

Abstract
Helicobacter pylori (H. pylori) infects the gastric mucosa and persists for the life of the host. Bacterial persistence may be due to the induction of regulatory T cells (Tregs) whichmay have protective effects against other diseases such as asthma. It has been shown that H. pylori modulates the T cell response through dendritic cell reprogramming but the molecular pathways involved are relatively unknown. The goal of this study was to identify critical elements of dendritic cell (DC) activation and evaluate potential influence on immune activation. Microarray analysis was used to demonstrate limited gene expression changes in H. pylori stimulated bone marrow derived DCs (BMDCs) compared to the BMDCs stimulated with E. coli. IRAK-M, a negative regulator of TLR signaling, was upregulated and we selectedit for investigation of its role in modulating the DC and T cell responses. IRAK-M−/− and wild type BMDC were compared for their response to H. pylori. Cells lacking IRAK-M produced significantly greater amounts of proinflammatory MIP-2 and reduced amounts of immunomodulatory IL-10 than wild type BMDC. IRAK-M−/− cells also demonstrated increased MHC II expression upon activation. However, IRAK-M−/− BMDCs were comparable to wild type BMDCs in inducing T-helper 17 (TH17) and Treg responses as demonstrated in vitro using BMDC CD4+ T cells co-culture assays,and in vivo though the adoptive transfer of CD4+ FoxP3-GFP T cells into H. pylori infected IRAK-M−/− mice. These results suggest that H. pylori infection leads to the upregulation of anti-inflammatory molecules like IRAK-M and that IRAK-M has a direct impact on innate functions in DCs such as cytokine and costimulation molecule upregulation but may not affect T cell skewing.
Introduction
Helicobacter pylori (H. pylori) colonizes the gastric mucosa of over half of the world's population [1]. Infection lasts for life and is associated with a variety of gastric diseases including peptic ulcer disease, gastric adenocarcinoma, and MALT lymphoma [1]–[7]. Greater than 80% of infected people do not develop disease but even asymptomatic individuals develop histologic gastritis [8], [9]. The lack of disease in most individuals was originally believed to be due in part to variations in bacterial virulence mechanisms between H. pylori strains. It is becoming increasingly evident however that limited disease is due in large part to host immunoregulatory mechanisms, a response that also favors bacterial persistence[10]–[17].
The development of histologic gastritis is T cell-dependent and is predominantly driven by a mix of TH1 and TH17 responses [18]–[23]. Despite the role of these T helper subsets in promoting inflammation, it has been shown that regulatory T cells (Tregs) accumulate in the gastric mucosa during chronic H. pylori infection and contribute to persistent H. pylori colonization [10], [13]–[15], [17]. The loss of regulatory T cell function in murine models of Helicobacter infection results in significantly increased inflammation and reduced bacterial loads, demonstrating that these H. pylori-mediated immunomodulatory effects may be beneficial to the host and the bacteria[10], [15], [16]. The benefits to the host extend beyond the stomach as H. pylori infection has been inversely correlated with esophageal cancer in adults and wheezing in children. The protective effects of H.pylori infection maybe dependent on Tregs [24]–[27].
Down regulation of the host immune response is mediated by regulatory T cells but the bacterial, environmental, and cellular factors that promote the activation of regulatory T cells remain ill-defined for H. pylori infection. Dendritic cells (DCs) are potent antigen-presenting cells that are critical for the induction of downstream adaptive immune responses [28], [29] and they have been demonstrated to play an important role in H. pylori infection. DCs sense H. pylori primarily through Toll-like receptors (TLR) 2 and 4 in a MyD88 dependent manner [30], [31]. H. pylori infection however may skew the DC response to favor the generation of Tregs cells via IL-18 dependent mechanisms [12], [27]. This Treg response, influenced by DCs, also protects against asthma in mice [32].
A better understanding of how H. pylori affects DC function and how DCs regulate downstream immune events may provide additional insight into H. pylori pathogenesis and persistence but may also enhance our understanding of the host response to mucosal bacteria in general. One of the mechanisms employed by the host to limit microbial induced activation of APCs is the expression of interleukin-1 receptor–associated kinase M (IRAK-M), a negative regulator or TLR [33]. IRAK-M expression has been demonstrated to limit immune activation to specific pathogens, and to play a role in maintaining immune homeostasis in the gut through its inducement by commensal bacteria [33]–[36]. We observed upregulation of IRAK-M in a transcriptome analysis of H. pylori stimulated DCs, one of only ten genes to be induced. The purpose of the present study therefore was to characterize the role of IRAK-M in H. pylori-activated DCs and to determine whether IRAK-M influences activation of the T cell response.We now report that IRAK-M expression in DCs is dependent upon TLR activation, and its expression is associated with limiting the innate proinflammatory activity of the DC, as well as maturation as measured by MHC II expression.
Methods
Ethics Statement
This study was carried out in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. The protocols were approved by the Institutional Animal Care and Use Committee of the University of Maryland in Baltimore (#0809004). All efforts were made to minimize pain and suffering.
Mice
Six- to thirteen-week-old C57BL/6, TLR2−/−, and TLR4−/− mice were obtained fromJackson Laboratory (Bar Harbor, ME). IRAK-M deficient mice were derived on a C57BL/6 background as described previously [33] and were bred homozygously at the University of Maryland School of Medicine (Baltimore, MD, USA). C57BL/6 FoxP3-GFP mice and C57BL/6 OT-II Foxp3-GFP mice were a generous gift from David Scott (Bethesda, MD, USA) [37]. All animals were housed under pathogen-free conditions in microisolater cages at the University of Maryland Baltimore animal facilities. Mice were euthanized for tissue collection by CO2 asphyxiation followed by thoracotomy.
Bacterial Strains and Infection
E. coli K12 was purchased from ATCC (#29425) (Manassas, VA) and grown on LB plates supplemented with amphotericin B (2.5 µg/ml). The mouse-adapted H. pylori Sydney Strain 1 (SS1) [38]and strain 26695 (ATCC #700392) were grown on Columbia agar (Difco, Detroit, MI) supplemented with7% horse blood and antibiotics at 37°C. For inoculation of mice, bacteria were transferred to 10 ml Brucella broth (Difco) supplemented with 10% fetal bovine serum (Invitrogen, Carlsbad, CA) and amphotericin B (2.5 µg/ml). Liquid cultures were established in T25 flasks and maintained at 37°C with 10% CO2. Infections with H. pylori SS1 were performed by delivering 1×107 CFU in 0.5 ml Brucella broth by oral gavage using a 20 G feeding needle attached to a 1cc syringe. Antigen lysates were prepared as previously described [39].
