had the same mutation in affected skin. In order to determine whether the detection of mutations in the CMN and neurological samples were simply because of a germline NRAS mutation, DNA from blood was sequenced in 11 of the 15 subjects who provided tissue for the study, and in 42 other patients with large CMN. All 53 blood DNA samples showed wild-type NRAS, even after three cycles of PCR/enzymatic digestion, suggesting that the codon 61 mutations were restricted to affected tissues. In support of this, no mutations were detected in unaffected skin of two subjects who provided nonlesional skin samples and in whom NRAS alterations were noted in their CMN (Table 1).
NRAS mutations in melanoma from CMN subjects
Of the 15 patients who provided tissue samples, 3 died of melanoma, with the primary tumor in the skin (case 5), leptomeninges (case 6), and cerebellum (case 12). Primary melanoma samples were only available for DNA analysis from cases 5 and 12. In case 5, pre- and postmalignant samples were available from the same cutaneous lesion, which revealed progression from heterozygosity to homozygosity for the mutation Q61K with the onset of malignancy (Figure 2). The only other case in which homozygosity for NRAS mutation was found (in this instance Q61R) was from case 6 where two histologically benign, but clinically highly proliferative, CMN samples were analyzed. At 3 months after the homozygous cutaneous sample was excised, the patient developed leptomeningeal melanocytic disease, which was histologically and clinically indistinguishable from malignant melanoma, and was fatal after spreading to the abdomen via the ventriculoperitoneal shunt. In case 12 in which NRAS was mutated in the CMN, there was no loss of heterozygosity for NRAS mutations in the primary CNS melanoma sample. Melanoma samples from cases 5 and 12 were negative for BRAF V600E mutations.
Figure 2.

Progression from NRAS Q61K heterozygosity to homozygosity with the onset of malignancy. (Above) Congenital proliferative nodule within a congenital melanocytic nevus (CMN). (From left to right) Clinical appearance, hematoxylin and eosin (H&E)-stained section at × 20 magnification, DNA sequence chromatogram (reverse) showing heterozygous Q61K mutation. (Below) Malignant melanoma arising within the same CMN in the same patient 5 years later. (From left to right) Clinical appearance, H&E-stained section at × 20 magnification, DNA sequence chromatogram showing homozygous Q61K mutation.
Array comparative genomic hybridization (CGH) findings in the melanoma samples were of multiple losses and gains of parts of chromosomes (Figure 3), consistent with the previously described pattern of malignant melanoma arising within CMN (Bastian et al., 2002). Notably, array CGH at 50 kb resolution in case 5 did not show a deletion encompassing NRAS, implying that the loss of heterozygosity was either due to an additional Q61K mutation in the normal allele or a very small deletion at this locus. In both cases 5 and 12, array CGH revealed a deletion in chromosome 9p (del chr9:45,724-40,026,947 and del chr9:45,724-31,673,803, respectively), with both deletions including the CDKN2A locus (Figure 3). In comparison, the array CGH from the clinically stable diffuse leptomeningeal melanocytosis from case 1 did not show any large losses or gains.
Figure 3.

Array comparative genomic hybridization (CGH) in melanoma samples. Array CGH findings in both available primary melanoma samples (cases 5 and 12 from Table 2) showing (a) a list of all gains and losses of ⩾30 Mb, and (b) heterozygous partial deletion of chromosome 9p, including the CDKN2A locus.
