

Abstract
Eukaryotic translation initiation factor 4E (eIF4E) is the rate-limiting factor for cap-dependent translation initiation, which is known to regulate oncogenesis. Elevated eIF4E and its negative impact on prognosis in human non-small cell lung cancer (NSCLC) have been reported previously. However, its potential as a therapeutic target and role in regulation of sensitivity to EGFR inhibitors is an area of ongoing investigations. In this study, we detected increased levels of eIF4E in 16 human NSCLC cell lines compared with their normal bronchial epithelial cells. Consistently, human tissue array analysis showed that eIF4E expression was significantly higher in human NSCLC tissues than normal tissues. Inhibition of eIF4E using eIF4E siRNA inhibited the growth and invasion of NSCLC cells. These data suggest that eIF4E overexpression plays a crucial role in positive regulation of the growth and invasion of NSCLC cells. By proteomics, we found that eIF4E levels were elevated in erlotinib-resistant cell lines compared with the sensitive parental cell line. In agreement, assembly of the eIF4F cap complex and several oncogenic proteins regulated by the cap-dependent translation mechanism, were also increased in erlotinib-resistant cells. Thus, erlotinib-resistant cells exhibit elevated eIF4E expression and cap-dependent translation. Inhibition of eIF4F with different means (e.g., gene knockdown) downregulated c-Met expression and partially restored cell sensitivity to erlotinib, suggesting that elevated eIF4E contributes to development of erlotinib resistance, likely through positive regulation of c-Met expression. Taken together, we suggest that elevated eIF4E in NSCLC cells is associated with proliferation, invasion and acquired erlotinib resistance.
Keywords: eIF4E, erlotinib, invasion, lung cancer, proliferation, resistance
Introduction
The long-established role of eukaryotic translation initiation factor 4E (eIF4E) in the cytoplasm is in the initiation of cap-dependent translation of cellular mRNAs. eIF4E is a cap-binding protein component of the eIF4F complex, which includes the RNA helicase eIF4A and the scaffolding protein eIF4G. Binding of eIF4E to the cap structure on the 5′ end of cellular mRNAs recruits the eIF4F complex to the mRNA. As a result, the eIF4F complex can scan from the 5′ cap through the untranslated region (5′-UTR), unwinding secondary structure to reveal the translation initiation codon, enable ribosome loading and facilitate final protein translation.1,2 Thus, recruitment of mRNA to the ribosomal apparatus constitutes a key event in the initiation of translation of mRNAs that are otherwise translationally repressed due to their long 5′-UTRs.
Because eIF4E is the least abundant among these initiation factors and is considered to be the rate-limiting factor for cap-dependent translation initiation, changes in the levels of eIF4E profoundly affect translation rates. While increasing global protein synthesis rates, higher levels of eIF4E preferentially enhance the synthesis of potent growth promoting proteins and oncogenic proteins (e.g., c-Myc, cyclin D1, HIF-1 and Mcl-1), which usually have lengthy, G/C-rich and highly structured 5′-UTRs in the mRNAs and, under normal cellular conditions, are translationally repressed. By this mechanism, cancer-related events such as transformation, tumorigenesis, angiogenesis, invasion and metastasis could be facilitated.1,3,4 It has been well documented that eIF4E expression is frequently elevated in many types of cancers and is associated with malignant progression. Inhibition of eIF4E effectively suppresses cellular transformation and tumor growth, invasiveness and metastasis.3,5,6
In human non-small cell lung cancer (NSCLC), elevated eIF4E expression has been documented in several previous studies.7-10 Moreover, elevated eIF4E expression is associated with short survival of patients with NSCLC.10-12 These results suggest that eIF4E may play an important role in positive regulation of the growth and other oncogenic phenotypes of NSCLC cells. However, whether eIF4E can serve as a good therapeutic target in NSCLC has not been demonstrated.
The epidermal growth factor receptor (EGFR) tyrosine kinase inhibitors (TKIs), erlotinib and gefitinib, are effective therapies for NSCLC patients with somatic mutations in EGFR. However, all patients eventually develop resistance (i.e., acquired resistance) to these agents.13 Thus, there is an urgent need to understand the mechanism(s) of acquired resistance to develop effective strategies to overcome the resistance. Until now, two different EGFR-TKI resistance mechanisms have been described: i.e., a secondary EGFR mutation-790M and amplification of the c-Met oncogene.13
eIF4E has been suggested to be involved in resistance to chemotherapy and androgen ablation (in prostate cancer cells).14,15 However, no study has linked eIF4E to EGFR-TKI resistance. Proteomics studies in comparing erlotinib-sensitive and resistant NSCLC cell lines uncovered an increase of eIF4E in erlotinib-resistant cells. Therefore, our present study analyzed eIF4E expression in human NSCLC cells and tissues, demonstrated its potential as a therapeutic target against NSCLC and elucidated its involvement in acquired EGFR-TKI resistance.
Results
Human NSCLC cells and tissues exhibit elevated eIF4E expression
We first examined eIF4E expression with western blotting in a panel of 16 NSCLC cell lines in comparison with two immortalized normal human bronchial epithelial (NHBE) cell lines (i.e., BEAS-2B and HBEC3KT). As presented in Figure 1A, all 16 NSCLC cell lines possessed much higher levels of eIF4E than both BEAS-2B and HBEC3KT cells, indicating that NSCLC cells exhibit elevated eIF4E expression. Moreover, we detected eIF4E expression with immunohistochemistry (IHC) in a tissue microarray (TMA) consisting of 40 cases of stage I-III lung cancer tissues (two cases of small cell lung cancer), 10 cases of metastatic cancer tissues from the primary lung cancer, and 9 cases of adjacent normal human lung tissues. In agreement with cell line data, we detected positive eIF4E staining in 71.1% (27/38) of NSCLC tissues, but only in 11.1% (1/9) of adjacent normal tissues (Fig. 1B and C). The eIF4E expression was significantly higher in NSCLC tissues than in adjacent normal tissues (p = 0.0016). Among these NSCLC tissues, we detected eIF4E expression in 92.3% (12/13) of squamous cell carcinoma, in 55.6% (10/18) of adenocarcinoma, and in 71.4% (5/7) of other NSCLC sub-types. Collectively, it is clear that eIF4E expression is elevated in human NSCLCs.