Generation of BMDCs and in Vitrostimulation Assays
Femurs and tibias were removed from 6–14 week old C57BL/6 WT, TLR2−/−, TLR4−/−, and IRAK-M−/− mice at necropsy. Bone marrow was flushed out with a syringe filled with RPMI 1640 and cells were cultured in RPMI medium supplemented with either 100 ηg/mL Flt3L (R&D Systems, Minneapolis, MN) or 7 ηg/ml GM-CSF, and 10% heat inactivated FBS. Bone marrow derived DC (BMDC) were recovered after 8–9 days and plated in 48 well plates at 1×106 cells/well. Stimulation of BMDCs was performed with 10 µg/mL of either H. pylori SS1 lysate, H. pylori 26695lysate or E. coli K12 lysate. For stimulation with live bacteria, bacterial density was determined by optical density at 450 ηm and used at a multiplicity of infection (MOI) of 10. Supernatants were collected at 4, 8, and 24 hours after addition of lysate for determination of TNFα, IL-10 and MIP-2 levels by quantitative enzyme linked immunosorbent assay (ELISA) using the relevant Duoset kits according to the manufacturer's instructions (R&D Systems).
T Cell Co-culture Experiments
Coculture experiments were performed by plating 1×105 BMDCs per well in 90 well U-bottom plates and stimulating with 10 µg/mL OVA peptide. CD4+ T cells were isolated from spleens from 6–14 week old C57BL/6 OT-II Foxp3-GFP mice using the CD4+ MagCellect Isolation Kit (R&D Systems) according to the manufacturer's instructions. T cells were added 5×105 cells per well to the BMDC in 96 well plates in the presence of either Treg promoting conditions (20 ng/mL TGF-β (R&D Systems) & 25 U of mIL-2 (E-bioscience, San Diego, CA), or TH17 promoting conditions (2 ng/mL TGF-β (R&D Systems) & 20 ng/mL mIL-6 (Gemini Bio-products, Sacramento, CA). Alternatively, 1×105 BMDCs were plated in 90 well U-bottom plates and stimulated with media alone or 10 µg/mL H. pylori SS1 antigen lysate. CD4+ T cells were isolated from spleens from 6–14 week old C57BL/6 mice infected with H. pylori SS1 and 5×105 T cells were added to the wells in the absence of any additional stimulation.
Flow Cytometry Analysis
T-cells were stained with anti-CD4-APC and anti-IL17A-PE (eBioscience). BMDCs were stained with anti-MHCII-Pacific Blue, anti-PD-L1 PE, anti-CD40 PE-Cy5, anti-CD86 PE-Cy5 (eBioscience). All cells were analysed using a LSRII flow cytometer (BD Biosciences, San Hose, CA). Data were analyzed by FlowJo7 software (Tree Star, Ashland, OR).
Adoptive Transfer Experiments
CD4+ T cells were isolated from the spleens of FoxP3-GFP mice using the MagCellect Mouse CD4+ T cell isolation kit (R&D Systems) and sorted for GFP negative cells using a BD FACSAria flow cytometer. A total of 2×106 CD4+, GFP− cells were transferred into WT and IRAK-M−/− recipients by tail vein injection. Animals were infected with SS1 on day 3 and animals were harvested at 8 weeks for analysis. RNA was isolated from gastric tissue using the RNeasy kit (Qiagen, Germantown, MD) and converted to cDNA for qPCR analysis of GFP expression.
RNA Isolation, Microarray Processing and Microarray Statistical Analysis
BMDCs were recovered following stimulation at the indicated timepoints and RNA was isolated using the RNeasy kit (Qiagen). cRNA was prepared from RNA using the Illumina TotalPrep RNA amplification kit. cRNA samples were hybridized onto Illumina MouseRef8_v2.0 bead array chips (San Diego, CA) containing 25,967 probes (n = 3 for antigen lysate treated BMDCs, n = 2 for media treated BMDCs). Analysis was performed using the limma R package to compare gene expression profiles among different stimulated groups compared to media-treated BMDCs. Only probes with a false discovery rate (FDR) of <0.1 were included. Raw and normalized data sets were deposited into the NCBI Gene Expression Omnibus database under accession number GSE 44954.
Quantitative real-time PCR
Quantitative PCR analysis was performed by first converting 1 µg RNA to cDNA using the Quantitect reverse transcription kit (Qiagen). cDNA was diluted 10-fold and PCR amplication was performed with an Eppendorf Realplex Instrument (Eppendorf AG, Hamburg, Germany) on 96 well plates with SYBR Green supermix (Fermentas, Glen Burnie, MD) in either duplicate or tripiclate. Each reaction mixture contained: 12.5 µL SYBR green mix, 11.1 µL dH2O, 0.2 µL each forward and reverse primers at 0.8 µM, and 1 µL cDNA. Primer sequences were as follows: IRAKM: TGAGCAACGGGACGCTTT (forward), GATTCGAACGTGCCAGGAA (reverse) [40]; GAPDH: CCAGGTTGTCTCCTGCGACTT (forward), CCTGTTGCTGTAGCCGTATTCA (reverse); GFP: CCTGAAGTTCATCTGCACCACC (forward), CTGCTGGTAGTGGT- CGGCGAGC (reverse). Relative gene expression changes were calculated using the ΔΔCT method, and expression normalization was done using the housekeeping gene glyceraldehyde 3-phosphate dehydrogenase (GAPDH).
Statistical Analysis
Statistical analysis was performed using GraphPad Prism for Macintosh 5.0 c (GraphPad Software, San Diego, CA). The Mann-Whitney test was used for analysis of statistical significance and a P value <0.05 was considered significant.
Results
H. pylori Stimulation of BMDCs Results in Gene Expression Changes in a Limited Set of Genes after 24 hours
To identify the molecular pathways activated by H. pylori stimulation in DCs, we performed microarray analysis on BMDCs stimulated for 24 hours with either E. coli or H. pylori bacterial lysate. As expected, E. coli-stimulated BMDCs (EC-BMDCs) exhibited gene expression changes in many genes (Figure 1A). More than half of 2162 impacted genes were downregulated. Surprisingly, H. pylori-stimulated BMDCs (HP-BMDCs) showed limited gene expression changes with only 10 genes significantly affected in their expression (Table 1). Six of the 10 genes are implicated in anti-inflammatory pathways. Since H. pylori has been demonstrated to activate DCs primarily through TLR2 and TLR4 [30], [31], we were particularly interested in the increased expression of IRAK-M, a negative regulator of TLR signaling[33]. Using a qPCR approach, we confirmed that IRAK-M was indeed upregulated in BMDCs following activation with H. pylori lysate, and that in contrast to EC-BMDCs, IRAK-M expression in HP-BMDCs remained significantly high at 24 hour (P<0.01; Figure 1B). IRAK-M was demonstrated to be significantly upregulated in H. pylori-stimulated BMDC generated with either Flt3L or GM-CSF (P<0.01; Figure 1C) and this response was consistent with using lysates from either the Hp SS1 or the common laboratory strain 26695. Live Hp SS1 was also demonstrated to induce significant levels of IRAK-M expression although less effective than lysate antigen (P<0.01).
