

Abstract
The present studies were designed to determine whether the multi-kinase inhibitor sorafenib (Nexavar) interacted with histone deacetylase inhibitors to kill glioblastoma and medulloblastoma cells. In a dose-dependent fashion sorafenib lethality was enhanced in multiple genetically disparate primary human glioblastoma isolates by the HDAC inhibitor sodium valproate (Depakote). Drug exposure reduced phosphorylation of p70 S6K and of mTOR. Similar data to that with valproate were also obtained using the HDAC inhibitor vorinostat (Zolinza). Sorafenib and valproate also interacted to kill medulloblastoma and PNET cell lines. Treatment with sorafenib and HDAC inhibitors radio-sensitized both GBM and medulloblastoma cell lines. Knock down of death receptor (CD95) expression protected GBM cells from the drug combination, as did overexpression of c-FLIP-s, BCL-XL and dominant negative caspase 9. Knock down of PDGFRα recapitulated the effect of sorafenib in combination with HDAC inhibitors. Collectively, our data demonstrate that the combination of sorafenib and HDAC inhibitors kills through activation of the extrinsic pathway, and could represent a useful approach to treat CNS-derived tumors.
Keywords: HDAC inhibitor, Sorafenib, apoptosis, glioma
Introduction
Glioblastoma multiforme (GBM) is a fatal malignancy with patient survival measured in months. There is an unmet need for new therapies to treat this disease and prolong patient survival. Reasons for the lethality of GBM are diverse and include the highly invasive nature of these tumor cells resulting in incomplete resection of the disease upon initial presentation, the relative resistance of GBM cells to chemotherapies in the brain micro-environment and the existence of the blood-brain barrier with associated drug efflux pumps.1,2 The presence of these pumps reduces the achievable steady-state levels of drugs within the tumor. GBM cells are also well known to exhibit gains in function of some oncogenes, e.g., the truncated activated EGFR vIII and PDGFRα, as well as loss of expression of tumor suppressors, e.g., PTEN and p53, which collectively results in a further level of therapeutic resistance.3
Sorafenib is a multi-kinase inhibitor originally developed to inhibit RAF-1 in the MEK1/2-ERK1/2 pathway. Subsequently sorafenib was shown to inhibit class III receptor tyrosine kinases including members of the VEGFR and PDGFR families.4 Recent studies by this laboratory in GI tumor cells have argued that sorafenib can activate SRC non-receptor tyrosine kinases and that this effect plays a role in the activation of death receptors, resulting in tumor cell death.5,6 Hypothetically sorafenib should be a useful drug in GBM owing to its anti-VEGFR / vascularization effects and sorafenib has undergone a number of clinical trials in GBM combined with established and more novel agents though has not proven highly effective.7
Histone deacetylase inhibitors (HDACIs) are a class of drugs, as their name suggests, that inhibit the enzymes that de-acetylate histones.8 By regulating histone acetylation chromatin condensation is changed and the levels of transcription may be altered. Many other proteins, however, that are cytosolic are also acetylated and it is probable that the actions of HDACIs involve both the regulation of gene expression, e.g., FAS-ligand as well as regulating acetylation of other cytosolic proteins, e.g., HSP90.9 In our studies in GI tumor cells HDAC inhibitors were shown to increase the levels of FAS-ligand as well as its receptor CD95, both of which played a role in HDACI toxicity.5,6 Based on our in vitro and in vivo studies a phase I trial combining sorafenib and vorinostat has opened at VCU-MCVH in liver cancer patients.
The present studies were designed to determine in primary adult GBM cells and in pediatric medulloblastoma cells whether sorafenib interacted with clinically relevant HDAC inhibitors to cause GBM and medulloblastoma-induced cell death. We discovered that in multiple cell isolates the sorafenib + HDAC inhibitor treatment, in a dose-dependent and greater-than-additive fashion killed CNS tumor cells. Drug exposure also radiosensitized tumor cells. Cell killing was dependent on the extrinsic apoptosis pathway. Collectively our findings argue that, in addition to GI tumor cells, tumor cells of the CNS are also sensitive to sorafenib + HDAC inhibitor treatment and that this approach warrants further exploration in the clinic.
Results
Prior studies from this laboratory have shown that sorafenib and vorinostat interact to kill a range of GI and GU tumor cells.5,6,10 The present studies were performed to assess whether sorafenib and HDAC inhibitors cooperate to kill tumor cell types derived from the CNS. Glioblastoma has a very poor survival rate and new approaches to treat the disease are needed. The glioblastoma isolates used in our studies include cells expressing mutant active forms of the EGF receptor (GBM6, EGFR vIII; GBM12, mutant active full-length EGFR); lacking PTEN function (GBM14); expressing a mutant active PI3 kinase and PDGFRα (GBM5) or with low levels of growth factor receptors (GBM15).