Figure 1.

eIF4E expression is elevated in human NSCLC cell lines (A) and tissues (B and C). (A) Whole-cell protein lysates were extracted from the indicated normal and NSCLC cell lines and used for detection of eIF4E expression with western blotting. (B and C) eIF4E expression in human NSCLC tissues was detected with IHC and scored as positive or negative expression (B). The representative images were also presented (C). SCC, squamous cell carcinoma; AD, adenocarcinoma; Normal, adjacent normal lung tissue.
siRNA-mediated knockdown of eIF4E inhibits the growth of NSCLC cells
If elevated eIF4E is critical for the growth of NSCLC, we hypothesized that downregulation of eIF4E would result in inhibition of the growth of NSCLC cells. To verify this, we used eIF4E siRNA to downregulate eIF4E expression and then determined its impact on the growth of NSCLC cells. As shown in Figures 2A, D and E, transfection of eIF4E siRNA into four NSCLC cell lines (i.e., H157, A549, 801C and 801D) substantially reduced the levels of eIF4E in comparison with control siRNA, indicating successful knockdown of eIF4E. Consequently, we found that all eIF4E siRNA-transfected cell lines grew much slower than cell lines transfected with the control siRNA (Fig. 2B), indicating that silencing of eIF4E inhibits the growth of NSCLC cells. Moreover, we tested the effects of eIF4E siRNA transfection on the growth of NSCLC colonies on soft agar. Again, we detected much less colonies in cells transfected with eIF4E siRNA than in control siRNA-transfected cells (Fig. 2C), further indicating that inhibition of eIF4E expression suppresses the growth of NSCLC cells. Using cleaved PARP as a readout of apoptosis, we further determined whether knockdown of eIF4E induces apoptosis in the tested cell lines. As presented in Figure 2D, we detected cleaved form of PARP in eIF4E siRNA-transfected 801D cells, but not in eIF4E siRNA-transfected H157 cells. As a positive control, tumor necrosis factor-related apoptosis-inducing ligand induced strong cleavage of PARP in the both cell lines. Thus, knockdown of eIF4E induces a cell line-dependent apoptosis.
Figure 2.

Knockdown of eIF4E (A, D and E) inhibits the growth of NSCLC cells (B and C) and induces apoptosis (D) with suppression of cap-dependent translation (E). (A and B) The indicated NSCLC cell lines were transfected with control (Ctrl) or eIF4E siRNA (20 nM) for 48 h and then subjected to western blot analysis for detection of eIF4E (A). The cells were also re-plated in 96-well plates. Cell numbers were estimated every 24 h with the SRB assay (B). The data are means ± SDs of four replicates. (C) 801-C and 801-D cells were transfected with control or eIF4E siRNA for overnight and equal numbers of cells were then used for soft agar in 35 mm diameter Petri dishes. After 14 d, colony numbers were counted and averaged from five random microscopic field or view. The final data are means ± SDs of triplicate independent determinations. The Student t-test was used to compare growth-inhibitory effects between two groups. (D and E) The indicated cell lines were transfected with control or eIF4E siRNA for 72 h (D) or 48 h (E). The cells were then harvested for preparation of whole-cell protein lysates and subsequent western blotting for detection of the given proteins. In (D) the control siRNA-transfected cells were exposed to 50 ng/ml tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) for 20 h before harvesting the cells. UD, undetected.
We also determined whether knockdown of eIF4E expression affected cap-dependent protein translation by detecting several proteins regulated by cap-dependent translation in eIF4E siRNA-transfected cells. As presented in Figure 2E, we detected reduced levels of c-Myc, cyclin D1, survivin and Mcl-1 in eIF4E siRNA-transfected H157 and 801D cells in comparison with control siRNA-transfected cells, suggesting that silencing of eIF4E expression in the tested cell systems inhibits cap-dependent translation.
Elevated eIF4E expression is associated with cell invasion
We detected that eIF4E levels were higher in 801D cells (a highly metastatic cell line) than in 801C cells (a low metastatic cell line) (Fig. 3A). Moreover, we noted that metastatic NSCLC tissues tended to have increased eIF4E staining rate than their matched primary tumor tissues (100 vs. 60%) (Fig. 3B). These data suggest that eIF4E may be involved in regulation of cancer metastasis. Therefore, we next determined whether inhibition of eIF4E expression impacted invasion of NSCLC cells. The matrigel chamber invasion assay showed that 801D cells had higher invasive capacity than 801C cells. Regardless, knockdown of eIF4E expression significantly reduced the number of invasive cells in both cell lines compared with control siRNA-transfected cells (Fig. 3C and D). Thus, inhibition of eIF4E expression suppresses the invasion of NSCLC cells, suggesting that elevated eIF4E expression is associated with positive regulation of cell invasion.
Figure 3.