Figure 1. Global gene expression level changes in BMDCs stimulated with media alone, H. pylori or E. coli after 24 h.

cRNA was hybridized onto Illumina Mouse Ref8_v2.0 chips with probes for >24,000 genes (n = 3 for antigen lysate treated BMDCs, n = 2 for media treated BMDCs). (A) The number of genes that were upregulated or downregulated in both treated groups compared to media alone. Data reflects probes with an FDR <0.1. (B) RT-PCR analysis confirmed that IRAK-M was significantly upregulated in BMDCS stimulated with either H. pylori or E. coli compared to media alone at 4 h, 8 h and 24 h. (C) At 24 h, both Flt3L and GM-CSF derived BMDCs upregulated IRAK-M expression after stimulation with either live SS1 bacteria (MOI 10), or SS1 and 26695 antigen lysate. **, P<0.01.
Table 1. H. pylori associated gene expression changes.
| Gene Name | Fold Change(H. pylori vs. Media alone) |
| Antimicrobial peptides | |
| Elastase 2, neutrophil (Ela2) | −4.76 |
| Cathelicidin antimicrobial peptide (CAMP) | −3.27 |
| Lipocalin 2 (Lcn2) | 2.09 |
| Anti-inflammatory molecules | |
| Zinc finger CCCH type containing 12A (Zc3h12a) | 1.46 |
| Acyloxyacyl hydrolase (Aoah) | 1.53 |
| Interleukin-1 receptor-associated kinase 3 (Irak3/IRAK-M) | 2.21 |
| Nuclear factor of kappa light polypeptide gene enhancer in B-cells inhibitor, zeta (Nfkbiz/IκB-ζ) | 2.71 |
| Tribbles homolog 3 (Drosophila) (Trib3) | 3.98 |
| Vanin 3 (Vnn3) | 4.06 |
| Trafficking Molecules | |
| Vesicle transport through interaction with t-SNAREs homolog 1A (yeast) (Vti1a) | 1.70 |
Lack of IRAK-M in BMDCs Results in a More Pro-inflammatory Phenotype in HP-BMDCs
IRAK-M has been shown to play a role in DC activation in a tumor vaccine and in a LPS endotoxin tolerance model of H. pylori activation. [41], [42] Therefore, we wanted to determine if IRAK-M expression affects cytokine production in HP-BMDCs by comparing WT and IRAK-M−/− BMDCs. IRAK-M−/− cells have previously been shown to have a more proinflammatory phenotype [33], [41]. BMDCs deficient in IRAK-M responded to H. pylori antigens by producing TNFα and MIP-2 as early as four hours post activation and peaking at eight hours when levels of both cytokines were significantly higher than for WT BMDCs (P<0.05; Figure 2A and 2B; Supplemental Figure 1).TNFα levels remained higher in IRAK-M−/− cells at 24 hours post stimulation (P<0.01). IL-12p70 was also measured but were undetectable (data not shown).This is consistent with our previous observations using H. felis activation of BMDC [43].Conversely, HP-BMDCs secreted significantly less IL-10 compared to WT HP-BMDCs at all time points although levels increased steadily over the 24-hour period (Figure 2C).
Figure 2. IRAK-M−/− BMDCs exhibited a more proinflammatory phenotype after H. pylori stimulation compared to WT BMDCs.
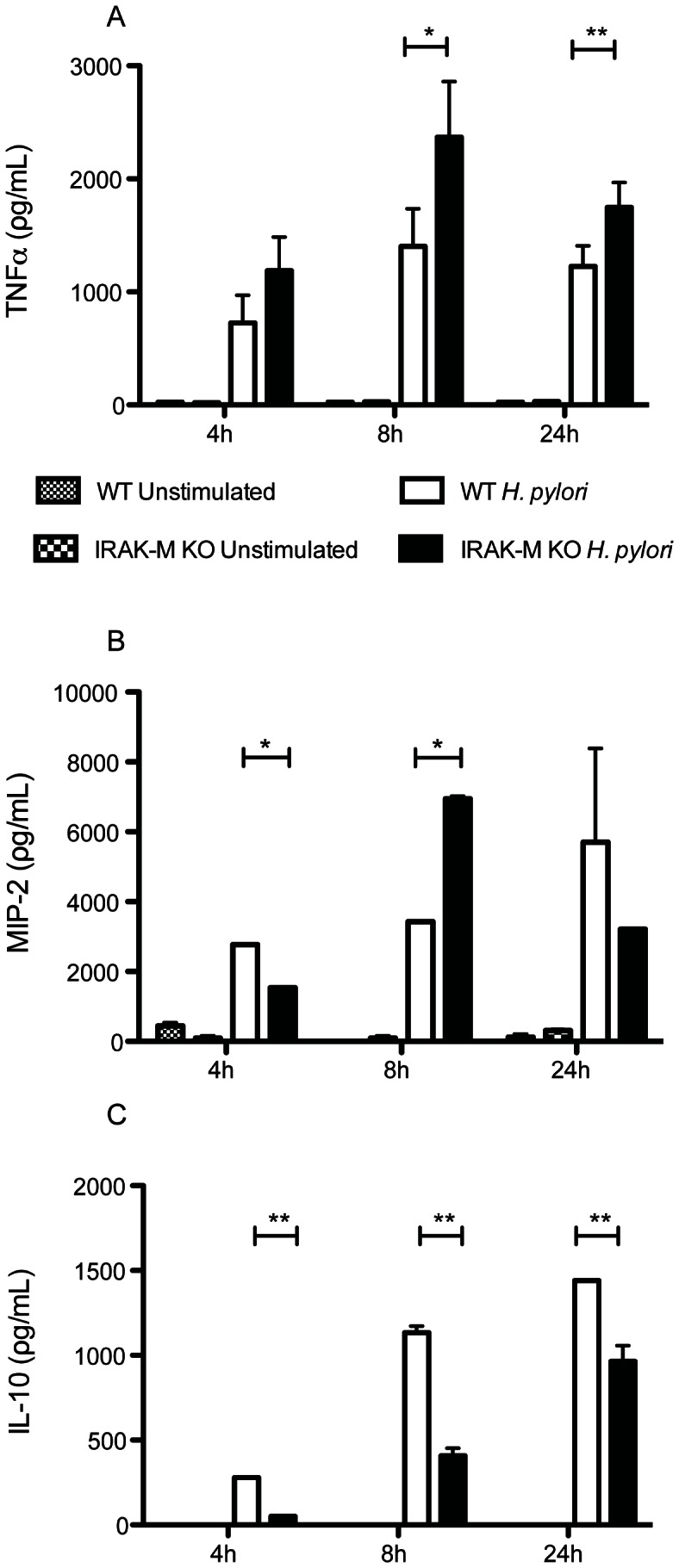
Supernatant from H. pylori antigen-stimulated WT and IRAK-M−/− BMDCs were collected at 4 h, 8 h and 24 h and used to determine (A) TNFα, (B) MIP-2, and (C) IL-10 levels by ELISA. Data reflects three independent experiments. Error bars indicate standard deviations. *, P<0.05; **, P<0.01.