DISCUSSION
As predicted by Happle (1987), somatic mosaicism for genes likely to be lethal in the germline has recently been found to be the cause of several conditions with severe clinical phenotypes involving the skin, including Proteus syndrome (Lindhurst et al., 2011), CLOVES syndrome (Kurek et al., 2012), and Schimmelpenning syndrome (Groesser et al., 2012). Our results from multiple CMN and neurocutaneous melanosis indicate that a similar somatic mosaicism is responsible for the phenotypic abnormalities in this condition, and suggest that the mutation probably occurs in the developing neural crest or neuroectoderm, although the exact cell lineage is not yet clear. Indeed, in the context of only 0.77% of neurological tumors in online databases being positive for NRAS mutations (COSMIC, 2012), our findings of NRAS alterations in the neurological as well as the skin lesions is highly supportive of a unifying causal mutation in these patients that affects pigmentary cells in the skin and pigmentary and/or non-pigmentary cells in the CNS. Furthermore, patients with CNS mutations also had the same mutation in affected skin, and patients with multiple cutaneous lesions harbored the same mutation in each lesion, but not in non-lesional skin. As all blood samples were negative for NRAS mutations, we hypothesize that this mutation may be lethal in the germline. The absence of codon 61 mutations in any samples from 3 of the 15 patients suggests that mosaic mutations in another NRAS codon or in another gene are likely to be responsible in this minority of cases.
NRAS is an extensively characterized oncogene involved in the control of key cell signaling pathways (Castellano and Downward, 2011; Pylayeva-Gupta et al., 2011). Transformation between inactive (guanosine diphosphate bound) and active (guanosine triphosphate bound) states allows RAS to act as a molecular switch (reviewed in Pylayeva-Gupta et al., 2011), controlling the signaling of RAF and phosphatidylinositol 3-kinase, and thereby the activation of the RAF/MEK/ERK pathway and Akt, respectively. Codon 61 in the guanosine triphosphate–binding site is crucial for normal inactivation, and mutations at this site lead to constitutive activation of NRAS. Our findings of NRAS codon 61 mutations in multiple CMN are supported by evidence from animal models, where injection of EGFP-NRASQ61K fusion protein into developing zebrafish leads to multiple cutaneous nevi, and transgenic zebrafish overexpressing NRASQ61K in developing melanocytes showed widespread hyperpigmentation (Dovey et al., 2009). A transgenic NRASQ61K murine model also developed CMN-like lesions (Ferguson et al., 2010). Furthermore, comparison of our results with the recent report of NRAS codon 12 mutation mosaicism in two patients with juvenile myelomonocytic leukemia, with no neurological or cutaneous features (Doisaki et al., 2012), suggests that the phenotype resulting from developmental mutations in NRAS are specific to the affected cell type and/or affected codon.
Germline mutations in the RAS/RAF/MEK/ERK pathway give rise to a group of conditions now termed RASopathies (Tidyman and Rauen, 2009). These are distinct but phenotypically related conditions including Neurofibromatosis type 1, Costello syndrome, Cardiofaciocutaneous syndrome, Noonan syndrome, and Leopard syndrome, most of which have a pigmentary component. Although the original description of RASopathy defined a germline genotypic abnormality (Tidyman and Rauen, 2009), our current findings and those in Schimmelpenning syndrome (Groesser et al., 2012) suggest that “mosaic RASopathies” could also be recognized as part of this spectrum. A recent finding in children with CMN is characteristic facial features (Kinsler et al., 2012a), namely wide or prominent forehead, apparent hypertelorism (the term used for hypertelorism described under the age of 15 years), eyebrow variants, periorbital fullness, small/short nose, narrow nasal ridge, broad nasal tip, broad or round face, full cheeks, prominent premaxilla, prominent/long philtrum, and everted lower lip. This finding has relevance as the neuroectoderm also contributes to the development of cartilage and bones of the face. The germline RASopathies all have characteristic facial features, demonstrating the effect of RAS/RAF/MEK/ERK pathway imbalance on facial development (Zenker, 2011). More specifically, germline mutations in other codons (i.e., not codon 61) of NRAS, such as those in a subset of Noonan syndrome, are known to affect facial development in humans (Cirstea et al., 2010) and in zebrafish (Runtuwene et al., 2011). Although speculative, it is feasible that the current finding of NRAS mutation mosaicism in individuals with multiple CMN could explain the facial similarities in this patient population as a result of a mutation in neuroectoderm cells affecting precursors involved in facial development.