Initial studies made use of the HDACI inhibitor sodium valproate (Depakote) that is used clinically as an anti-convulsive drug, and that can cross the blood-brain barrier.11 In a dose-dependent fashion sorafenib toxicity was enhanced by valproate in GBM6 cells (Fig. 1A). GBM12 cells were apparently more resistant to the drug combination than GBM6 cells (Fig. 1B). GBM15 and GBM14 were both sensitive to the drug combination (Fig. 1C, lower graph). These events correlated with reduced phosphorylation of mTOR and of p70 S6K (Fig. 1C, upper blot). The cells that were most sensitive to the sorafenib and valproate drug combination were GBM5 cells; cells that overexpress one of the reported growth factor receptor targets of sorafenib, PDGFRα (Fig. 1D). These findings, from short-term assays, were also reflected in long-term colony formation assays where cells were treated with the drug and then permitted to form colonies in drug free media (Fig. 1E).

Figure 1. Sorafenib and Sodium Valproate interact to kill GBM cells. (A) GBM6 cells were treated with vehicle or Sodium Valproate (1 mM). Thirty minutes later cells were treated with vehicle or sorafenib (3 μM; 6 μM). Cells were isolated 24 h and 48 h later. Viability was determined by trypan blue exclusion (n = 3, ± SEM) *p < 0.05 greater than corresponding vehicle control treated cells. (B) GBM12 cells were treated with vehicle or Sodium Valproate (1 mM). Thirty minutes later cells were treated with vehicle or sorafenib (3 μM; 6 μM). Cells were isolated 48 h later. Viability was determined by trypan blue exclusion (n = 3, ± SEM) *p < 0.05 greater than corresponding vehicle control treated cells. (C) GBM15 and GBM14 cells were treated with vehicle or Sodium Valproate (1 mM). Thirty minutes later cells were treated with vehicle or sorafenib (3 μM; 6 μM). Cells were isolated 48 h later. Viability was determined by trypan blue exclusion (n = 3, ± SEM) *p < 0.05 greater than corresponding vehicle control treated cells. Upper blot: GBM15 cells were treated with the indicated drugs and cells isolated 24 h later for determination of P-p70 S6K and P-mTOR levels. (D) GBM5 cells were treated with vehicle or Sodium Valproate (1 mM). Thirty minutes later cells were treated with vehicle or sorafenib (1 μM; 2 μM; 3 μM; 6 μM). Cells were isolated 24 h and 48 h later. Viability was determined by trypan blue exclusion (n = 3, ± SEM) *p < 0.05 greater than corresponding vehicle control treated cells. (E) GBM5 and GBM6 cells plated as single cells were treated with vehicle or Sodium Valproate (1 mM). Thirty minutes later cells were treated with vehicle or sorafenib (2 μM–6 μM, as shown). The media was changed 48 h later to drug free media and colonies permitted to form for the next 14 d. (n = 3, ± SEM) *p < 0.05 greater killing corresponding vehicle control treated cells.
We next determined whether other clinically relevant HDAC inhibitors, specifically vorinostat (Zolinza), also enhanced sorafenib toxicity in GBM cells. In a similar fashion to our findings using sodium valproate, vorinostat enhanced sorafenib lethality in GBM5 and GBM14 cells (Fig. 2A and B). As with valproate, the sorafenib and vorinostat combination more weakly killed GBM12 cells (Fig. 2C).