Cell surface analysis of activated cells showed that IRAK-M−/− HP-BMDCs expressed higher levels of MHC II (P<0.01;Figure 3A), suggesting that IRAK-M normally limits DC activation as measured by MHC II expression in response to H. pylori stimulation. Conversely, expression of the down regulatory co-receptor PD-L1 was significantly reduced in activated IRAK-M−/− BMDC compared to WT cells (P<0.05; Figure 3B), indicating that IRAK-M normally limits the potential of DC to activate Th cells upon activation with H. pylori. Co-receptors CD86 and CD40 however remained comparable between activated IRAK-M−/− and WT BMDC (Figures 3C and 3D). Together, these data suggest that in response to H. pylori stimulation, IRAK-M expression contributes to a lack of DC maturation and promotes a regulatory phenotype exemplified by IL-10 production.
Figure 3. BMDCs stimulated for 24 h with H. pylori antigen were collected for flow cytometry analysis to determine (A) MHC-II expression and (B) PD-L1 expression compared to cells in media alone and isotype controls.

Graphical representation of mean ± SD from data collected from three individual experiments performed in duplicate is shown on the right. Graphical representation of surface expression (C) CD86 and (D) CD40 determined by flow cytometry analysis *, P<0.05; **, P<0.01.
IRAK-M upregulation in HP-BMDCs is dependent on both TLR2 and TLR4
TLR2 and TLR4 have been shown to play an important role in H. pylori sensing by DCs [31]. We therefore sought to determine if either TLR2 or TLR4 might be important in IRAK-M upregulation by comparing HP-BMDCs from WT, TLR2−/− and TLR4−/− mice. Whereas WT HP-BMDC displayed a 15-fold increase in IRAK-M expression by eight hours that remained high at 24 hours, Figure 4A illustrates that IRAK-M upregulation in HP-BMDCs is dependent on both TLR2 and TLR4 expression, and that abrogation of either TLR results in a reduction in IRAK-M expression(P<0.05).Additional evidence for the importance of both TLR2 and TLR4 in H. pylori lysate induced activation of DC is demonstrated in Figure 4B and 4C where expression of IL-10 and MIP-2 respectively are shown to be significantly reduced compared to BMDC from WT mice (P<0.01). Although expression of both cytokines increased by 24 hours for TLR4 KO cells, these cytokines were largely absent in the cells from TLR2 deficient mice.
Figure 4.

(A)IRAK-M induction in BMDCs after H. pylori stimulation is dependent on TLR expression. RNA from WT, TLR2−/−, TLR4−/− BMDCs was collected at 4 h, 8 h and 24 h and converted to cDNA. qPCR was used to determine relative IRAK-M expression levels. The diagram represents mean ± SD from data collected from two individual experiments performed in duplicate. (B) Supernatants collected from H. pylori antigen-stimulated WT, TLR2−/− and TLR4−/ BMDCs were collected at 4 h, 8 h and 24 h and used to determine (B) IL-10 and (C) MIP-2 levels by ELISA. *, P<0.05; **, P<0.01.
IRAK-M expression in DCs does not affect TH17 differentiation in T cells
Since TH17 cells have been shown to contribute to the gastritis seen in H. pylori infection as well as to protection against H. pylori in experimental murine vaccine models [21], [44]–[46], we sought to determine whether the proinflammatory phenotype of IRAK-M−/− BMDCs might increase TH17 activation using a DC-T cell coculture system. Studies using H. pylori stimulated BMDC cells to stimulate splenic CD4+ cells from mice infected with H. pylori showed no increase in either IFNγ or IL-17 producing cells from either WT or IRAK-M−/− mice (Supplemental Figure 2). This is consistent with the suppression that occurs in the H. pylori-specific T cell response in infected hosts. T cells from transgenic mice with a TCR specific for the OVA antigen were used to increase the frequency of responsive cells. IRAK-M−/− BMDCs were similar to WT BMDCs in their ability to generate IL-17A+CD4+ T cells (Figure 5A and 5B). There was no difference in the number of IL-17A+ T cells following OVA exposure when H. pylori activated DC from WT and IRAK-M−/− were used as APC cells.
Figure 5. IRAK-M−/− BMDCs do not increase TH17 induction in vitro.

(A) BMDCs isolated from WT and IRAK-M−/− mice were plated and pulsed with OVA for 2 hours before CD4+ T cells isolated from OT-II Foxp3-GFP animals were added to the wells in the presence of IL-6 and TGFβ for 72 hours. Cells were restimulated with PMA and ionomycin in the presence of monesin, and production of IL-17A in CD4+ T cells was measured by flow cytometry. (B) Data are representative of three independent experiments. Bar graph represents mean ± SD from data collected from three individual experiments performed in duplicate.
IRAK-M−/− BMDCs are Comparable to WT BMDCs in Generating Tregs
Since the balance of TH17/Tregs cells contributes to the extent of the inflammatory response in H. pylori infection [12], we also sought to determine if Treg generation is affected by the lack of IRAK-M in BMDCs using the DC-T cell co-culture system described above. The OVA TCR transgenic mice are also transgenic of FoxP3-GFP expression, providing a convenient marker for FoxP3. HP-BMDC were co-cultured with these T cells and stimulated with OVA and the activated T cells were assessed by flow cytometery for GFP (Figure 6A and6B). WT and IRAK-M−/− BMDCs did not differ in their ability to generate Tregs. To determine whether IRAK-M expression influences Treginduction in response to H. pylori in vivo, we sorted CD4+ GFP− T cells from Foxp3-GFP C57BL/6 animals to eliminate natural Treg cells and any preexisting iTreg cells. These GFP negative cells were used for adoptive transfer into WT and IRAK-M−/− recipients. Recipient mice were subsequently infected with H. pylori and the amount of new FoxP3-GFP expression was determined four weeks later by isolating gastric tissue and using qPCR analysis. H. pylori infection in WT animals resulted in the induction of Foxp3 expression in the gastric mucosa (Figure 7, P<0.05). Gastric tissue from IRAK-M−/− animals also had increased Foxp3 expression after H. pylori infection but levels were comparable to those observed in the gastric tissue of WT animals. Together, this data suggests that the proinflammatory phenotype of IRAK-M−/− BMDCs does not affect Treg generation.