An interesting additional question is how these somatic NRAS mutations are related to the recent finding of a higher frequency of two MC1R variant alleles in the germline of individuals with CMN, and a phenotype exacerbating the effect of certain alleles (Kinsler et al., 2012b). Interactions between germline MC1R genotype and somatic mutations in the mitogen-activated protein kinase pathway (BRAF/NRAS) have been reported; however the data have been conflicting (Landi et al., 2006; Hacker et al., 2010; Scherer et al., 2010). A possible explanation would be that reduced or altered signaling via MC1R could promote clonal growth of NRAS mutated cells, as has been reported for p53 clonal patches (Robinson et al., 2010), but further studies will be required to test this hypothesis.
In conclusion, our data suggest that multiple CMN and neuromelanosis (including nonmelanocytic CNS lesions) are caused by somatic mosaicism for NRAS codon 61 mutations in a progenitor cell within the neuroectoderm in patients with this condition. Loss of heterozygosity was associated with the timing of progression to malignancy in two cases, suggesting a central role for the mosaic mutation in a multistep model of melanoma in this condition.
MATERIALS AND METHODS
Subjects
This study was approved by the Great Ormond Street Hospital for Children and the Institute of Child Health Research Ethics Committee, and complied with the Declaration of Helsinki Principles. Samples were obtained from 57 patients with multiple CMN recruited prospectively from the Paediatric Dermatology outpatient department of the Great Ormond Street Hospital for Children between 2006 and 2012, and from 2 patients recruited retrospectively, who had neurological or malignant samples stored in the Pathology department. All patients recruited prospectively had a blood sample taken for DNA extraction. Where prospectively recruited patients underwent routine cutaneous or neurological surgery for clinical reasons during the study period, tissue samples were obtained for DNA extraction subject to an extra level of written consent. A subset of these patients also consented to a punch biopsy of unaffected skin being taken for this research. This method of collecting tissue in only those who were having surgery for cutaneous or neurological reasons was chosen to be the least invasive for participants, and had the effect of increasing the collection of neurological and malignant samples. This was desirable in the investigation of mosaicism within the CNS.
A single experienced assessor (VAK) performed clinical phenotyping (Table 2). The severity of cutaneous lesions was classified by the total number of lesions, and the projected adult size of the largest lesion, which is the best-available and the most widely used classification of CMN (Ruiz-Maldonado, 2004; Krengel et al., 2011). Magnetic resonance imaging scans of the CNS were performed where clinically indicated (as per published protocols; Kinsler et al., 2008).
Table 2. Clinical phenotype of 15 patients from whom tissue was obtained.
| Case number | Age at the time of study (years) | PAS | Total number of nevi at enrollment | Routine CNS MRI findings in the first year of life | Additional subsequent CNS MRI progression | Clinical CNS findings | Melanoma | At least three characteristic facial features |
|---|---|---|---|---|---|---|---|---|
| 1 | 8.29 | 40–60 cm | >200 | Parenchymal neuromelanosis, two congenital spinal neurocristic hamartomata, Dandy–Walker malformation | Hydrocephalus, spinal syrinx, diffuse leptomeningeal melanocytosis stable for 7 y | Wheelchair bound, developmental delay, loss of sensation in one arm, seizures | No | Yes |
| 2 | 16.28 | 40–60 cm | 20–50 | Normal | Not repeated | Speech delay, seizures at puberty, all resolved | No | No |
| 3 | 12.48 | 20–40 cm | 100–200 | Normal | Not repeated | None | No | Yes |
| 4 | 2.45 | >60 cm | 100–200 | Parenchymal neuromelanosis | Not repeated | None | No | Yes |
| 5 | Deceased age 7 y | 10–20 cm | 50–100 | Normal | Normal at 7 y | None | Primary in largest CMN, metastatic to lymph nodes | Yes |