eIF4E expression is increased in metastatic NSCLC cells (A) and tissues (B) and is associated with cell invasion (C and D). (A) eIF4E expression in 801C and 801D cells was detected with western blot analysis. (B) eIF4E expression in primary and matched metastatic NSCLC tissues was detected with IHC. (C and D) Both 801C and 801D cells were transfected with control (Ctrl) or eIF4E siRNA for 48 h and then subjected to matrigel chamber invasion assay. After 36 h, the non-invaded cells and collagen matrix on top of the membranes were removed. Invasive cells on the bottoms of the membranes were counted and normalized by the live cells cultured under the same conditions (C). Representative images of invasive cells on the membranes were also shown (D). The data are means ± SDs of triplicate determinations. The student t-test was used to compare inhibitory effects on invasion between two groups.
EGFR-TKI-resistant NSCLC cells possess elevated eIF4E expression and cap-dependent translation
In an effort to understand the biology of acquired EGFR-TKI resistance, we conducted proteomics by comparing HCC827/ER (derived from HCC827 with acquired resistance to erlotinib) with HCC827 cells using SILAC (stable isotope labeling with amino acids in cell culture) technique. Interestingly, eIF4E was among the proteins that were increased in HCC827/ER cells. By western blot analysis, we further confirmed increased eIF4E expression in HCC827/ER cells. In agreement, PC-9/GR cell also showed increased levels of eIF4E compared with PC-9 cells (Fig. 4A). Erlotinib treatment did not alter the expression of eIF4E both in HCC827 and HCC827/ER cells (Fig. 4B). By RT-PCR, we detected increased levels of eIF4E mRNA in HCC827/ER cells (Fig. 4C). Transfection of eIF4E promoter reporter plasmid (i.e., pGL3-eIF4E-luc) resulted in much higher luciferase activity in HCC827/ER cell than in HCC827 cells (Fig. 4D), indicating that HCC827/ER cells possess increased transcriptional activity of eIF4E. Thus, it appears that increased eIF4E in HCC827/ER cells occurs at the transcriptional level. Collectively, these data clearly demonstrate that eIF4E expression is upregulated in EGFR-TKI-resistant NSCLC cells.
Figure 4.

Erlotinib-resistant NSCLC cells possess elevated levels of eIF4E protein (A and B) and mRNA (C and D). (A and B) Whole-cell protein lysates were prepared from the indicated cell lines (A) or cell lines exposed to different concentrations of erlotinib for 6 h (B) and then used for western blot analysis to detect the given proteins as indicated. (C) Total cellular RNA was isolated from both parental and HCC827/ER cells for detection of eIF4E mRNA by RT-PCR. (D) eIF4E promoter activities in the given cell lines were performed with transfection of the given reporter constructs into HCC827 or HCC827/ER cells followed with a luciferase activity assay after 48 h. Each column represents the mean ± SD of triplicate determinations.
Moreover, we analyzed whether EGFR-TKI resistant cells exhibit elevated cap-dependent translation by examining the formation of eIF4F complex and expression of proteins regulated by cap-dependent translation. Interestingly we found that both PC-9/GR and HCC827/ER cells expressed higher levels of eIF4G in addition to eIF4E than their corresponding counterparts. Accordingly, we detected more eIF4G bound to m7GTP in both PC-9/GR and particularly HCC827/ER cells than their respective parent cells in our m7GTP-pull down assay (Fig. 5A). This result indicates that EGFR-TKI-resistant NSCLC cells possess elevated eIF4F assembly. Furthermore, we detected higher levels of HIF1α, c-Myc and Mcl-1, which are typical proteins subject to regulation by the cap-dependent translation, in HCC827/ER cells than in HCC827 cells (Fig. 5B). Taken together we suggest that EGFR-TKI-resistant cells possess elevated cap-dependent translation.
Figure 5.
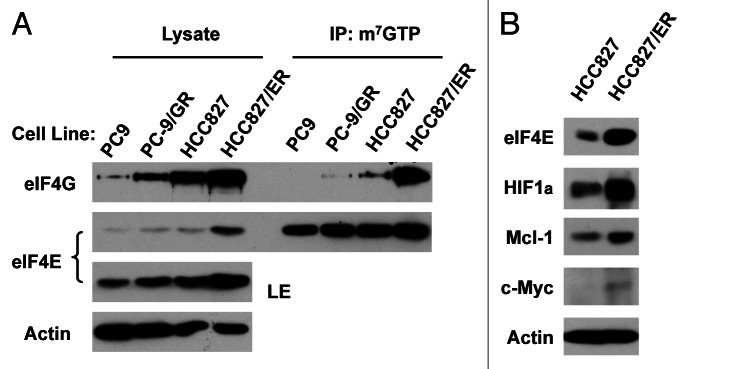
Erlotinib-resistance cells exhibit elevated eIF4F assembly (A) and expression of oncogenic proteins regulated by the cap-dependent translation (B). (A) Whole-cell protein lysates prepared from the given cell lines were used for m7GTP pull-down assay followed with western blot analysis to detect the indicated proteins. LE, long exposure. (B) Whole-cell protein lysates were prepared from the indicated cell lines and then used for western blot analysis to detect the given proteins as indicated.
Inhibition of eIF4E partially restores sensitivity of EGFR-TKI-resistant cells to erlotinib
If elevated eIF4E is involved in development of acquired resistance to EGFR-TKIs, we speculated that inhibition of eIF4E would overcome EGFR-TKI-resistance and restore the sensitivity of to EGFR-TKIs. To test this hypothesis, we used eIF4E siRNA to knock down eIF4E expression in HCC827/ER cells and then examined its impact on cell response to erlotinib. As presented in Figure 6A, erlotinib at 2 μM inhibited the growth of control siRNA-transfected HCC827/ER cells only by < 15%; however, it suppressed the growth of eIF4E siRNA-transfected HCC827/ER cells by > 55%, which was also greater than that cause by knockdown of eIF4E alone (< 35% growth inhibition). Thus, it is clear that silencing of eIF4E enhances the growth-inhibitory effect of erlotinib in HCC827/ER cells.