Figure 6. IRAK-M−/− BMDCs do not affect Treg induction in vitro.

(A) BMDCs isolated from WT and IRAK-M−/− mice were plated and pulsed with OVA for 2 hours before CD4+ T cells isolated from OT-II Foxp3-GFP animals were added to the wells in the presence of IL-2 and TGFβ for 72 hours. Cells were restimulated with PMA and ionomycin in the presence of monesin, and Foxp3-GFP expression in CD4+ T cells was measured by flow cytometry.Data are representative of three independent experiments. (B) Bar graph represents mean ± SD from data collected from three individual experiments performed in duplicate.
Figure 7. IRAK-M deficiency does not affect iTreg generation in vivo.
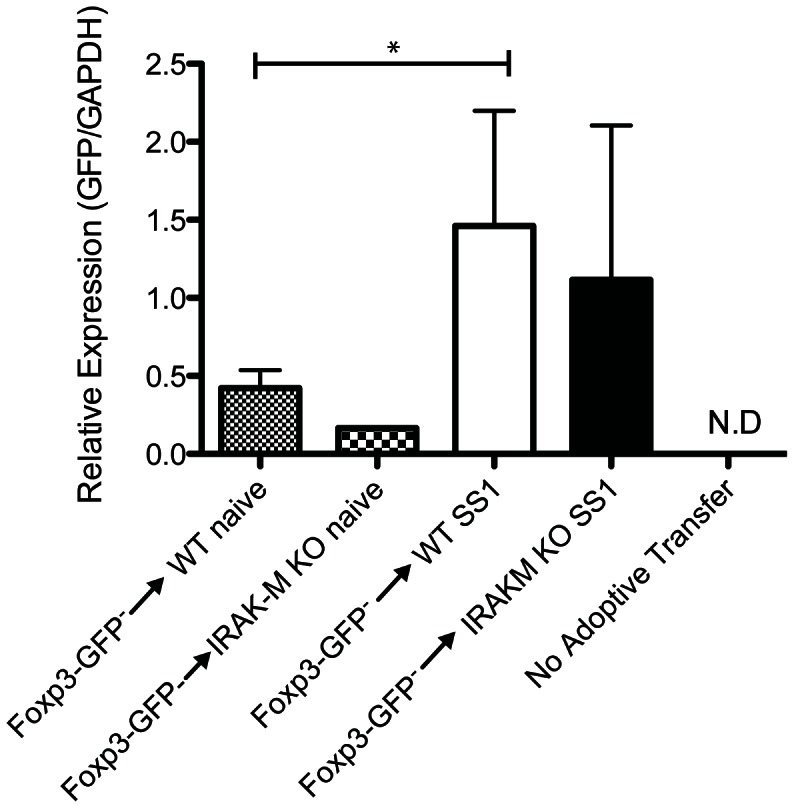
GFP−CD4+ T cells were isolated from Foxp3-GFP mice and sorted forlack of GFPexpression. 2×105 GFP− cells were transferred to the indicated recipient mice i.v. Mice were left untreated or infected with H. pylori SS1 for 8 weeks. RNA was isolated from gastric tissue to determine relative GFP expression in the stomach. Each group contained 3-6 mice. Bar graph represents mean ± SD. N.D = not detectable. *, P<0.05.
Discussion
Recent studies have demonstrated that DCs play an important immunoregulatory role in H. pylori infection and may even impact susceptibility or severity of other diseases such as asthma development [11], [12], [27], [32], [47]. An understanding of the molecular pathways that are activated in DCs by H. pylori, therefore, could provide significant insight into how immunoregulatory and inflammatory pathways are controlled during the course of infection and how these mechanisms may act more broadly. In the present study, we used a microarray approach to identify molecules in DCs whose expression is changed most significantly by H. pylori. We identified IRAK-M as a potential important regulatory protein for further characterization.
By comparing HP-BMDCs to EC-BMDCs in our microarray study, H. pylori appeared to be weakly immunogenic as only 10 gene expression changes were apparent after 24 hours. Although this contrasted significantly with the 2162 gene expression changes seen in the EC-BMDCs, our data are consistent with previous microarray analyses on H. pylori-activated cells. One study conducted a BMDC microarray following H. pylori exposure observed 126 gene expression changes after six hours [30]. A more recent study using H. pylori LPS stimulation of HEK293 cells reported only three significant gene expression changes after 24 hours [48]. The low number of gene expression changes may be a reflection of H. pylori pathogen associated molecular patterns (PAMPs)having reduced TLR stimulating activity and that after 24 hours, the induced expression of early genes may no longer be evident. The LPS is well documented to lack endotoxin activity, and structural studies confirm that the length and number of its lipid chains do not favor binding to TLR4 [49]–[51]. The flagellin protein has also been shown to lack the consensus sequence typically associated with binding to TLR5 [52].
Among the TLRs, TLR2 has been demonstrated to play a dominant role on the activation of APC by H. pylori [31]. We therefore investigated IRAK-M, a negative regulator of TLR signaling, in greater detail [33]. IRAK-M lacks kinase activity and its expression pattern was initially thought to be restricted to the monocyte/macrophage lineage [53]. It is now known that IRAK-M may also be expressed in dendritic cells and epithelial cells and is a key factor in endotoxin tolerance [33], [41], [54], [55]. IRAK-M−/− BMDCs secrete increased levels of TH1 cytokines such as IFNγ, and skew the T cell response in vivotowards a more proinflammatory phenotype to prolong survival in a tumor vaccine model [41]. In a separate study, IRAK-M−/− BMDCs that were tolerized to LPS exhibited higher levels of MHCII and lower levels of IL-10 [42].