| 6 | Deceased age 2 y | >60 cm | 50–100 | Multiple foci of parenchymal neuromelanosis | Diffuse progressive leptomeningeal melanocytosis, hydrocephalus | Progressive spinal cord compression | Leptomeningeal disease metastatic to abdomen via ventriculoperitoneal shunt | Yes |
| 7 | 9.12 | 40–60 cm | >200 | Parenchymal neuromelanosis | Not repeated | None | No | No |
| 8 | 2.94 | No single larger lesion | 10–20 | Parenchymal neuromelanosis nonmelanocytic dural deposits | No change on annual scans | None | No | Yes |
| 9 | 18.07 | 20–40 cm | >200 | Parenchymal neuromelanosis | No change over time | Seizures, mild developmental delay | No | Yes |
| 10 | 2.04 | 10–20 cm | <10 | Normal | Not repeated | None | No | Yes |
| 11 | 4.24 | 10–20 cm | 2 | Frontal lobe meningioma | Postsurgical changes only | None pre- or post-resection | No | Yes |
| 12 | Deceased age 10 y | >60 cm | 100–200 | Not done | Cerebellar melanoma, diffuse leptomeningeal melanocytosis | None before melanoma, raised intracranial pressure and progressive spinal cord compression | Primary in cerebellum, diffuse progressive leptomeningeal melanocytosis, metastatic to liver | Yes |
| 13 | 22.98 | 10–20 cm | 2 | Not done | Hydrocephalus, choroid plexus papilloma | Of raised intracranial pressure pre-resection, none post-resection | No | Not done |
| 14 | 17.06 | >60 cm | >200 | Normal | Not repeated | None | No | Yes |
| 15 | 2.79 | No single larger lesion | 100–200 | Parenchymal neuromelanosis | Not repeated | None | No | Yes |
Abbreviations: CMN, congenital melanocytic nevus; CNS, central nervous system; MRI, magnetic resonance imaging; PAS, projected adult size of largest CMN; y, year.
Total number of nevi includes the largest CMN.
Selective amplification of mutant alleles
DNA was extracted from fresh tissue using the DNeasy Blood and Tissue Kit (Qiagen) and from paraffin-embedded tissue using Ambion RecoverAll total nucleic acid extraction kit for FFPE (Life Technologies). Site-directed mutagenesis of two forward primers was used to introduce a single-nucleotide change at chr1:115256535 C>G or chr1:115256532 G>T, creating recognition sites GTNNAC and TGTACA only in the wild-type sequence, for restriction enzymes Hpy166II and BsrG1, respectively (New England Biolabs, Ipswich, MA). Standard PCR (94 °C for 2 minutes, 35 cycles of 94 °C for 30 seconds/54 °C for 1 minute/72 °C for 1 minute, 72 °C for 2 minutes) was followed by enzymatic digestion and a subsequent hemi-nested amplification (94 °C for 2 minutes, 25 cycles of 94 °C for 30 seconds/60 °C for 30 seconds/72 °C for 30 seconds, 72 °C for 2 minutes), with Sanger sequencing with the reverse primer after each PCR. Where wild-type results were obtained, two further cycles of digestion and nested PCR were done, as used in the detection of mutant GNAS alleles in mosaic fibrous dysplasia (Candeliere et al., 1997). NRAS primer sequences are shown in Supplementary Table S1 online. Enzymatic digestion used 5 μl PCR product, 1 μl enzyme (1,000 U ml−1), and 45 μl New England Biolabs Buffer 4, incubated at 37 °C for 15 minutes. Then, 2 μl of this solution was used in the ensuing 20 μl PCR reaction. Enzymatic digestion of the wild-type allele significantly increased NRAS mutation detection (Supplementary Figure S1online); however, clear detection of the mutant allele was possible in all cases after the first cycle of digestion, with no mutations discovered with subsequent cycles. All sequencing was performed on an ABI3130XL Sequencer (Applied Biosystems). Sequencing data were analyzed using Sequencher (Gene Codes, Ann Arbor, MI) without access to the sample identifiers.
Quantification of mosaicism
Quantification of mosaicism was performed using Mutation Surveyor software (Soft Genetics, State College, PA) to calculate the simplified allele ratio. Using this method the software looks at the relative fluorescent units of the normal peak and the mutant peak, and uses the following formula to work out the mutant percentage:
100 × (Mutant peak intensity/(Mutant peak intensity+Normal peak intensity)).