Figure 6.
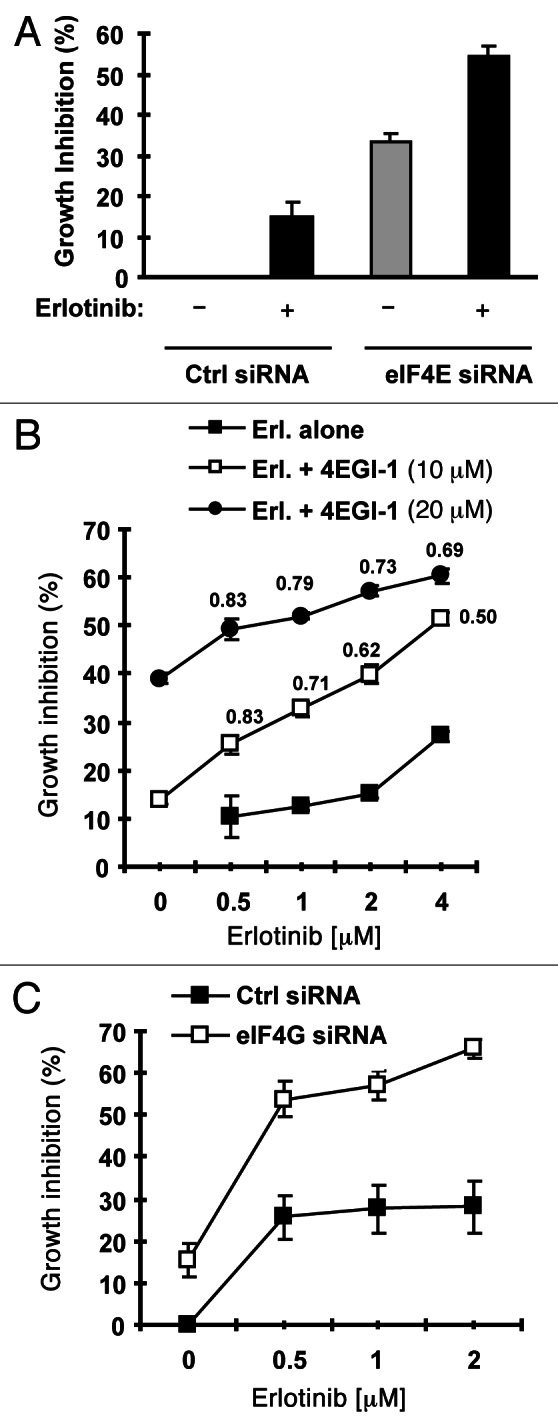
Inhibition of eIF4F formation by knocking down eIF4E (A) or eIF4G (C) or by inhibiting eIF4E and eIF4G interaction with 4EGI-1 (B) sensitizes erlotinib-resistant cells to erlotinib. (A) HCC828/ER cells were transfected with control (Ctrl) or eIF4E siRNA for overnight and then exposed to 2 μM erlotinib for 3 d. B, HCC828/ER cells were treated with indicated concentrations of erlotinib in the absence and presence of 4EGI-1 for 3 d. (C) HCC828/ER cells were transfected with control (Ctrl) or eIF4G siRNA for overnight and then exposed to the indicated concentrations of erlotinib for 3 d. After the aforementioned treatments, the cell numbers were estimated by the SRB assay. The data are means ± SDs of four replicate determinations. The numbers by the lines in (B) are combination indexes for the combinations of erlotinib and 4EGI-1.
4EGI-1 is a small molecule that inhibits eIF4E and eIF4G interaction and cap-dependent translation.16 Thus, we further determined whether addition of 4EGI-1 would enhance the growth inhibitory effects of erlotinib on HCC827/ER cells. In a 3 d assay, the combination of erlotinib and 4EGI-1 was more potent than either agent alone in inhibiting the growth of HCC827/ER cells. The combination indexes were < 1 for all combination treatments (Fig. 6B), indicating synergy between erlotinib and 4EGI-1 in inhibiting the growth of HCC827/ER cells. In agreement, the long-term colony formation assay generated similar results as presented in Fig. S1. The presence of 4EGI-1 enhanced the ability of erlotinib to inhibit the formation and growth of the colonies of HCC827/ER cells. Taken together, these results indicate that the combination of erlotinib and 4EGI-1 synergistically inhibits the growth of HCC827/ER cells.
eIF4G is also the major component in the eIF4F complex. Thus, we further knocked down eIF4G in HCC827/ER cells and analyzed its impact on cell sensitivity to erlotinib. As presented in Figure 6C, erlotinib at up 2 μM inhibited the growth of control siRNA-transfected HCC827/ER cells by approximately 25%, but the growth of eIF4G-transfected HCC827/ER cells by about 70%. Thus, the knockdown of eIF4G greatly sensitizes HCC827/ER cells to erlotinib, furthering the notion that inhibition of eIF4F cap complex restores TKI-resistant cells to TKIs.
Elevated eIF4E is associated with increased Met expression in TKI-resistant cells
c-Met amplification represents one of the major mechanisms accounting for EGFR TKI-resistance.13 In HCC827/ER cells, c-Met expression is elevated compared with their parent HCC827 cells (Fig. S2). Since eIF4E is primarily involved in regulation of cap-dependent protein translation, we then asked whether elevated eIF4E enhances c-Met translation. To this end, we knocked down eIF4E and eIF4G, respectively, and then examined their impact on c-Met expression. Indeed, knockdown of either eIF4E or eIF4G reduced the levels of c-Met protein (Fig. 7A and B). Similarly, treatment of HCC827/ER cells with 4EGI-1 also reduced c-Met levels (Fig. 7C). These data collectively indicate that inhibition of eIF4F cap complex inhibits c-Met expression.