In our study, IRAK-M−/− BMDCs stimulated with H. pylori also displayed a more pro-inflammatory phenotype compared to WT BMDCs. IRAK-M−/− BMDCs displayed increased MHC II expression and higher MIP-2, TNFα production, and also produced less immunoregulatory IL-10 and PD-L1. IRAK-M expression was shown to be dependent on TLR2 and TLR4 activation. Both TLR2 and TLR4 have been shown to be important in H. pylori infection [31]. Despite the impact of the IRAK-M deficiency on MHC II and cytokine balance in HP-BMDCs, IRAK-M−/− DCs failed to change the balance of T cell subsets induced in vitro. DCs have been shown to affect the balance of TH17 and Treg cells and to influence the outcome of H. pylori infection [12]. IRAK-M−/− DCs however were comparable to WT DCs in generating TH17 and Treg in vitro. Additionally, the IRAK-M deficiency in mice did not affect iTreg generation in vivo.
IRAK-M appears to play different roles in other infections and can be either beneficial or deleterious to the host [35], [36], [40], [54], [56]. For example, IRAK-M deficiency led to improved bacterial clearance and host survival in response to both Streptococcus pneumoniae and Klebsiella pneumoniae infections [35], [36]. In contrast, IRAK-M deficiency is deleterious to the host after influenza infection because the pro-inflammatory phenotype leads to more extensive lung injury in the host [40]. These studies all indicate that IRAK-M helps limit inflammation against pathogenic microbes, an event that can be beneficial to the host, as in the case of influenza infection. Interestingly, gut commensals have also been shown to activate IRAK-M expression and in the absence of both IRAK-M and IL-10, mice were more prone to developing colitis [34].
Of possible relation to H. pylori infection, in human genetic studies, IRAK-M has also been associated with asthma in an Italian cohort [57]. The association was not observed in either Japanese or German groups [58], [59]. Given the link between H. pylori infection and the reduced incidence of asthma in a variety of studies [24], [27], [32], it will be interesting to further dissect how IRAK-M affects the host response in H. pylori infection, and whether it has consequences at other mucosal sites such as the lung. We are currently working on further elucidating the role of IRAK-M in H. pylori infection and looking at parameters of the immune response outside of DCs activation.
In summary, we present data to demonstrate that H. pylori upregulates IRAK-M expression in DCs. We also show that IRAK-M normally functions to downregulate events associated with immune activation such as MHCII expression and MIP-2 production, and promotes regulatory activity such as the production of IL-10 and expression of PD-L1. IRAK-M expression as well as the activities associated with IRAK-M were dependent upon TLR2, and to a lesser extent TLR4 activation. However, we were unable to demonstrate that IRAK-M plays a role in skewing the balance between TH17 and Treg cells. Thus, the manifestation of IRAK-M expression may be in limitations in acute or innate host responses. It will be noteworthy to explore how IRAK-M may affect the variety of disease outcomes in H. pylori infection and whether there may be any therapeutic potential in modulating IRAK-M expression.
Supporting Information
GM-CSF BMDCs and Flt3L BMDCs share similar cytokine profiles when IRAK-M is deficient. Supernatant from WT and IRAK-M−/− BMDCs generated by the two different methods stimulated with either live H. pylori SS1 (MOI 10) or SS1 and 26695 antigen lysate were collected at 24 h and used to determine TNFα and IL-10 levels by ELISA. Data reflects two independent experiments. Error bars indicate standard deviations. *, P<0.05.
(TIF)
WT and IRAK-M deficient BMDCs have similar T cell differentiation capabilities in the presence of H. pylori stimulation. BMDCs isolated from WT and IRAK-M−/− mice were plated and pulsed with either media or H. pylori SS1 lysate for 2 hours before CD4+ T cells isolated from SS1 infected C56BL/6 animals were added to the wells for 72 hours. Cells were restimulated with PMA and ionomycin in the presence of monesin, and production of (A) IFNγ, (B) IL-17A or (C) Foxp3 in CD4+ T cells was measured by flow cytometry.
(TIF)
Funding Statement
This work was supported by National Institutes of Health grants AI-055710, and DK-46461, and Canadian Institutes of Health Research grant DFSA-236840. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Marshall BJ (1995) The 1995 Albert Lasker Medical Research Award. Helicobacter pylori. The etiologic agent for peptic ulcer. J Am Med Assoc 274: 1064–1066. [DOI] [PubMed] [Google Scholar]
- 2. Genta RM, Hamner HW, Graham DY (1993) Gastric lymphoid follicles in Helicobacter pylori infection: frequency, distribution, and response to triple therapy. Hum Pathol 24: 577–583. [DOI] [PubMed] [Google Scholar]
- 3. Marshall BJ, Warren JR (1984) Unidentified curved bacilli in the stomach of patients with gastritis and peptic ulceration. Lancet 1: 1311–1315. [DOI] [PubMed] [Google Scholar]
- 4. NIH Consensus Conference (1994) Helicobacter pylori in peptic ulcer disease. J Am Med Assoc 272: 65–69. [PubMed] [Google Scholar]
- 5. Parsonnet J, Friedman GD, Vandersteen DP, Chang Y, Vogelman JH, et al. (1991) Helicobacter pylori infection and the risk of gastric carcinoma. N Engl J Med 325: 1127–1131. [DOI] [PubMed] [Google Scholar]
- 6.World Health Organization (1994) Infection with Helicobacter pylori Schistosomes, Liver Flukes and Helicobacter pylori. Lyon: International Agency for Research on Cancer. 177–241. [Google Scholar]
- 7. Wotherspoon AC, Ortiz-Hidalgo C, Falzon MR, Isaacson PG (1991) Helicobacter pylori-associated gastritis and primary B-cell gastric lymphoma. Lancet 338: 1175–1176. [DOI] [PubMed] [Google Scholar]
- 8. Dooley CP, Cohen H, Fitzgibbons PL, Bauer M, Appleman MD, et al. (1989) Prevalence of Helicobacter pylori infection and histologic gastritis in asymptomatic persons. N Engl J Med 321: 1562–1566. [DOI] [PubMed] [Google Scholar]
- 9. Suerbaum S, Michetti P (2002) Helicobacter pylori infection. N Engl J Med 347: 1175–1186. [DOI] [PubMed] [Google Scholar]
- 10. Anderson KM, Czinn SJ, Redline RW, Blanchard TG (2006) Induction of CTLA-4-mediated anergy contributes to persistent colonization in the murine model of gastric Helicobacter pylori infection. J Immunol 176: 5306–5313. [DOI] [PubMed] [Google Scholar]