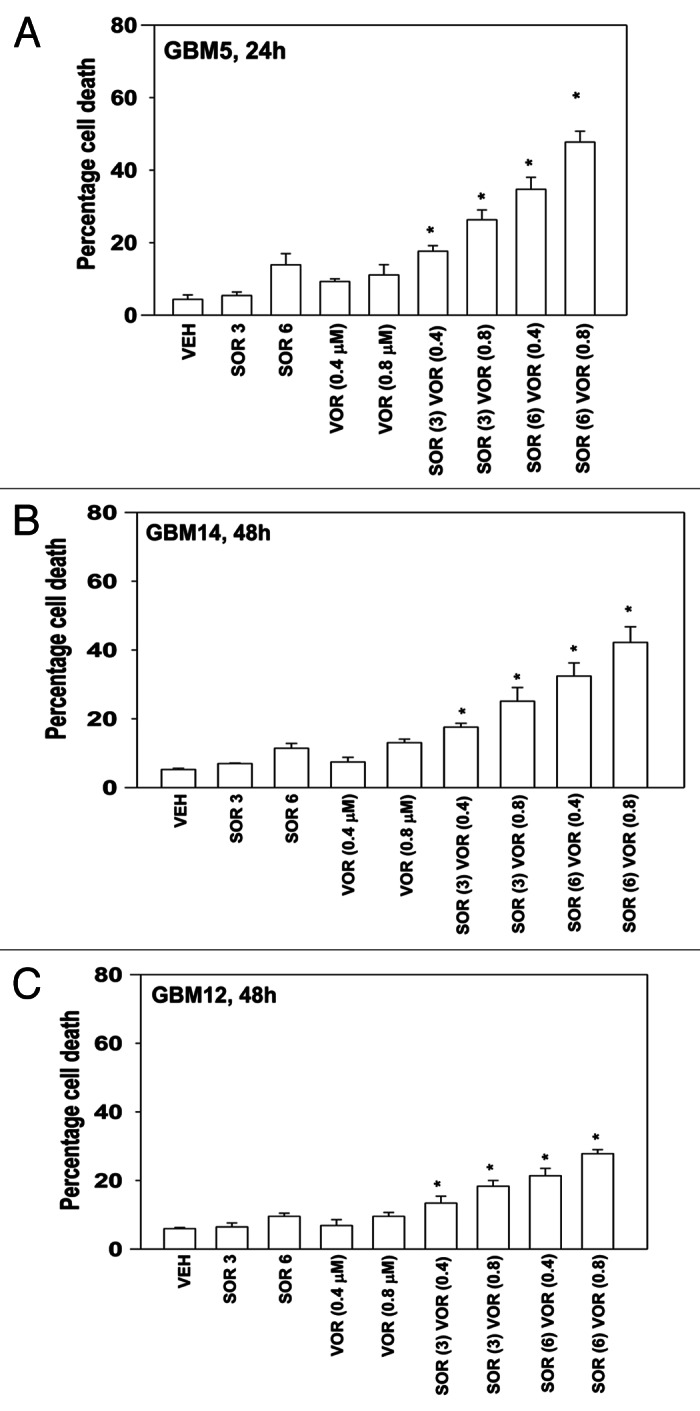
Figure 2. Sorafenib and Vorinostat interact to kill GBM cells. (A) GBM5 cells were treated with vehicle or Vorinostat (0.4 μM; 0.8 μM). Thirty minutes later cells were treated with vehicle or sorafenib (3 μM; 6 μM). Cells were isolated 24 h later. Viability was determined by trypan blue exclusion (n = 3, ± SEM) *p < 0.05 greater than corresponding vehicle control treated cells. (B) GBM14 cells were treated with vehicle or Vorinostat (0.4 μM; 0.8 μM). Thirty minutes later cells were treated with vehicle or sorafenib (3 μM; 6 μM). Cells were isolated 24 h later. Viability was determined by trypan blue exclusion (n = 3, ± SEM) *p < 0.05 greater than corresponding vehicle control treated cells. (C) GBM12 cells were treated with vehicle or Vorinostat (0.4 μM; 0.8 μM). Thirty minutes later cells were treated with vehicle or sorafenib (3 μM; 6 μM). Cells were isolated 24 h later. Viability was determined by trypan blue exclusion (n = 3, ± SEM) *p < 0.05 greater than corresponding vehicle control treated cells.
Medulloblastoma is a form of infra-tentorial primitive neuro-ectodermal tumor that develops in the brain stem of children, close to the cerebellum.12 In contrast, what is called a primitive neuroectodermal tumor (PNET) is a pediatric supra-tentorial neural crest tumor. With the use of high dose multi-modal chemotherapy and radio-therapy response rates of 60–80% have been observed in children for both tumor types, although this also results in significant long-term negative sequelae for the affected child. Clearly, new ways of treating these diseases are required. In contrast to our findings in adult GBM cells, pediatric medulloblastoma and PNET cells were appeared to be relatively more sensitive to sorafenib as a single agent (Fig. 3A–C). However, sorafenib treated medulloblastoma/PNET cells still interacted with valproate, or with vorinostat, as effectively as they did GBM cells to cause enhanced tumor cell death. (Fig. 3A–C, data not shown).