Figure 7.
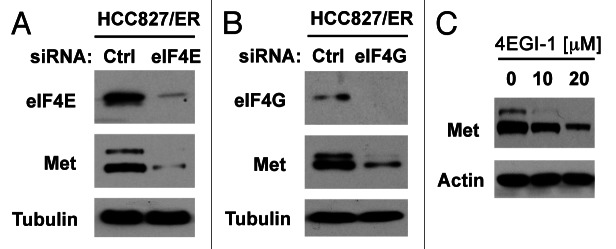
Inhibition of eIF4F formation by knocking down eIF4E (A) or eIF4G (B) or by inhibiting eIF4E and eIF4G interaction with 4EGI-1 (C) reduces c-Met protein levels. (A and B) HCC828/ER cells were transfected with control (Ctrl), eIF4E or eIF4G siRNA for 48 h. (C) HCC828/ER cells were treated with the indicated concentrations of 4EGI-1 for 24 h. After the aforementioned treatments, the cells were harvested for preparation of whole-cell protein lysates and subsequent western blotting.
Discussion
In this study, we have shown that human NSCLC cell lines and tissues possess significantly elevated expression of eIF4E in comparison with their normal counterparts (Fig. 1). These findings are in agreement with previous observations.7-10 In variance with a report that eIF4E is rarely increased in squamous cell carcinoma of lung,7 we detected eIF4E expression in 92% (12/13) of squamous cell carcinoma. Nonetheless, our current and previous studies together clearly indicate that NSCLCs exhibit elevated eIF4E expression.
Given that elevated eIF4E expression is significantly associated with short survival of NSCLC patients,10-12 it is plausible to speculate a role of eIF4E in positive regulation of the growth of NSCLC cells. Indeed, knockdown of eIF4E expression by siRNA in our study substantially inhibited the growth of NSCLC cells (Fig. 2), suggesting that eIF4E plays a critical role in mediating the growth of NSCLC cells. In this study, we found that knockdown of eIF4E induced apoptosis in 801D cells, but not in H157 cells although it effectively inhibited the growth of both cell lines, suggesting that inhibition of eIF4E inhibit the growth of cancer cells through growth arrest or both growth arrest and apoptosis. It has been recently shown that eIF4E-specific antisense oligonucleotides effectively inhibit the growth of cancer xenografts in mice with minimal toxicity,17 hence providing robust validation for eIF4E-targeted cancer therapy. Our results also support eIF4E as a promising target for therapy of NSCLCs.
We noted that eIF4E knockdown potently reduced the levels of Mcl-1 in 801D cells, but only minimally in H157 cells even though it effectively decreased survivin levels in the both cell lines (Fig. 2E). Coincidentally, knockdown of eIF4E induced apoptosis in 801D cells, but not in H157 cells (Fig. 2D). Whether this suggests that Mcl-1 downregulation plays a critical role in mediating eIF4E inhibition-induced apoptosis needs further investigation.
The early work with antisense of eIF4E in Ras-transformed rat embryo fibroblasts showed that cells with reduced levels of eIF4E had delayed and reduced invasiveness and decreased experimental metastasis,18 suggesting that eIF4E plays a role in regulation of invasion and metastasis.1 In our study, we detected elevated eIF4E expression in metastatic NSCLC cells and tissues. Moreover, knockdown of eIF4E significantly inhibited invasion of NSCLC cells (Fig. 3), suggesting that elevated eIF4E expression is associated with positive regulation of invasion of NSCLC cells. Thus, our findings support the notion that eIF4E is involved in regulation of cancer invasion and metastasis.
Acquired resistance to EGFR-TKIs is a major obstacle and challenge in the treatment of NSCLCs with EGFR-TKIs.13 Our exciting finding in this study that eIF4E expression is elevated in NSCLC cells with acquired resistance to EGFR-TKIs (e.g., HCC827/ER and PC-9/GR) is significant for future efforts to overcome EGFR-TKI resistance. Moreover we have shown that these EGFR-TKI-resistant NSCLC cells possess increased capacity of eIF4F assembly and elevated expression of oncogenic proteins known to be regulated by the cap-dependent translation mechanism (e.g., HIF1α, c-Myc and Mcl-1) (Figs. 4 and 5). These results together indicate that eIF4E expression and cap-dependent translation are elevated in EGFR-TKI-resistant NSCLC cells. To the best of our knowledge, this is the first study that links eIF4E and cap-dependent translation to the acquired EGFR-TKI resistance of NSCLCs.
In our study, inhibition of eIF4F assembly by knocking down of eIF4E or eIF4G with eIF4E or eIF4G siRNA enhanced the effect of erlotinib against the growth of HCC827/ER cells. Moreover, the combination of erlotinib and 4EGI-1, an inhibitor of eIF4E and eIF4G interaction, synergistically inhibited the growth of HCC827/ER cells (Figs. 6and S2). These data collectively suggest that elevated eIF4E expression is involved in development of acquired EGFR-TKI resistance. However, we noted that inhibition of eIF4F with the aforementioned approaches did not fully restore the sensitivity of HCC827/ER cell to erlotinib. Thus, we suggest that elevated eIF4E alone may not be sufficient to confer cell full resistance to EGFR-TKIs although it does contribute to development of acquired EGFR-TKI resistance.