- 11. Arnold IC, Lee JY, Amieva MR, Roers A, Flavell RA, et al. (2011) Tolerance rather than immunity protects from Helicobacter pylori-induced gastric preneoplasia. Gastroenterol 140: 199–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kao JY, Zhang M, Miller MJ, Mills JC, Wang B, et al. (2010) Helicobacter pylori immune escape is mediated by dendritic cell-induced Treg skewing and Th17 suppression in mice. Gastroenterol 138: 1046–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lundgren A, Stromberg E, Sjoling A, Lindholm C, Enarsson K, et al. (2005) Mucosal FOXP3-expressing CD4+ CD25high regulatory T cells in Helicobacter pylori-infected patients. Infect Immun 73: 523–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lundgren A, Suri-Payer E, Enarsson K, Svennerholm AM, Lundin BS (2003) Helicobacter pylori-specific CD4+ CD25high regulatory T cells suppress memory T-cell responses to H. pylori in infected individuals. Infect Immun 71: 1755–1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Rad R, Brenner L, Bauer S, Schwendy S, Layland L, et al. (2006) CD25+/Foxp3+ T cells regulate gastric inflammation and Helicobacter pylori colonization in vivo. Gastroenterol 131: 525–537. [DOI] [PubMed] [Google Scholar]
- 16. Raghavan S, Fredriksson M, Svennerholm AM, Holmgren J, Suri-Payer E (2003) Absence of CD4+CD25+ regulatory T cells is associated with a loss of regulation leading to increased pathology in Helicobacter pylori-infected mice. Clin Exp Immunol 132: 393–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Raghavan S, Suri-Payer E, Holmgren J (2004) Antigen-specific in vitro suppression of murine Helicobacter pylori-reactive immunopathological T cells by CD4CD25 regulatory T cells. Scand J Immunol 60: 82–88. [DOI] [PubMed] [Google Scholar]
- 18. Bamford KB, Fan X, Crowe SE, Leary JF, Gourley WK, et al. (1998) Lymphocytes in the human gastric mucosa during Helicobacter pylori have a T helper cell 1 phenotype. Gastroenterol 114: 482–492. [DOI] [PubMed] [Google Scholar]
- 19. Eaton KA, Mefford M, Thevenot T (2001) The role of T cell subsets and cytokines in the pathogenesis of Helicobacter pylori gastritis in mice. J Immunol 166: 7456–7461. [DOI] [PubMed] [Google Scholar]
- 20. Fan XJ, Chua A, Shahi CN, McDevitt J, Keeling PWN, et al. (1994) Gastric T lymphocyte responses to Helicobacter pylori colonisation. Gut 35: 1379–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Horvath DJ Jr, Washington MK, Cope VA, Algood HM (2012) IL-23 contributes to control of chronic Helicobacter pylori iInfection and the development of T helper responses in a mouse model. Front Immunol 3: 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Karttunen RA, Karttunen TJ, Yousfi MM, El-Zimaity H, Graham DY, et al. (1997) Expression of mRNA for interferon-gamma, interleukin-10, and interleukin-12 (p40) in normal gastric mucosa and in mucosa infected with Helicobacter pylori . Scand J Gastroenterol 32: 22–27. [DOI] [PubMed] [Google Scholar]
- 23. Roth KA, Kapadia SB, Martin SM, Lorenz RG (1999) Cellular immune responses are essential for the development of Helicobacter felis-associated gastric pathology. J Immunol 163: 1490–1497. [PubMed] [Google Scholar]
- 24. Holster IL, Vila AM, Caudri D, den Hoed CM, Perez-Perez GI, et al. (2012) The impact of Helicobacter pylori on atopic disorders in childhood. Helicobacter 17: 232–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Blaser MJ (2010) Helicobacter pylori and esophageal disease: wake-up call? Gastroenterol 139: 1819–1822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sonnenberg A, Lash RH, Genta RM (2010) A national study of Helicobactor pylori infection in gastric biopsy specimens. Gastroenterol 139: : 1901–1894 e1892; quiz e1812. [DOI] [PubMed] [Google Scholar]
- 27. Oertli M, Sundquist M, Hitzler I, Engler DB, Arnold IC, et al. (2012) DC-derived IL-18 drives Treg differentiation, murine Helicobacter pylori-specific immune tolerance, and asthma protection. J Clin Invest 122: 1082–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Steinman RM (2012) Decisions about dendritic cells: past, present, and future. Annu Rev Immunol 30: 1–22. [DOI] [PubMed] [Google Scholar]
- 29. Steinman RM, Cohn ZA (1973) Identification of a novel cell type in peripheral lymphoid organs of mice. I. Morphology, quantitation, tissue distribution. J Exp Med 137: 1142–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rad R, Brenner L, Krug A, Voland P, Mages J, et al.. (2007) Toll-like receptor-dependent activation of antigen-presenting cells affects adaptive immunity to Helicobacter pylori. Gastroenterol 133: : 150–163 e153. [DOI] [PubMed] [Google Scholar]
- 31. Rad R, Ballhorn W, Voland P, Eisenacher K, Mages J, et al. (2009) Extracellular and intracellular pattern recognition receptors cooperate in the recognition of Helicobacter pylori . Gastroenterol 136: 2247–2257. [DOI] [PubMed] [Google Scholar]
- 32. Arnold IC, Dehzad N, Reuter S, Martin H, Becher B, et al. (2011) Helicobacter pylori infection prevents allergic asthma in mouse models through the induction of regulatory T cells. J Clin Invest 121: 3088–3093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kobayashi K, Hernandez LD, Galan JE, Janeway CA Jr, Medzhitov R, et al. (2002) IRAK-M is a negative regulator of Toll-like receptor signaling. Cell 110: 191–202. [DOI] [PubMed] [Google Scholar]
- 34. Biswas A, Wilmanski J, Forsman H, Hrncir T, Hao L, et al. (2011) Negative regulation of Toll-like receptor signaling plays an essential role in homeostasis of the intestine. Eur J Immunol 41: 182–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hoogerwerf JJ, van der Windt GJ, Blok DC, Hoogendijk AJ, De Vos AF, et al. (2012) Interleukin-1 receptor-associated kinase M-deficient mice demonstrate an improved host defense during Gram-negative pneumonia. Mol Med 18: 1067–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. van der Windt GJ, Blok DC, Hoogerwerf JJ, Lammers AJ, de Vos AF, et al. (2012) Interleukin 1 receptor-associated kinase m impairs host defense during pneumococcal pneumonia. J Infect Dis 205: 1849–1857. [DOI] [PubMed] [Google Scholar]