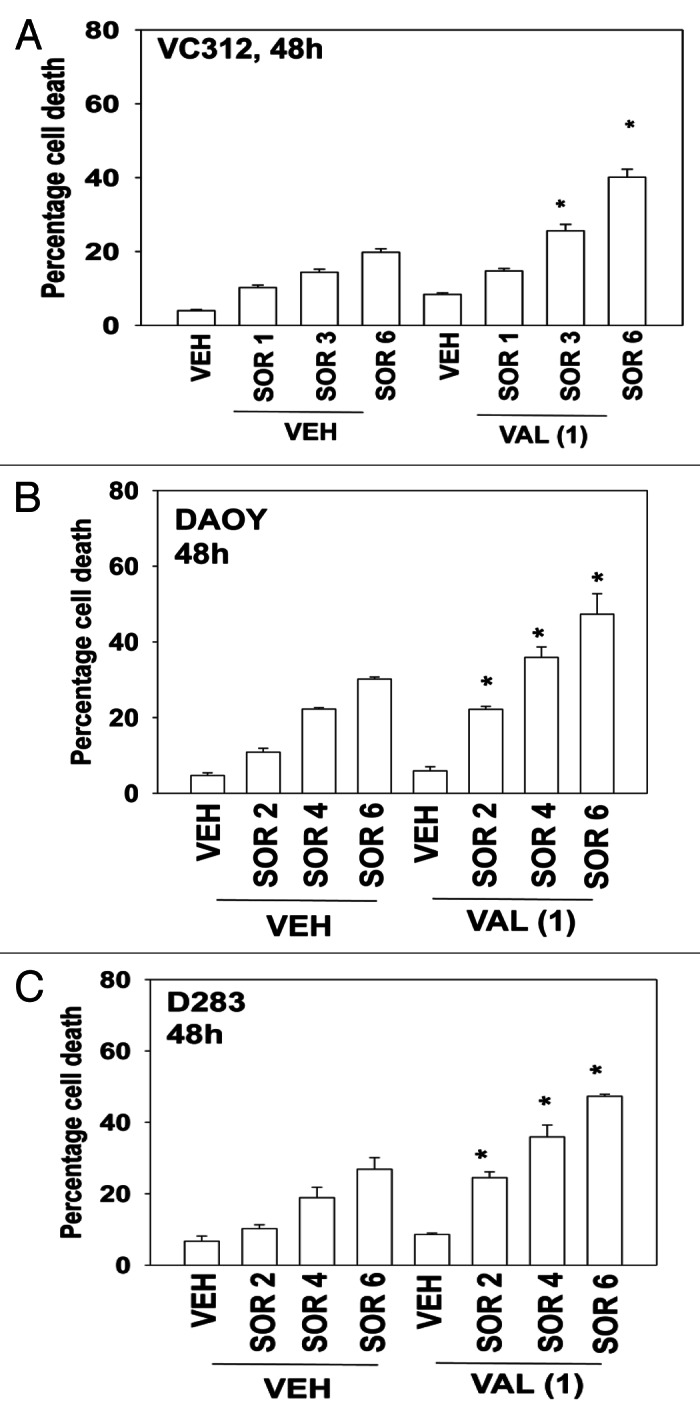
Figure 3. Sorafenib and Vorinostat interact to kill medulloblastoma and PNET cells. (A) VC312 cells were treated with vehicle or Sodium Valproate (1 mM). Thirty minutes later cells were treated with vehicle or sorafenib (1 μM; 3 μM; 6 μM). Cells were isolated 48 h later. Viability was determined by trypan blue exclusion (n = 3, ± SEM) *p < 0.05 greater than corresponding vehicle control treated cells. (B) DAOY cells were treated with vehicle or Sodium Valproate (1 mM). Thirty minutes later cells were treated with vehicle or sorafenib (2 μM; 4 μM; 6 μM). Cells were isolated 48 h later. Viability was determined by trypan blue exclusion (n = 3, ± SEM) *p < 0.05 greater than corresponding vehicle control treated cells. (C) D283 cells were treated with vehicle or Sodium Valproate (1 mM). Thirty minutes later cells were treated with vehicle or sorafenib (2 μM; 4 μM; 6 μM). Cells were isolated 48 h later. Viability was determined by trypan blue exclusion (n = 3, ± SEM) *p < 0.05 greater than corresponding vehicle control treated cells.