One of known mechanisms underlying acquired EGFR-TKI resistance is c-Met amplification.13 Here, we showed that HCC827/ER cells possessed elevated expression of c-Met (Fig. S2). Importantly, we found that inhibition of eIF4F cap complex with either eIF4E or eIF4G siRNA or the small molecule 4EGI-1 reduced c-Met protein levels (Fig. 7), indicating that elevated eIF4E and cap-dependent cap initiation regulates c-Met expression. Thus it is likely that elevated eIF4E contributes to development of acquired EGFR-TKI resistance through facilitating c-Met expression in addition to gene amplification. Our findings warrant further investigation in this direction.
Since we detected increased levels of eIF4E mRNA and transcriptional activity in HCC827/ER cells (Fig. 4), it appears that elevated eIF4E expression in EGFR-TKI-resistant cells occurs at the transcriptional level. Thus, our current findings warrant further study to fully elucidate the mechanisms by which eIF4E expression is upregulated in EGFR-TKI-resistant cells.
In summary, the current study has demonstrated that eIF4E expression is elevated in human NSCLCs. The elevated eIF4e expression is associated with positive regulation of cell proliferation and invasion of NSCLC cells and contributes to development of acquired resistance to EGFR-TKIs.
Materials and Methods
Reagents
Erlotinib and gefitinib were purchased from LC Laboratories. 4EGI-1 was purchased from EMD Chemicals, Inc. or Calbiochem. They were dissolved in DMSO at the concentration of 20 or 100 mM, and aliquots were stored at -80°C. Stock solutions were diluted to the appropriate concentrations with growth medium immediately before use. eIF4E, eIF4G, Mcl-1, survivin, PARP, p-EGFR (Tyr1068), EGFR, p-Erb3 (Tyr1289), p-Met (Tyr1234/1235), Met and Akt antibodies were purchased from Cell Signaling Technology, Inc. Rabbit anti-HIF-1α and p-Akt (S473) were purchased from Epitomics. Mouse monoclonal anti-c-Myc and rabbit anti-Erb3 antibodies were purchased from Santa Cruz Biotechnology, Inc. Mouse monoclonal cyclin D1 antibody (clone DCS-6) was purchased from Dako. Mouse monoclonal anti-actin and anti-tubulin antibodies were purchased from Sigma Chemical Co. 7-methyl GTP (m7GTP)-sepharose 4B was purchased from GE Healthcare Biosciences.
Cell lines and cell culture
H1975, HCC827 and H1650 were purchased from the American Type Culture Collection. The SV40-immortalized normal human bronchial epithelial (NHBE) cell line, BEAS-2B,19 and other NSCLC cell lines were generously provided by Dr. R. Lotan (MD Anderson Cancer Center). The Cdk4/hTERT-immortalized NHBE cell line, HBEC3KT,20 was obtained from Dr. J. Minna (University of Texas Southwestern Medical Center). PLA-801C and PLA-801D cell lines21 were obtained from Dr. Y. L. Lu (Institute of Basic Medical Science, Academy of Military Medical Sciences). PC-9 and gefitinib-resistant PC-9 (PC-9/GR) cell lines were provided by Dr. P. A. Jänne (Dana Farber Cancer Institute). All NSCLC cell lines were cultured with RPMI 1640 containing 5% fetal bovine serum and HBEC3KT and BEAS-2B cells were cultured with K-SFM medium containing 50 μg/mL bovine pituitary extract and 5 ng/mL EGF (Life Technologies) at 37°C in a humidified atmosphere of 5% CO2 and 95% air.
Establishment of an erlotinib-resistant NSCLC cell line
The erlotinib-resistant HCC827 cell line (HCC827/ER) was established by exposing HCC827 cells to 3.5 μM erlotinib for 2 mo followed with one more month of exposure to 7.5 μM with 5 d drug on and 5 d drug off cycle. The resistant cell population was then routinely cultured with medium containing 1 μM erlotinib. HCC827/ER cell is also cross-resistant to gefitinib (Fig. S2A). Compared with HCC827 cells, HCC827/ER cells have downregulated EGFR/p-EGFR and elevated levels of Met/p-Met, Akt/p-Akt and ERKs/p-ERKs, which are resistant to modulation by erlotinib (Fig. S2B). The resistance remains unchanged after withdrawal of erlotinib from culture medium for 6 mo, suggesting an irreversible phenotype (Fig. S2C).
Growth inhibition assay
Cell number in monolayer culture in 96-well plates was estimated by the sulforhodamine B (SRB) assay and the growth inhibition was calculated as previously described.22 Combination index (CI) for drug interaction (e.g., synergy) was calculated using the CompuSyn software (ComboSyn, Inc.).
Colony formation assays
Colony formation assay on plastic surface was conducted in 6-well plate (approximately 600/well) as described previously.23 To perform colony formation assay on soft agar, 0.5% bottom agar and 0.35% top agar were prepared and used for each 35 mm Petri dish. The top agar contained 5,000 cells. The dishes were cultured for 14 d and then stained with 0.005% crystal violet for 30 min. The colonies were then counted under a microscope.
Cell invasion assay
Cell invasion assay were performed using BD BioCoat™ Matrigel™ Invasion (BD Biosciences) coated with BD Matrigel Basement Membrane Matrix in a working concentration of 350 µg/ml. For each coated chamber, 25,000 cells in 500 μl of serum-free medium were seeded in the cell insert and pre-cultured for 8 h. After that, 750 μl complete medium supplemented with 10% fetal bovine serum was added to each lower chamber and culture for another 36 h. The invasive cells on the bottoms of the membranes were then counted after staining with Fisher Hema 3 Manual Staining System (Fisher Scientific) and normalized by live cells (determined by trypan blue) cultured at the same condition.