- 37. Skupsky J, Zhang AH, Su Y, Scott DW (2010) B-cell-delivered gene therapy induces functional T regulatory cells and leads to a loss of antigen-specific effector cells. Mol Ther 18: 1527–1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Lee A, O′Rourke J, De Ungria MC, Robertson B, Daskalopoulos G, et al. (1997) A standardized mouse model of Helicobacter pylori infection: introducing the Sydney strain. Gastroenterol 112: 1386–1397. [DOI] [PubMed] [Google Scholar]
- 39. Rahn W, Redline RW, Blanchard TG (2004) Molecular analysis of Helicobacter pylori-associated gastric inflammation in naive versus previously immunized mice. Vaccine 23: 807–818. [DOI] [PubMed] [Google Scholar]
- 40. Seki M, Kohno S, Newstead MW, Zeng X, Bhan U, et al. (2010) Critical role of IL-1 receptor-associated kinase-M in regulating chemokine-dependent deleterious inflammation in murine influenza pneumonia. J Immunol 184: 1410–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Turnis ME, Song XT, Bear A, Foster AE, Gottschalk S, et al. (2010) IRAK-M removal counteracts dendritic cell vaccine deficits in migration and longevity. J Immunol 185: 4223–4232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cole TS, Zhang M, Standiford TJ, Newstead M, Luther J, et al. (2012) IRAK-M modulates expression of IL-10 and cell surface markers CD80 and MHC II after bacterial re-stimulation of tolerized dendritic cells. Immunol Lett 144: 49–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Drakes ML, Czinn SJ, Blanchard TG (2006) Regulation of murine dendritic cell immune responses by Helicobacter felis antigen. Infect Immun 74: 4624–4633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. DeLyria ES, Redline RW, Blanchard TG (2009) Vaccination of mice against H pylori induces a strong Th-17 response and immunity that is neutrophil dependent. Gastroenterol 136: 247–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Shiomi S, Toriie A, Imamura S, Konishi H, Mitsufuji S, et al. (2008) IL-17 is involved in Helicobacter pylori-induced gastric inflammatory responses in a mouse model. Helicobacter 13: 518–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Velin D, Favre L, Bernasconi E, Bachmann D, Pythoud C, et al.. (2009) Interleukin-17 is a critical mediator of vaccine-induced reduction of Helicobacter infection in the mouse model. Gastroenterol 136: : 2237–2246 e2231. [DOI] [PubMed] [Google Scholar]
- 47.Hitzler I, Oertli M, Becher B, Agger EM, Muller A (2011) Dendritic cells prevent rather than promote immunity conferred by a helicobacter vaccine using a mycobacterial adjuvant. Gastroenterol 141: : 186–196, 196 e181. [DOI] [PubMed] [Google Scholar]
- 48. Smith SM, Moran AP, Duggan SP, Ahmed SE, Mohamed AS, et al. (2011) Tribbles 3: a novel regulator of TLR2-mediated signaling in response to Helicobacter pylori lipopolysaccharide. J Immunol 186: 2462–2471. [DOI] [PubMed] [Google Scholar]
- 49. Mandell L, Moran AP, Cocchiarella A, Houghton J, Taylor N, et al. (2004) Intact gram-negative Helicobacter pylori, Helicobacter felis, and Helicobacter hepaticus bacteria activate innate immunity via toll-like receptor 2 but not toll-like receptor 4. Infect Immun 72: 6446–6454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Suda Y, Ogawa T, Kashihara W, Oikawa M, Shimoyama T, et al. (1997) Chemical structure of lipid A from Helicobacter pylori strain 206-1 lipopolysaccharide. J Biochem 121: 1129–1133. [DOI] [PubMed] [Google Scholar]
- 51. Tran AX, Stead CM, Trent MS (2005) Remodeling of Helicobacter pylori lipopolysaccharide. J Endotoxin Res 11: 161–166. [DOI] [PubMed] [Google Scholar]
- 52. Andersen-Nissen E, Smith KD, Strobe KL, Barrett SL, Cookson BT, et al. (2005) Evasion of Toll-like receptor 5 by flagellated bacteria. Proc Natl Acad Sci U S A 102: 9247–9252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Wesche H, Gao X, Li X, Kirschning CJ, Stark GR, et al. (1999) IRAK-M is a novel member of the Pelle/interleukin-1 receptor-associated kinase (IRAK) family. J Biol Chem 274: 19403–19410. [DOI] [PubMed] [Google Scholar]
- 54. Takahashi N, Honda T, Domon H, Nakajima T, Tabeta K, et al. (2010) Interleukin-1 receptor-associated kinase-M in gingival epithelial cells attenuates the inflammatory response elicited by Porphyromonas gingivalis . J Periodontal Res 45: 512–519. [DOI] [PubMed] [Google Scholar]
- 55. van 't Veer C, van den Pangaart PS, van Zoelen MA, de Kruif M, Birjmohun RS, et al. (2007) Induction of IRAK-M is associated with lipopolysaccharide tolerance in a human endotoxemia model. J Immunol 179: 7110–7120. [DOI] [PubMed] [Google Scholar]
- 56. Deng JC, Cheng G, Newstead MW, Zeng X, Kobayashi K, et al. (2006) Sepsis-induced suppression of lung innate immunity is mediated by IRAK-M. J Clin Invest 116: 2532–2542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Balaci L, Spada MC, Olla N, Sole G, Loddo L, et al. (2007) IRAK-M is involved in the pathogenesis of early-onset persistent asthma. Am J Hum Genet 80: 1103–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Beygo J, Parwez Q, Petrasch-Parwez E, Epplen JT, Hoffjan S (2009) No evidence of an association between polymorphisms in the IRAK-M gene and atopic dermatitis in a German cohort. Mol Cell Probes 23: 16–19. [DOI] [PubMed] [Google Scholar]
- 59. Nakashima K, Hirota T, Obara K, Shimizu M, Jodo A, et al. (2006) An association study of asthma and related phenotypes with polymorphisms in negative regulator molecules of the TLR signaling pathway. J Hum Genet 51: 284–291. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
GM-CSF BMDCs and Flt3L BMDCs share similar cytokine profiles when IRAK-M is deficient. Supernatant from WT and IRAK-M−/− BMDCs generated by the two different methods stimulated with either live H. pylori SS1 (MOI 10) or SS1 and 26695 antigen lysate were collected at 24 h and used to determine TNFα and IL-10 levels by ELISA. Data reflects two independent experiments. Error bars indicate standard deviations. *, P<0.05.
(TIF)
WT and IRAK-M deficient BMDCs have similar T cell differentiation capabilities in the presence of H. pylori stimulation. BMDCs isolated from WT and IRAK-M−/− mice were plated and pulsed with either media or H. pylori SS1 lysate for 2 hours before CD4+ T cells isolated from SS1 infected C56BL/6 animals were added to the wells for 72 hours. Cells were restimulated with PMA and ionomycin in the presence of monesin, and production of (A) IFNγ, (B) IL-17A or (C) Foxp3 in CD4+ T cells was measured by flow cytometry.
(TIF)