Radiotherapy is a standard of care approach for treating GBM and medulloblastoma and we next determined whether sorafenib + valproate treatment radiosensitized GBM cells. Using a relatively low dose of sorafenib + valproate, GBM6 cells were radiosensitized in short-term cell viability assays (Fig. 4A). Similar data were also obtained in GBM5 cells and in DAOY cells (Fig. 4B and C). In long-term colony formation assays, where cells were transiently exposed to drugs, sorafenib + valproate treatment also enhanced the toxicity of radiation exposure (Fig. 4D and E).

Figure 4. Sorafenib and HDAC inhibitors radiosensitize GBM cells. (A) GBM6 cells were treated with vehicle or Sodium Valproate (1 mM). Thirty minutes later cells were treated with vehicle or sorafenib (1 μM). Thirty minutes later cells were exposed to ionizing radiation (0–6 Gy). Cells were isolated 48 h later. Viability was determined by trypan blue exclusion (n = 3, ± SEM) *p < 0.05 greater than corresponding vehicle control treated cells. (B) GBM5 cells were treated with vehicle or Sodium Valproate (1 mM). Thirty minutes later cells were treated with vehicle or sorafenib (1 μM). Thirty minutes later cells were exposed to ionizing radiation (0–6 Gy). Cells were isolated 48 h later. Viability was determined by trypan blue exclusion (n = 3, ± SEM) *p < 0.05 greater than corresponding vehicle control treated cells. (C) DAOY cells were treated with vehicle or Sodium Valproate (1 mM). Thirty minutes later cells were treated with vehicle or sorafenib (1 μM). Thirty minutes later cells were exposed to ionizing radiation (0–6 Gy). Cells were isolated 48 h later. Viability was determined by trypan blue exclusion (n = 3, ± SEM) *p < 0.05 greater than corresponding vehicle control treated cells. (D) GBM5 and GBM6 cells plated as single cells were treated with vehicle or Sodium Valproate (1 mM). Thirty minutes later cells were treated with vehicle or sorafenib (1 μM). Thirty minutes later cells were irradiated. The media was changed 48 h later to drug free media and colonies permitted to form for the next 14 d (n = 3, ± SEM). (E) DAOY and VC312 cells plated as single cells were treated with vehicle or Sodium Valproate (1 mM). Thirty minutes later cells were treated with vehicle or sorafenib (1 μM). Thirty minutes later cells were irradiated. The media was changed 48 h later to drug free media and colonies permitted to form for the next 14 d (n = 3, ± SEM).
We next defined the molecular mechanisms by which sorafenib and HDAC inhibitors interacted in CNS tumor cells to cause cell death. Knock down of the death receptor CD95 protected cells from sorafenib and valproate treatment (Fig. 5A). Molecular inhibition of the extrinsic pathway, by overexpressing c-FLIP-s also reduced drug-induced killing (Fig. 5B). Overexpression of BCL-XL or expression of dominant caspase 9 were as effective as c-FLIP-s in suppressing drug lethality. Overexpression of c-FLIP-s also suppressed, but did not abolish, the toxic interaction between sorafenib + valproate and radiation (Fig. 5C and D). GBM5 cells overexpress the PDGFRα receptor, a receptor that is a known target for the multi-kinase inhibitor sorafenib. Hence we determined whether this receptor was a major target for sorafenib in our system. Knock down of PDGFRα enhanced the toxicity of both valproate and vorinostat in GBM cells in a dose-dependent fashion (Fig. 5E and F).

Figure 5. Death receptor signaling is essential for sorafenib and valproate toxicity. (A) GBM5 and GBM6 cells were transfected with either scrambled control (siSCR) or an siRNA to knock down CD95 expression. Twenty-four h after transfection cells were treated with vehicle or valproate (1 mM) and sorafenib (3 μM). Cells were isolated 48 h later. Viability was determined by trypan blue exclusion (n = 3, ± SEM) #p < 0.05 less than corresponding vehicle control treated cells. (B) GBM5 cells were infected with the indicated recombinant adenoviruses (50 min.o.i.). Twenty-four h after infection as shown portions of cells were treated with the pan-caspase inhibitor zVAD (25 μM). Cells were isolated 48 h later. Viability was determined by trypan blue exclusion (n = 3, ± SEM). (C and D) GBM6 (C) and GBM5 (D) cells were infected with an empty vector virus or a virus to express c-FLIP-s. Twenty-four h after infection cells were treated with vehicle or valproate (1 mM) and sorafenib (3 μM). Thirty minutes later cells were irradiated (4 Gy). Cells were isolated 48 h later. Viability was determined by trypan blue exclusion (n = 3, ± SEM). (E and F) GBM5 cells were transfected with scrambled siRNA (siSCR) or an siRNA to knock down PDGFRα. Twenty-four h after transfection cells were treated with Sodium Valproate or Vorinostat. Cells were isolated 48 h later. Viability was determined by trypan blue exclusion (n = 3, ± SEM).