To verify the accuracy of this method of quantification, TA cloning of heterozygous Q61K samples was performed using TOPO TA Cloning Kit (Life Technologies), and homozygous and wild-type alleles isolated and confirmed by sequencing. Homozygous and wild-type alleles were mixed in known quantities to produce samples of 100, 50, 25, 10, 5, and 1% mosaicism for Q61K. These were sequenced in triplicate and the percentage mosaicism measured blind using the method described above. The correlation between the measured and known percentages of mosaicism on linear regression was high (r2=0.990, P<0.001) (Supplementary Figure S1 online).
Array CGH
Array CGH was performed on five samples: two melanoma samples and two proliferative nodule samples from patients 5 and 12, and one of nonmalignant clinically stable diffuse leptomeningeal melanocytosis sample from patient 1 (patient details are in Table 2). Then, 1–3 μg of patient and pooled-control DNA was labeled with fluorescent dyes Cy3 and Cy5, respectively, and hybridized to NimbleGen (Madison, WI) 135K oligonucleotide arrays. Data were analyzed using InfoQuant (London, UK) CGHFusion (version 5.7.0). The melanoma samples were also sequenced for BRAF V600E mutations using primers forward and reverse 5′-GCTCGCCCAGGAGTGCCAAG-3′/ 5′-TGGCCCTGAGATGCTGCTGA-3′.
Acknowledgments
This research was funded by the Wellcome Trust, Caring Matters Now charity, and patient support group. We are grateful to all the patients and families who participated in this research. GEM leads the fetal development and growth research group funded by Wellbeing of Women, MRC, and the Wellcome Trust. PS is supported by Great Ormond Street Hospital Children's Charity.
Glossary
- CGH
comparative genomic hybridization
- CMN
congenital melanocytic nevi
- CNS
central nervous system
- MC1R
melanocortin 1 receptor
The authors state no conflict of interest.
Footnotes
SUPPLEMENTARY MATERIAL
Supplementary material is linked to the online version of the paper at http://www.nature.com/jid
Supplementary Material
References
- Adjei AA, Cohen RB, Franklin W, et al. Phase I pharmacokinetic and pharmacodynamic study of the oral, small-molecule mitogen-activated protein kinase kinase 1/2 inhibitor AZD6244 (ARRY-142886) in patients with advanced cancers. J Clin Oncol. 2008;26:2139–2146. doi: 10.1200/JCO.2007.14.4956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agero AL, Benvenuto-Andrade C, Dusza SW, et al. Asymptomatic neurocutaneous melanocytosis in patients with large congenital melanocytic nevi: a study of cases from an Internet-based registry. J Am Acad Dermatol. 2005;53:959–965. doi: 10.1016/j.jaad.2005.07.046. [DOI] [PubMed] [Google Scholar]
- Amir J, Metzker A, Nitzan M. Giant pigmented nevus occurring in one identical twin. Arch Dermatol. 1982;118:188–189. [PubMed] [Google Scholar]
- Bastian BC, Xiong J, Frieden IJ, et al. Genetic changes in neoplasms arising in congenital melanocytic nevi: differences between nodular proliferations and melanomas. Am J Pathol. 2002;161:1163–1169. doi: 10.1016/S0002-9440(10)64393-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer J, Curtin JA, Pinkel D, et al. Congenital melanocytic nevi frequently harbor NRAS mutations but no BRAF mutations. J Invest Dermatol. 2007;127:179–182. doi: 10.1038/sj.jid.5700490. [DOI] [PubMed] [Google Scholar]