Western blot analysis
The procedures for preparation of whole-cell protein lysates and for western blotting were the same as described previously.24
IHC
Human lung cancer TMA was purchased from Imgenex (IMH-358). The TMA was stained with IHC using the EnVisionTM + Dual Link System-HRP Kit (Dako) following the protocol. The rabbit polyclonal antibody against eIF4E (9742) was purchased from Cell signaling and used at 1:100 dilutions. eIF4E staining was scored as negative (< 10% staining) and positive staining (≥ 10% staining), respectively.
m7GTP pull-down for analysis of eIF4F complex
eIF4F complex in cell extracts was detected using affinity chromatography m7GTP-sepharose as described previously.25
Small interfering RNA (siRNA)-mediated eIF4E and eIF4G knockdown
The siRNA duplexes for non-silencing control and eIF4E and their transfections were described previously.25 eIF4G siRNA (sc-35286) was purchased from Santa Cruz Biotechnology.
Detection of eIF4E mRNA
The forward primer 5′-GGTTGCTAACCCAGAACAC-3′ and reverse primer 5′-CACTTCGTCTCTGCTGTTTG-3′ were used for the RT-PCR to detect the eIF4E mRNA level. Forward primer 5′-GAAACTACCTTCAACTCCATC-3′ and reverse primer 5′-CTAGAAGCATTTGCGGTGGACGATGGAGGGGCC-5′ were used to detect actin mRNA level as an internal control.
Construction of eIF4E reporter plasmid and luciferase activity assay
To make an eIF4E reporter construct, RT-PCR was used to amplify eIF4E promoter region (-1507 to +72) from genomic DNA extracted from H157 cells using the following primers: forward 5′-GCGGGTACCGCACAGGCAGCCTGCATACA-3′ and reverse 5′-CCCAAGCTTTCTCCTCTTCTGTAGTCGGGGG-3′. The final PCR product was then cloned into pGL3-basic luciferase reporter vector (Promega Inc.) through KpnI and Hind III cloning sites to generate pGL3-eIF4E-luc construct. The transient transfection and subsequent luciferase assay have been described previously.26
Statistical analyses
The statistical significance between two groups was analyzed with two-sided unpaired Student t-tests or with Fisher’s exact test. All of these analyses were done by use of Graphpad InStat 3 software (GraphPad Software). Results were considered to be statistically significant at p < 0.05.
Supplementary Material
Acknowledgment
This study was supported by the Georgia Cancer Coalition Distinguished Cancer Scholar award (to S-Y.S.), NIH R01 CA118450 (S-Y.S.) and P01 CA116676 (Project 1 to F.R.K. and S-Y.S.), DOD BATTLE award W81XWH-06–1-0303 (Project 4 to F.R.K. and S-Y.S.) and Emory Winship Cancer Institute Cancer Cell Biology seed grant. S.S.R., T.K.O., F.R.K. and S-Y.S. are Georgia Cancer Coalition Distinguished Cancer Scholars.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/cbt/article/18923
References
- 1.De Benedetti A, Graff JR. eIF-4E expression and its role in malignancies and metastases. Oncogene. 2004;23:3189–99. doi: 10.1038/sj.onc.1207545. [DOI] [PubMed] [Google Scholar]
- 2.Goodfellow IG, Roberts LO. Eukaryotic initiation factor 4E. Int J Biochem Cell Biol. 2008;40:2675–80. doi: 10.1016/j.biocel.2007.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thumma SC, Kratzke RA. Translational control: a target for cancer therapy. Cancer Lett. 2007;258:1–8. doi: 10.1016/j.canlet.2007.08.022. [DOI] [PubMed] [Google Scholar]
- 4.Graff JR, Zimmer SG. Translational control and metastatic progression: enhanced activity of the mRNA cap-binding protein eIF-4E selectively enhances translation of metastasis-related mRNAs. Clin Exp Metastasis. 2003;20:265–73. doi: 10.1023/A:1022943419011. [DOI] [PubMed] [Google Scholar]
- 5.Graff JR, Konicek BW, Carter JH, Marcusson EG. Targeting the eukaryotic translation initiation factor 4E for cancer therapy. Cancer Res. 2008;68:631–4. doi: 10.1158/0008-5472.CAN-07-5635. [DOI] [PubMed] [Google Scholar]
- 6.Clemens MJ. Targets and mechanisms for the regulation of translation in malignant transformation. Oncogene. 2004;23:3180–8. doi: 10.1038/sj.onc.1207544. [DOI] [PubMed] [Google Scholar]
- 7.Rosenwald IB, Hutzler MJ, Wang S, Savas L, Fraire AE. Expression of eukaryotic translation initiation factors 4E and 2alpha is increased frequently in bronchioloalveolar but not in squamous cell carcinomas of the lung. Cancer. 2001;92:2164–71. doi: 10.1002/1097-0142(20011015)92:8<2164::AID-CNCR1559>3.0.CO;2-A. [DOI] [PubMed] [Google Scholar]
- 8.Seki N, Takasu T, Mandai K, Nakata M, Saeki H, Heike Y, et al. Expression of eukaryotic initiation factor 4E in atypical adenomatous hyperplasia and adenocarcinoma of the human peripheral lung. Clin Cancer Res. 2002;8:3046–53. [PubMed] [Google Scholar]
- 9.Yang SX, Hewitt SM, Steinberg SM, Liewehr DJ, Swain SM. Expression levels of eIF4E, VEGF, and cyclin D1, and correlation of eIF4E with VEGF and cyclin D1 in multi-tumor tissue microarray. Oncol Rep. 2007;17:281–7. [PubMed] [Google Scholar]