Discussion
The present studies were to determine whether the multi-kinase inhibitor sorafenib interacted with HDAC inhibitors to kill a broad range of CNS tumor cell types. Based on testing in five primary human GBM cell isolates and three established medulloblastoma/PNET cell lines we discovered that sorafenib and multiple HDAC inhibitors interacted in a greater than additive fashion to kill tumor cells. This occurred in tumor cells overexpressing oncogenes, e.g., EGFR vIII or lacking expression of tumor suppressor genes, e.g., PTEN. The mechanism of drug-induced killing required death receptor (CD95) function and inhibition of the CD95 intracellular effector caspase 8 also suppressed drug combination lethality. That both overexpression of BCL-XL and expression of dominant negative caspase 9 suppressed killing argues that death receptor signaling required mitochondrial dysfunction and the actions of caspase 9, rather than caspase 8 directly cleaving and activating pro-caspase 3.
Radiotherapy is a mainstay of therapeutic intervention in both GBM and medulloblastoma and additional studies indicated that GBM and medulloblastoma cells were radiosensitized in short-term viability and long-term colony formation assays by the drug combination. The toxic interaction between radiation and sorafenib + HDAC inhibitor treatment also was significantly reduced by overexpression of c-FLIP-s, arguing that death receptor signaling was essential for the radiosensitization effect. These findings also argue that other death receptors, such as TRAIL, are not mechanistically involved in tumor cell killing.
Sorafenib has a number of cellular targets. The drug was originally designed to inhibit RAF family kinases, particularly RAF-1, that would in turn inhibit the ERK1/2 pathway. Data from multiple laboratories has shown that in addition to inhibiting RAF-1, sorafenib also inhibits a number of class III receptor tyrosine kinases. In our prior studies in GI and GU tumor cells we had noted that inhibition of the PDGFRβ receptor caused activation of SRK kinases that promoted death receptor-induced tumor cell killing.5,6 In the present studies in GBM cells we found that knock down of PDGFRα enhanced HDAC inhibitor lethality, in agreement with PDGFRα being a target of sorafenib in GBM cells. Other validated sorafenib targets include Flt3 and other members of the VEGF receptor family. Such receptors are expressed in GBM tumor endothelial cells at generally high levels in concordance with GBM tumors being highly vascularized. Thus it is possible in vivo that not only will sorafenib and HDAC inhibitor treatment kill tumor cells directly but may also have collateral anti-endothelial cell effects.
HDAC inhibitors have pleiotropic targets based on their actions at altering protein acetylation. Although histones are the most recognized targets for these agents other proteins, for example HSP90, are also regulated by acetylation. We have found in GI and GU tumor cells that HDAC inhibitors facilitate the generation of reactive oxygen species that interact with sorafenib to promote tumor cell killing.5,6 However, the precise target(s) by which HDAC inhibitors generate ROS and interact with sorafenib are at present unclear. The chemically different HDAC inhibitors chosen for the present studies, sodium valproate and vorinostat, can both penetrate the blood brain barrier.12,13 Sorafenib can also cross the blood brain barrier.14
Glioblastoma is a malignancy with a highly invasive phenotype, however, established commercially available GBM cell lines do not generally exhibit the diffuse invasive nature of the disease in a patient. Our studies have made use of GBM isolates that are passaged in mice and retain their invasive characteristics. As our data demonstrates in multiple invasive cell isolates that sorafenib and HDAC inhibitors interact this, together with the use of drugs that can cross the blood brain barrier, suggests this drug combination could have utility in an in vivo/patient setting. And, based on the data in this manuscript, by summer 2012 a Phase I trial combining sorafenib and valproate for recurrent GBM patients will open at VCU-MCVH.
Materials and Methods
Materials
Phospho-/total-antibodies, were purchased from Cell Signaling Technologies. All the secondary antibodies were purchased from Santa Cruz Biotechnology. Commercially available validated short hairpin RNA molecules to knock down RNA/protein levels were from Qiagen. Primary human GBM cells and information on the genetic background of such cells were very kindly supplied for our use by Dr. C. David James, University of California San Francisco, CA. Medulloblastoma DAOY and D283 cells were obtained from the ATCC. VC312 PNET cells were obtained with informed consent from a patient treated at VCU Medical Center. Other reagents and techniques were as described in refs 5, 6 and 10.