- Candeliere GA, Roughley PJ, Glorieux FH. Polymerase chain reaction-based technique for the selective enrichment and analysis of mosaic arg201 mutations in G alpha s from patients with fibrous dysplasia of bone. Bone. 1997;21:201–206. doi: 10.1016/s8756-3282(97)00107-5. [DOI] [PubMed] [Google Scholar]
- Castellano E, Downward J. RAS interaction with PI3K: more than just another effector pathway. Genes Cancer. 2011;2:261–274. doi: 10.1177/1947601911408079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castilla EE, da Graca DM, Orioli-Parreiras IM. Epidemiology of congenital pigmented naevi: I. Incidence rates and relative frequencies. Br J Dermatol. 1981;104:307–315. doi: 10.1111/j.1365-2133.1981.tb00954.x. [DOI] [PubMed] [Google Scholar]
- Cirstea IC, Kutsche K, Dvorsky R, et al. A restricted spectrum of NRAS mutations causes Noonan syndrome. Nat Genet. 2010;42:27–29. doi: 10.1038/ng.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COSMIC 2012 ) http://www.sanger.ac.uk/genetics/CGP/cosmic/
- de Wijn RS, Zaal LH, Hennekam RC, et al. Familial clustering of giant congenital melanocytic nevi. J Plast Reconstr Aesthet Surg. 2010;63:906–913. doi: 10.1016/j.bjps.2009.02.090. [DOI] [PubMed] [Google Scholar]
- Dessars B, De Raeve LE, El HH, et al. Chromosomal translocations as a mechanism of BRAF activation in two cases of large congenital melanocytic nevi. J Invest Dermatol. 2007;127:1468–1470. doi: 10.1038/sj.jid.5700725. [DOI] [PubMed] [Google Scholar]
- Dessars B, De Raeve LE, Morandini R, et al. Genotypic and gene expression studies in congenital melanocytic nevi: insight into initial steps of melanotumorigenesis. J Invest Dermatol. 2009;129:139–147. doi: 10.1038/jid.2008.203. [DOI] [PubMed] [Google Scholar]
- Doisaki S, Muramatsu H, Shimada A, et al. Somatic mosaicism for oncogenic NRAS mutations in juvenile myelomonocytic leukemia. Blood. 2012;120:1485–1488. doi: 10.1182/blood-2012-02-406090. [DOI] [PubMed] [Google Scholar]
- Dovey M, White RM, Zon LI. Oncogenic NRAS cooperates with p53 loss to generate melanoma in zebrafish. Zebrafish. 2009;6:397–404. doi: 10.1089/zeb.2009.0606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferguson B, Konrad Muller H, Handoko HY, et al. Differential roles of the pRb and Arf/p53 pathways in murine naevus and melanoma genesis. Pigment Cell Melanoma Res. 2010;23:771–780. doi: 10.1111/j.1755-148X.2010.00752.x. [DOI] [PubMed] [Google Scholar]
- Foster RD, Williams ML, Barkovich AJ, et al. Giant congenital melanocytic nevi: the significance of neurocutaneous melanosis in neurologically asymptomatic children. Plast Reconstr Surg. 2001;107:933–941. doi: 10.1097/00006534-200104010-00005. [DOI] [PubMed] [Google Scholar]
- Frieden IJ, Williams ML. Familial site-specific congenital melanocytic nevus: report of two families. Arch Dermatol. 1994;130:1075–1076. [PubMed] [Google Scholar]
- Frieden IJ, Williams ML, Barkovich AJ. Giant congenital melanocytic nevi: brain magnetic resonance findings in neurologically asymptomatic children. J Am Acad Dermatol. 1994;31:423–429. doi: 10.1016/s0190-9622(94)70204-7. [DOI] [PubMed] [Google Scholar]
- Groesser L, Herschberger E, Ruetten A, et al. Postzygotic HRAS and KRAS mutations cause nevus sebaceous and Schimmelpenning syndrome. Nat Genet. 2012;44:783–787. doi: 10.1038/ng.2316. [DOI] [PubMed] [Google Scholar]
- Hacker E, Hayward NK, Dumenil T, et al. The association between MC1R genotype and BRAF mutation status in cutaneous melanoma: findings from an Australian population. J Invest Dermatol. 2010;130:241–248. doi: 10.1038/jid.2009.182. [DOI] [PubMed] [Google Scholar]