- 10.Wang R, Geng J, Wang JH, Chu XY, Geng HC, Chen LB. Overexpression of eukaryotic initiation factor 4E (eIF4E) and its clinical significance in lung adenocarcinoma. Lung Cancer. 2009;66:237–44. doi: 10.1016/j.lungcan.2009.02.001. [DOI] [PubMed] [Google Scholar]
- 11.Seki N, Takasu T, Sawada S, Nakata M, Nishimura R, Segawa Y, et al. Prognostic significance of expression of eukaryotic initiation factor 4E and 4E binding protein 1 in patients with pathological stage I invasive lung adenocarcinoma. Lung Cancer. 2010;70:329–34. doi: 10.1016/j.lungcan.2010.03.006. [DOI] [PubMed] [Google Scholar]
- 12.Khoury T, Alrawi S, Ramnath N, Li Q, Grimm M, Black J, et al. Eukaryotic initiation factor-4E and cyclin D1 expression associated with patient survival in lung cancer. Clin Lung Cancer. 2009;10:58–66. doi: 10.3816/CLC.2009.n.009. [DOI] [PubMed] [Google Scholar]
- 13.Engelman JA, Janne PA. Mechanisms of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non-small cell lung cancer. Clin Cancer Res. 2008;14:2895–9. doi: 10.1158/1078-0432.CCR-07-2248. [DOI] [PubMed] [Google Scholar]
- 14.Wendel HG, De Stanchina E, Fridman JS, Malina A, Ray S, Kogan S, et al. Survival signalling by Akt and eIF4E in oncogenesis and cancer therapy. Nature. 2004;428:332–7. doi: 10.1038/nature02369. [DOI] [PubMed] [Google Scholar]
- 15.Andrieu C, Taieb D, Baylot V, Ettinger S, Soubeyran P, De-Thonel A, et al. Heat shock protein 27 confers resistance to androgen ablation and chemotherapy in prostate cancer cells through eIF4E. Oncogene. 2010;29:1883–96. doi: 10.1038/onc.2009.479. [DOI] [PubMed] [Google Scholar]
- 16.Moerke NJ, Aktas H, Chen H, Cantel S, Reibarkh MY, Fahmy A, et al. Small-molecule inhibition of the interaction between the translation initiation factors eIF4E and eIF4G. Cell. 2007;128:257–67. doi: 10.1016/j.cell.2006.11.046. [DOI] [PubMed] [Google Scholar]
- 17.Graff JR, Konicek BW, Vincent TM, Lynch RL, Monteith D, Weir SN, et al. Therapeutic suppression of translation initiation factor eIF4E expression reduces tumor growth without toxicity. J Clin Invest. 2007;117:2638–48. doi: 10.1172/JCI32044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Graff JR, Boghaert ER, De Benedetti A, Tudor DL, Zimmer CC, Chan SK, et al. Reduction of translation initiation factor 4E decreases the malignancy of ras-transformed cloned rat embryo fibroblasts. Int J Cancer. 1995;60:255–63. doi: 10.1002/ijc.2910600221. [DOI] [PubMed] [Google Scholar]
- 19.Sun SY, Kurie JM, Yue P, Dawson MI, Shroot B, Chandraratna RA, et al. Differential responses of normal, premalignant, and malignant human bronchial epithelial cells to receptor-selective retinoids. Clin Cancer Res. 1999;5:431–7. [PubMed] [Google Scholar]
- 20.Ramirez RD, Sheridan S, Girard L, Sato M, Kim Y, Pollack J, et al. Immortalization of human bronchial epithelial cells in the absence of viral oncoproteins. Cancer Res. 2004;64:9027–34. doi: 10.1158/0008-5472.CAN-04-3703. [DOI] [PubMed] [Google Scholar]
- 21.Zhang JQ, Wang Y, Wang T, Du ZY, Xu YJ, Lu YL. Differentially expressed genes in human giant-cell lung cancer lines with different metastatic potentials. Zhonghua Zhong Liu Za Zhi. 2004;26:590–3. [PubMed] [Google Scholar]
- 22.Sun SY, Yue P, Dawson MI, Shroot B, Michel S, Lamph WW, et al. Differential effects of synthetic nuclear retinoid receptor-selective retinoids on the growth of human non-small cell lung carcinoma cells. Cancer Res. 1997;57:4931–9. [PubMed] [Google Scholar]
- 23.Wang X, Hawk N, Yue P, Kauh J, Ramalingam SS, Fu H, et al. Overcoming mTOR inhibition-induced paradoxical activation of survival signaling pathways enhances mTOR inhibitors' anticancer efficacy. Cancer Biol Ther. 2008;7:1952–8. doi: 10.4161/cbt.7.12.6944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sun SY, Rosenberg LM, Wang X, Zhou Z, Yue P, Fu H, et al. Activation of Akt and eIF4E Survival Pathways by Rapamycin-Mediated Mammalian Target of Rapamycin Inhibition. Cancer Res. 2005;65:7052–8. doi: 10.1158/0008-5472.CAN-05-0917. [DOI] [PubMed] [Google Scholar]
- 25.Fan S, Li Y, Yue P, Khuri FR, Sun SY. The eIF4E/eIF4G interaction inhibitor 4EGI-1 augments TRAIL-mediated apoptosis through c-FLIP Down-regulation and DR5 induction independent of inhibition of cap-dependent protein translation. Neoplasia. 2010;12:346–56. doi: 10.1593/neo.10144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sun SY, Liu X, Zou W, Yue P, Marcus AI, Khuri FR. The Farnesyltransferase Inhibitor Lonafarnib Induces CCAAT/Enhancer-binding Protein Homologous Protein-dependent Expression of Death Receptor 5, Leading to Induction of Apoptosis in Human Cancer Cells. J Biol Chem. 2007;282:18800–9. doi: 10.1074/jbc.M611438200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.