Methods
Culture and in vitro exposure of cells to drugs
All established cell lines were cultured at 37°C 5% (v/v) CO2 in vitro using RPMI supplemented with 5% (v/v) fetal calf serum and 10% (v/v) non-essential amino acids. Primary human glioma cells were cultured in 2% (v/v) fetal calf serum to prevent growth of contaminating rodent fibroblasts during in vitro analyses. For short-term cell killing assays and immunoblotting studies, cells were plated at a density of 3 × 103 per cm2 (~2 × 105 cells per well of a 12 well plate) and 48 h after plating treated with various drugs, as indicated. In vitro OSU-03012 treatment was from a 100 mM stock solution of each drug and the maximal concentration of Vehicle (DMSO) in media was 0.02% (v/v). Cells were not cultured in reduced serum media during any study in this manuscript.
In vitro cell treatments, microscopy, SDS-PAGE and western blot analysis
For in vitro analyses of short-term cell death effects, cells were treated with Vehicle/sorafenib/Na Valproate/vorinostat for the indicated times in the Figure legends. For apoptosis assays where indicated, cells were pre-treated with vehicle (VEH, DMSO), zVAD (50 μM); cells were isolated at the indicated times, and subjected to trypan blue cell viability assay by counting in a light microscope.
For SDS PAGE and immunoblotting, cells were plated and treated with drugs at the indicated concentrations and after the indicated time of treatment, lysed in whole-cell lysis buffer (0.5 M TRIS-HCl, pH 6.8, 2% SDS, 10% glycerol, 1% β-mercaptoethanol, 0.02% bromophenol blue), and the samples were boiled for 30 min. The boiled samples were loaded onto 10–14% SDS-PAGE and electrophoresis was run overnight. Proteins were electrophoretically transferred onto 0.22 μm nitrocellulose, and immunoblotted with various primary antibodies against different proteins. All immunoblots were visualized using fluorescent secondary antibodies and a Li-Cor Odyssey Infrared imaging machine.
Infection of cells with recombinant adenoviruses
Cells were plated at 3 × 103 per cm2 in each well of a 12 well, 6 well or 60 mm plate. After plating (24 h), cells were infected (at a multiplicity of infection of 50) with a control empty vector virus (CMV) or the recombinant adenoviruses as indicated (Vector Biolabs). Twenty-four hours after infection cells were treated with the indicated concentrations of vehicle and/or drugs, and cell survival or changes in expression/protein phosphorylation determined 0–48 h after drug treatment by trypan blue assay and immunoblotting, respectively.
Transfection with siRNA
Cells were plated in 60 mm dishes from a fresh culture growing in log phase as described above, and 24 h after plating transfected. Prior to transfection, the medium was aspirated and 1 ml serum-free medium was added to each plate. For transfection, 10 nM of the annealed siRNA, the positive sense control doubled stranded siRNA targeting GAPDH or the negative control (a “scrambled” sequence with no significant homology to any known gene sequences from mouse, rat or human cell lines) were used (predominantly Qiagen; occasional alternate siRNA molecules were purchased from Ambion, Inc.). Ten nM siRNA (scrambled or experimental) was diluted in serum-free media. Four μl Hiperfect (Qiagen) was added to this mixture and the solution was mixed by pipetting up and down several times. This solution was incubated at room temp for 10 min, then added dropwise to each dish. The medium in each dish was swirled gently to mix, then incubated at 37°C for 2 h. One ml of 10% (v/v) serum-containing medium was added to each plate, and cells were incubated at 37°C for 24–48 h before re-plating (50 × 103 cells each) onto 12-well plates. Cells were allowed to attach overnight, then treated with drugs (0–48 h). Trypan blue exclusion assays and SDS PAGE/immunoblotting analyses were then performed at the indicated time points.
Data analysis
Comparison of the effects of various treatments was performed following ANOVA using the Student t-test. Differences with a p-value of < 0.05 were considered statistically significant. Experiments shown are the means of multiple individual points (± SEM).