- Hale EK, Stein J, Ben-Porat L, et al. Association of melanoma and neurocutaneous melanocytosis with large congenital melanocytic naevi--results from the NYU-LCMN registry. Br J Dermatol. 2005;152:512–517. doi: 10.1111/j.1365-2133.2005.06316.x. [DOI] [PubMed] [Google Scholar]
- Happle R. Lethal genes surviving by mosaicism: a possible explanation for sporadic birth defects involving the skin. J Am Acad Dermatol. 1987;16:899–906. doi: 10.1016/s0190-9622(87)80249-9. [DOI] [PubMed] [Google Scholar]
- Ichii-Nakato N, Takata M, Takayanagi S, et al. High frequency of BRAFV600E mutation in acquired nevi and small congenital nevi, but low frequency of mutation in medium-sized congenital nevi. J Invest Dermatol. 2006;126:2111–2118. doi: 10.1038/sj.jid.5700366. [DOI] [PubMed] [Google Scholar]
- Kadonaga JN, Frieden IJ. Neurocutaneous melanosis: definition and review of the literature. J Am Acad Dermatol. 1991;24:747–755. doi: 10.1016/0190-9622(91)70115-i. [DOI] [PubMed] [Google Scholar]
- Kinsler V, Shaw AC, Merks JH, et al. The face in congenital melanocytic nevus syndrome. Am J Med Genet A. 2012a;158A:1014–1019. doi: 10.1002/ajmg.a.34217. [DOI] [PubMed] [Google Scholar]
- Kinsler VA, Abu-Amero S, Budd P, et al. Germline melanocortin-1-receptor genotype is associated with severity of cutaneous phenotype in congenital melanocytic nevi: a role for MC1R in human fetal development. J Invest Dermatol. 2012b;132:2026–2032. doi: 10.1038/jid.2012.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinsler VA, Birley J, Atherton DJ. Great Ormond Street Hospital for Children Registry for congenital melanocytic naevi: prospective study 1988-2007. Part 1-epidemiology, phenotype and outcomes. Br J Dermatol. 2009;160:143–150. doi: 10.1111/j.1365-2133.2008.08849.x. [DOI] [PubMed] [Google Scholar]
- Kinsler VA, Chong WK, Aylett SE, et al. Complications of congenital melanocytic naevi in children: analysis of 16 years' experience and clinical practice. Br J Dermatol. 2008;159:907–914. doi: 10.1111/j.1365-2133.2008.08775.x. [DOI] [PubMed] [Google Scholar]
- Kinsler VA, Paine SM, Anderson GW, et al. Neuropathology of neurocutaneous melanosis: histological foci of melanotic neurones and glia may be undetectable on MRI. Acta Neuropathol. 2012c;123:453–456. doi: 10.1007/s00401-012-0945-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krengel S, Breuninger H, Beckwith M, et al. Meeting report from the 2011 International Expert Meeting on Large Congenital Melanocytic Nevi and Neurocutaneous Melanocytosis, Tubingen. Pigment Cell Melanoma Res. 2011;24:E1–E6. doi: 10.1111/j.1755-148X.2011.00875.x. [DOI] [PubMed] [Google Scholar]
- Krengel S, Hauschild A, Schafer T. Melanoma risk in congenital melanocytic naevi: a systematic review. Br J Dermatol. 2006;155:1–8. doi: 10.1111/j.1365-2133.2006.07218.x. [DOI] [PubMed] [Google Scholar]
- Kumar R, Angelini S, Snellman E, et al. BRAF mutations are common somatic events in melanocytic nevi. J Invest Dermatol. 2004;122:342–348. doi: 10.1046/j.0022-202X.2004.22225.x. [DOI] [PubMed] [Google Scholar]
- Kurek KC, Luks VL, Ayturk UM, et al. Somatic mosaic activating mutations in PIK3CA cause CLOVES syndrome. Am J Hum Genet. 2012;90:1108–1115. doi: 10.1016/j.ajhg.2012.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landi MT, Bauer J, Pfeiffer RM, et al. MC1R germline variants confer risk for BRAF-mutant melanoma. Science. 2006;313:521–522. doi: 10.1126/science.1127515. [DOI] [PubMed] [Google Scholar]
- Lindhurst MJ, Sapp JC, Teer JK, et al. A mosaic activating mutation in AKT1 associated with the Proteus syndrome. N Engl J Med. 2011;365:611–619. doi: 10.1056/NEJMoa1104017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papp T, Pemsel H, Zimmermann R, et al. Mutational analysis of the N-ras, p53, p16INK4a, CDK4, and MC1R genes in human congenital melanocytic naevi. J Med Genet. 1999;36:610–614. [PMC free article] [PubMed] [Google Scholar]
- Papp T, Schipper H, Kumar K, et al. Mutational analysis of the BRAF gene in human congenital and dysplastic melanocytic naevi. Melanoma Res. 2005;15:401–407. doi: 10.1097/00008390-200510000-00008. [DOI] [PubMed] [Google Scholar]
- Phadke PA, Rakheja D, Le LP, et al. Proliferative nodules arising within congenital melanocytic nevi: a histologic, immunohistochemical, and molecular analyses of 43 cases. Am J Surg Pathol. 2011;35:656–669. doi: 10.1097/PAS.0b013e31821375ea. [DOI] [PubMed] [Google Scholar]
- Pollock PM, Harper UL, Hansen KS, et al. High frequency of BRAF mutations in nevi. Nat Genet. 2003;33:19–20. doi: 10.1038/ng1054. [DOI] [PubMed] [Google Scholar]
- Pylayeva-Gupta Y, Grabocka E, Bar-Sagi D. RAS oncogenes: weaving a tumorigenic web. Nat Rev Cancer. 2011;11:761–774. doi: 10.1038/nrc3106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramaswamy V, Delaney H, Haque S, et al. Spectrum of central nervous system abnormalities in neurocutaneous melanocytosis. Dev Med Child Neurol. 2012;54:563–568. doi: 10.1111/j.1469-8749.2012.04275.x. [DOI] [PubMed] [Google Scholar]
- Robinson S, Dixon S, August S, et al. Protection against UVR involves MC1R-mediated non-pigmentary and pigmentary mechanisms in vivo. J Invest Dermatol. 2010;130:1904–1913. doi: 10.1038/jid.2010.48. [DOI] [PubMed] [Google Scholar]
- Ruiz-Maldonado R. Measuring congenital melanocytic nevi. Pediatr Dermatol. 2004;21:178–179. doi: 10.1111/j.0736-8046.2004.21222.x. [DOI] [PubMed] [Google Scholar]
- Ruiz-Maldonado R, del Rosario Barona-Mazuera M, Hidalgo-Galvan LR, et al. Giant congenital melanocytic nevi, neurocutaneous melanosis and neurological alterations. Dermatology. 1997;195:125–128. doi: 10.1159/000245713. [DOI] [PubMed] [Google Scholar]
- Runtuwene V, van Eekelen M, Overvoorde J, et al. Noonan syndrome gain-of-function mutations in NRAS cause zebrafish gastrulation defects. Dis Model Mech. 2011;4:393–399. doi: 10.1242/dmm.007112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherer D, Rachakonda PS, Angelini S, et al. Association between the germline MC1R variants and somatic BRAF/NRAS mutations in melanoma tumors. J Invest Dermatol. 2010;130:2844–2848. doi: 10.1038/jid.2010.242. [DOI] [PubMed] [Google Scholar]
- Tidyman WE, Rauen KA. The RASopathies: developmental syndromes of Ras/MAPK pathway dysregulation. Curr Opin Genet Dev. 2009;19:230–236. doi: 10.1016/j.gde.2009.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu D, Wang M, Wang X, et al. Lack of BRAF(V600E) mutations in giant congenital melanocytic nevi in a Chinese population. Am J Dermatopathol. 2011;33:341–344. doi: 10.1097/DAD.0b013e3181fb5bc7. [DOI] [PubMed] [Google Scholar]
- Zenker M. Clinical manifestations of mutations in RAS and related intracellular signal transduction factors. Curr Opin Pediatr. 2011;23:443–451. doi: 10.1097/MOP.0b013e32834881dd. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.