Glossary
Abbreviations:
- ERK
extracellular regulated kinase
- MEK
mitogen activated extracellular regulated kinase
- JNK
c-Jun NH2-terminal kinase
- PI3K
phosphatidyl inositol 3 kinase
- MAPK
mitogen activated protein kinase
- ca
constitutively active
- dn
dominant negative
- EGFR
epidermal growth factor receptor
- CMV
empty vector plasmid or virus
- si
small interfering
- SCR
scrambled
- IP
immunoprecipitation
- Ad
adenovirus
- TUNEL
terminal deoxynucleotidyl transferase UTP nick end labeling
- VEH
vehicle
- HDACI
histone deacetylase inhibitor
- SOR
sorafenib
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Footnotes
Previously published online: www.landesbioscience.com/journals/cbt/article/19771
References
- 1.Squatrito M, Holland EC. DNA damage response and growth factor signaling pathways in gliomagenesis and therapeutic resistance. Cancer Res. 2011;71:5945–9. doi: 10.1158/0008-5472.CAN-11-1245. [DOI] [PubMed] [Google Scholar]
- 2.Buonerba C, Di Lorenzo G, Marinelli A, Federico P, Palmieri G, Imbimbo M, et al. A comprehensive outlook on intracerebral therapy of malignant gliomas. Crit Rev Oncol Hematol. 2011;80:54–68. doi: 10.1016/j.critrevonc.2010.09.001. [DOI] [PubMed] [Google Scholar]
- 3.Bonavia R, Inda MM, Cavenee WK, Furnari FB. Heterogeneity maintenance in glioblastoma: a social network. Cancer Res. 2011;71:4055–60. doi: 10.1158/0008-5472.CAN-11-0153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Liu KW, Hu B, Cheng SY. Platelet-derived growth factor receptor alpha in glioma: a bad seed. Chin J Cancer. 2011;30:590–602. doi: 10.5732/cjc.011.10236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Park MA, Reinehr R, Häussinger D, Voelkel-Johnson C, Ogretmen B, Yacoub A, et al. Sorafenib activates CD95 and promotes autophagy and cell death via Src family kinases in gastrointestinal tumor cells. Mol Cancer Ther. 2010;9:2220–31. doi: 10.1158/1535-7163.MCT-10-0274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Park MA, Mitchell C, Zhang G, Yacoub A, Allegood J, Häussinger D, et al. Vorinostat and sorafenib increase CD95 activation in gastrointestinal tumor cells through a Ca(2+)-de novo ceramide-PP2A-reactive oxygen species-dependent signaling pathway. Cancer Res. 2010;70:6313–24. doi: 10.1158/0008-5472.CAN-10-0999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Reardon DA, Vredenburgh JJ, Desjardins A, Peters K, Gururangan S, Sampson JH, et al. Effect of CYP3A-inducing anti-epileptics on sorafenib exposure: results of a phase II study of sorafenib plus daily temozolomide in adults with recurrent glioblastoma. J Neurooncol. 2011;101:57–66. doi: 10.1007/s11060-010-0217-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Venugopal B, Evans TR. Developing histone deacetylase inhibitors as anti-cancer therapeutics. Curr Med Chem. 2011;18:1658–71. doi: 10.2174/092986711795471284. [DOI] [PubMed] [Google Scholar]
- 9.Reikvam H, Ersvaer E, Bruserud O. Heat shock protein 90 - a potential target in the treatment of human acute myelogenous leukemia. Curr Cancer Drug Targets. 2009;9:761–76. doi: 10.2174/156800909789271486. [DOI] [PubMed] [Google Scholar]
- 10.Zhang G, Park MA, Mitchell C, Hamed H, Rahmani M, Martin AP, et al. Vorinostat and sorafenib synergistically kill tumor cells via FLIP suppression and CD95 activation. Clin Cancer Res. 2008;14:5385–99. doi: 10.1158/1078-0432.CCR-08-0469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bolaños JP, Medina JM. Effect of valproate on the metabolism of the central nervous system. Life Sci. 1997;60:1933–42. doi: 10.1016/S0024-3205(96)00687-X. [DOI] [PubMed] [Google Scholar]
- 12.Pfister SM, Korshunov A, Kool M, Hasselblatt M, Eberhart C, Taylor MD. Molecular diagnostics of CNS embryonal tumors. Acta Neuropathol. 2010;120:553–66. doi: 10.1007/s00401-010-0751-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hockly E, Richon VM, Woodman B, Smith DL, Zhou X, Rosa E, et al. Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor, ameliorates motor deficits in a mouse model of Huntington’s disease. Proc Natl Acad Sci U S A. 2003;100:2041–6. doi: 10.1073/pnas.0437870100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Agarwal S, Sane R, Ohlfest JR, Elmquist WF. The role of the breast cancer resistance protein (ABCG2) in the distribution of sorafenib to the brain. J Pharmacol Exp Ther. 2011;336:223–33. doi: 10.1124/jpet.110.175034. [DOI] [PMC free article] [PubMed] [Google Scholar]